Abstract
7-Bromo-4-chloro-1H-indazol-3-amine is a heterocyclic fragment used in the synthesis of Lenacapavir, a potent capsid inhibitor for the treatment of HIV-1 infections. In this manuscript, we describe a new approach to synthesizing 7-bromo-4-chloro-1H-indazol-3-amine from inexpensive 2,6-dichlorobenzonitrile. This synthetic method utilizes a two-step sequence including regioselective bromination and heterocycle formation with hydrazine to give the desired product in an overall isolated yield of 38–45%. The new protocol has been successfully demonstrated on hundred-gram scales without the need for column chromatography purification. This new synthesis provides a potential economical route to the large-scale production of this heterocyclic fragment of Lenacapavir.
1. Introduction
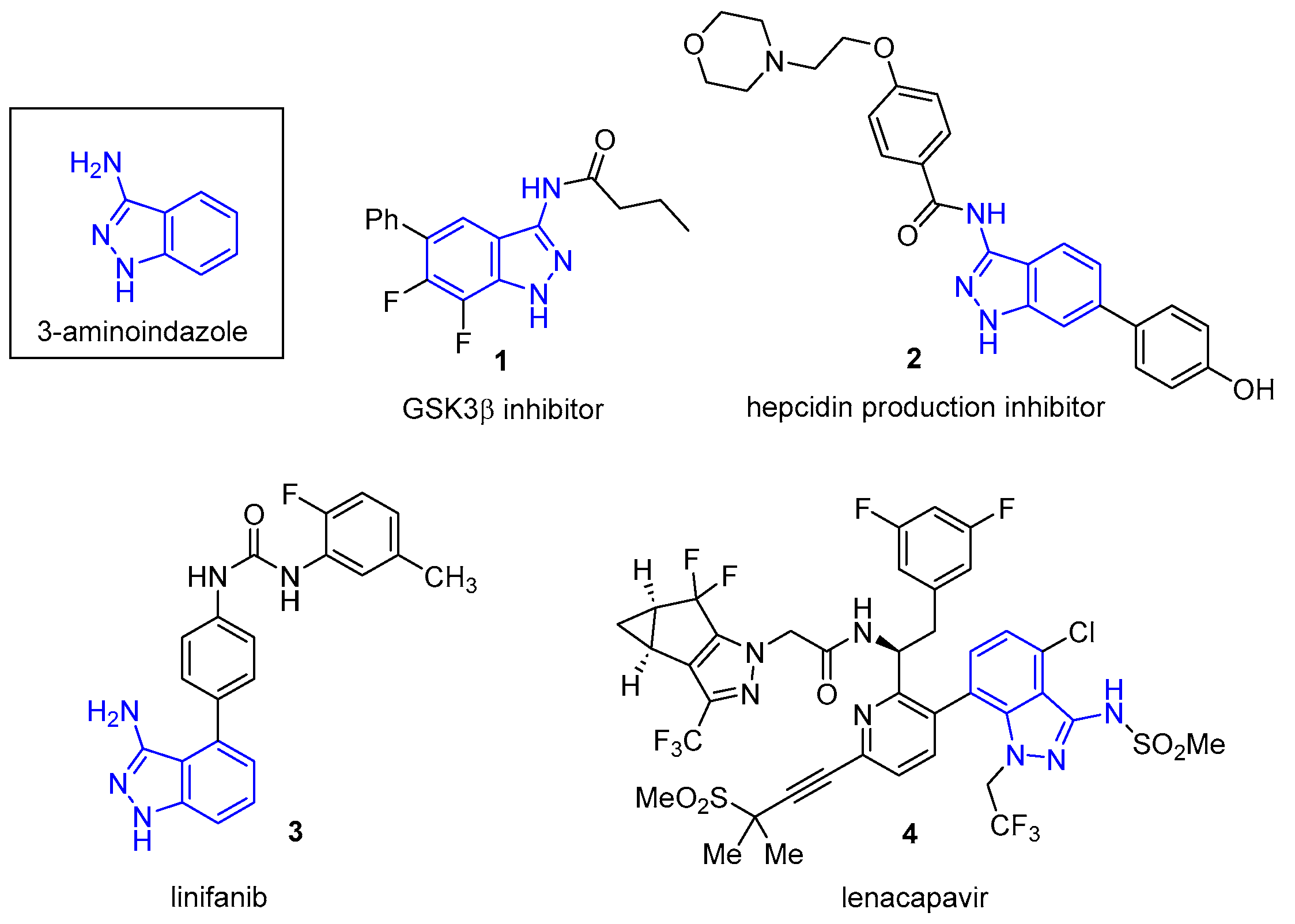
3-Aminoindazoles are a privileged class of heterocyclic structures common to many biologically active compounds. For example, this structure is found in the glycogen synthase 3β inhibitor 1, a substance having potential for the treatment of Alzheimer’s disease (Figure 1) [1]. The 3-aminoindazole (2) was developed as a possible treatment for iron deficiency [2], while linifanib (3) is a potent tyrosine kinase receptor inhibitor used to suppress tumor growth [3]. Recently, Gilead developed Lenacapavir (4), which exhibits high potency in the treatment of human immunodeficiency virus (HIV) [4,5]. Among the synthetic routes used to prepare 3-aminoindazoles, SNAr chemistry has often been utilized. For example, in the synthesis of Lenacapavir, 3-bromo-6-chloro-2-fluorobenzonitrile (5) reacts with hydrazine to provide the 3-aminoindazole (6) in a 90% yield (Equation (1)) [4].
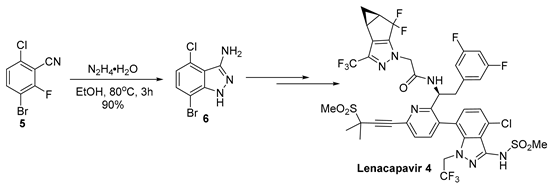
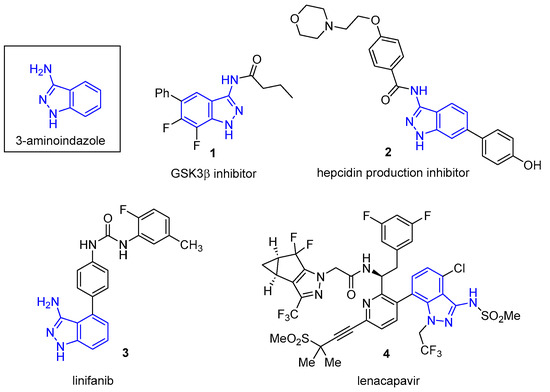
Figure 1.
Biologically active 3-aminoindazoles.
A similar transformation is achieved with 2,6-dichlorobenzonitrile (7) and hydrazine to form the 3-aminoindazole leading to linifanib (3) [6]. While 2-chloro and 2-fluorobenzonitriles are commonly used in this method, 2-bromobenzonitriles have provided 3-aminoindazoles through transition metal-catalyzed reactions [7]. The 3-aminoindazole scaffold has also been prepared from SNAr chemistry with hydrazine and a 2-nitrobenzonitrile [8]. Besides these approaches, synthetic routes to 3-aminoindazoles have utilized 2-fluorocarboxylic acids (via thioamides), ortho-haloaryl hydrazines in Pd-catalyzed cyclization, the C-N coupling of halogenated indazoles, intramolecular N-N coupling reactions, and others [9,10,11].
With the promising results from the clinical trials of Lenacapavir, it is vitally important that this substance can be prepared economically on a large scale [12]. While the 3-aminoindazole fragment of Lenacapavir has been prepared on a pilot plant scale (Equation (1)), the synthetic route utilizes a costly 3-bromo-6-chloro-2-fluorobenzonitrile (5) [13] and the heterocycle synthesis leads to the elimination of HF [6]. We envisioned that 3-bromo-2,6-dichlorobenzonitrile (8) might be used as precursor to synthesize 3-aminoindazole (6) using a similar SNAr cyclization with hydrazine, perhaps allowing for the use of the less expensive 2,6-dichlorobenzonitrile as the starting material. In the present manuscript, we describe our work in the preparation of the 3-aminoindazole fragment of Lenacapavir. The new chemistry leverages a highly regioselective bromination and selective cyclization step to provide an efficient route to the functionalized heterocycle.
2. Results
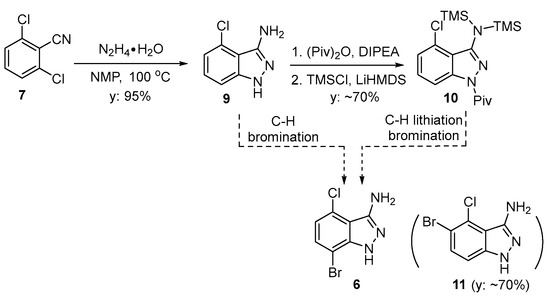
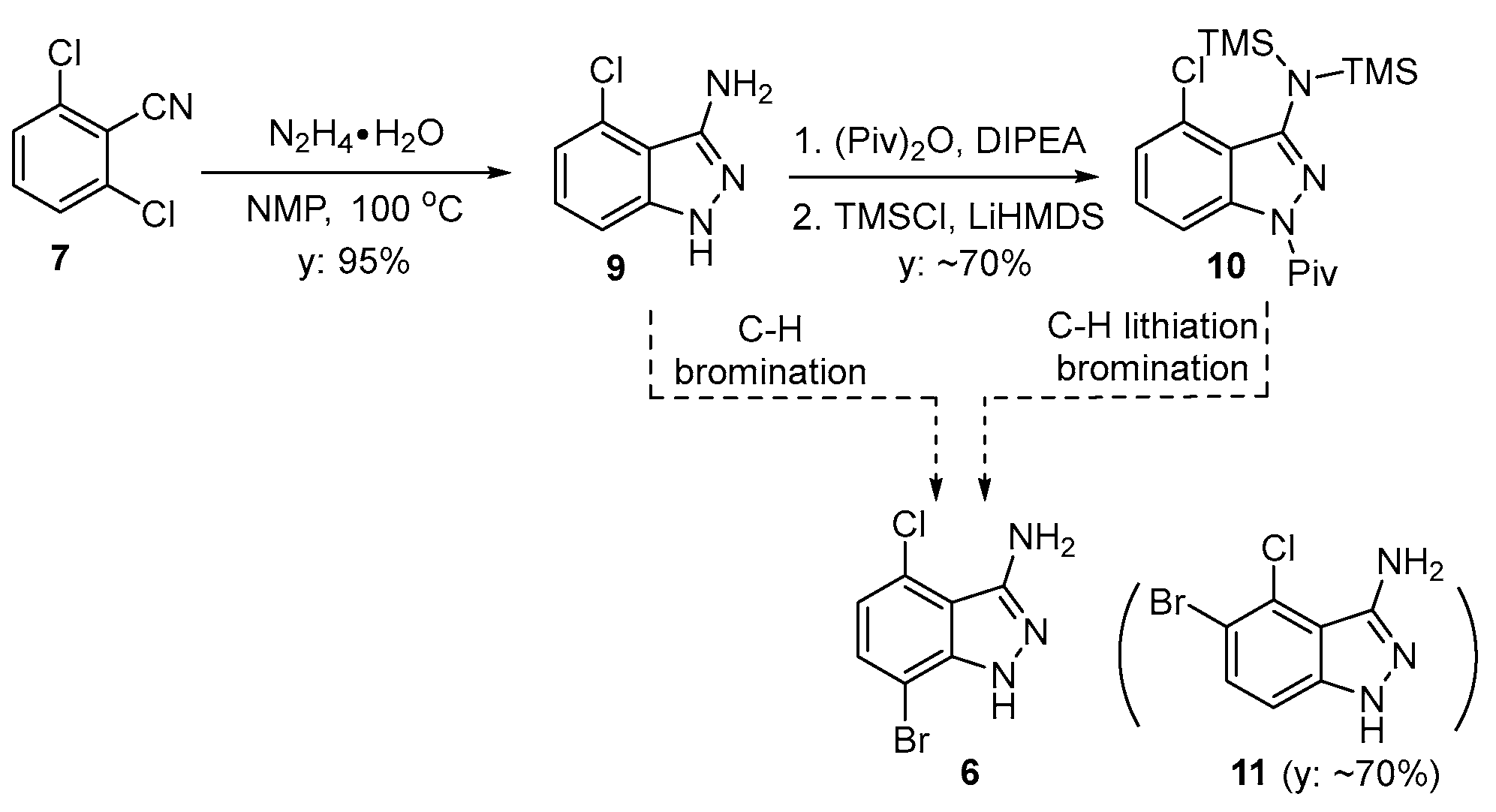
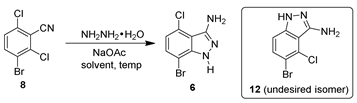
Our initial efforts to produce 7-bromo-4-chloro-1H-indazol-3-amine (6) involved forming the 3-aminoindazole ring first and then brominating the heterocyclic product. In accordance with the literature, the 3-aminoindazole (9) was prepared from the condensation of 7 with hydrazine hydrate in a 95% yield (Scheme 1) [6]. The direct bromination of compound 9 was not successful. The treatment of 9 with NBS afforded the undesired regioisomer 11 as the major product (based on 1HNMR, GCMS).
Scheme 1.
Initial attempt at synthesis of 6.
As another approach to functionalize the 7-position, we sought to carry out the regioselective functionalization of protected 3-aminoindazole derivative 10. Treating 10 with different organolithium reagents, such as n-butyllithium (BuLi), lithium diisopropylamide (LDA), or lithium bis(trimethylsilyl)amide (LiHMDS), followed by treatment with bromine, resulted in a complex mixture of products. The functionalization of the 7-position was also attempted with the borylation of the lithiated products of 10, using B(OMe)3 and B(OiPr)3 after a reaction with the organolithium base. This approach also failed to yield any desired product with functionalization at the 7-position, but rather resulted in recovering the starting material 3-aminoindazole.
2.1. Synthesis of 3-Bromo-2,6-dichlorobenzonitrile
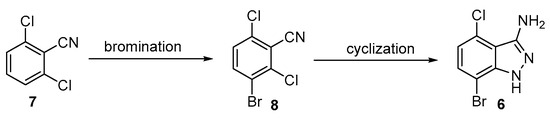
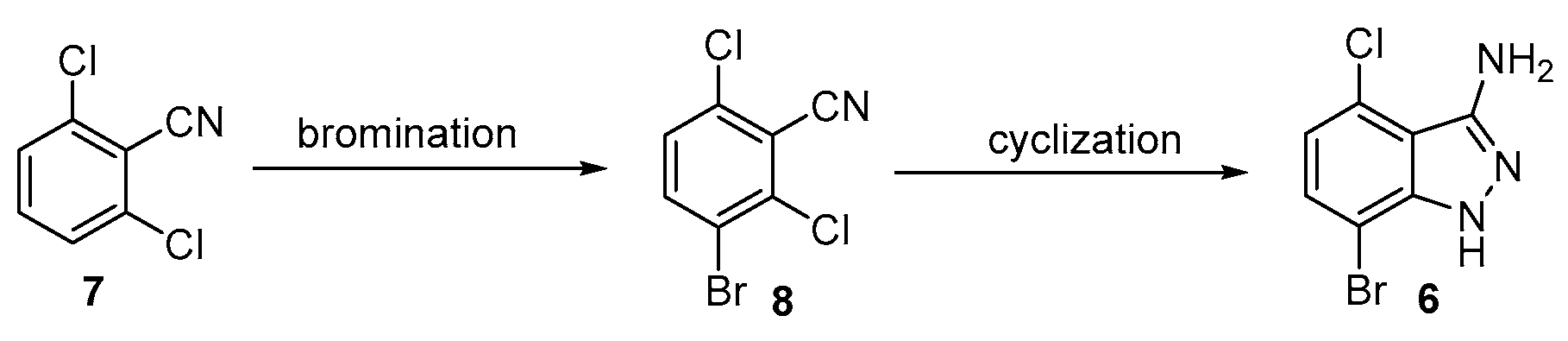
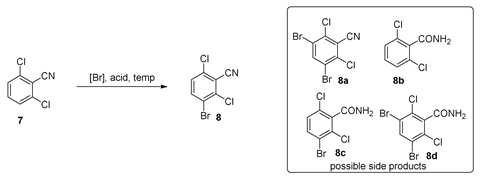
As an alternative approach, it was hypothesized that the bromination of 2,6-dichlorobenzonitrile could provide compound 8 and this derivative could afford the desired 3-aminoindazole 6 (Scheme 2). 3-Bromo-2,6-dichlorobenzonitrile 8 was initially prepared according to a reported method in which the treatment of 7 with potassium bromate and sulfuric acid afforded 8 in a ~60% yield after column purification [14]. Unfortunately, this bromination method possessed potential safety issues as it was an extremely exothermic reaction. The conversion was also accompanied by side reactions, such as over-bromination and the hydrolysis of the cyano group. Decreasing the reaction temperature to 0 °C and −10 °C resulted in lower conversion with similarly low product purity.
Scheme 2.
Synthesis of indazole 6 through regioselective cyclization of 8.
To identify more practical and safer bromination conditions to prepare 3-aminoindazole 8, alternative brominating reagents and conditions were explored (Table 1). Two commonly used brominating reagents, Br2 and NBS, were investigated. Reactions with bromine resulted in the hydration of the cyano group to an amide. More hydrolyzed side products were observed under elevated temperatures (Table 1, entries 1–3). N-Bromosuccinimide (NBS) was identified as the optimal brominating reagent for this transformation. The initial reaction of 7 with 1.2 equivalents of NBS at 25 °C afforded product 8 in 75 A%, along with 12 A% of 7 and 12 A% of dibrominated product 8a detected by GCMS total ion chromatography (TIC) (Table 1, entry 4). With an increase in the reaction temperature, the hydration of the nitrile group and over-bromination dominated (Table 1, entries 5–6). By decreasing the equivalents of NBS to 1.07 (25 °C), the reaction furnished 8 in >93 A% without the hydration of the nitrile group or over-bromination (Table 1, entry 7). An investigation of the equivalents and concentration of H2SO4 identified that 10–11 equivalents of 96% H2SO4 was optimal for the bromination (Table 1, entries 8–12). The use of 11 equivalents of 96% H2SO4 gave only 1 A% more 8, compared to the use of 10 equivalents; thus, 10 equivalents of 96% H2SO4 was used for scale-up. Notably, swapping H2SO4 with other acids, such as TFA or acetic acid, resulted in no reaction (Table 1, entries 13–14). As a result, the optimal bromination conditions were identified for scale-up (NBS (1.07 eq.), 96% H2SO4 (10 eq.), 25 °C, 18 h). The optimized conditions were demonstrated in hundred-gram scale (25–300 g) reactions. After the completion of the reaction, the product mixture was poured into 15 volumes of ice-cold water and the resulting precipitate was collected by filtration. The filter cake was washed with 3 volumes of ethyl acetate to obtain the desired 8 in a 75–80% isolated yield with 95–96% purity (qNMR) (Table 1, entries 15–17).

Table 1.
Bromination of 2,6-dichlorobenzonitrile 7.
2.2. Synthesis of 7-Bromo-4-chloro-1H-indazol-3-amine
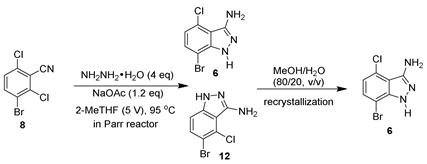
With 3-bromo-2,6-dichlorobenzonitrile (8) in hand, cyclization with hydrazine hydrate was investigated (Table 2). As shown in Table 2, a variety of solvents, such as aprotic polar solvents, protic solvents, basic solvents, and other organic solvents, were screened in the cyclization chemistry. Cyclization in aprotic polar solvents, such as NMP and DMSO, was performed smoothly under mild conditions (60 °C, 2 eq. of hydrazine hydrate and 1.2 eq. of NaOAc) [15]. Unfortunately, both chloro groups of 8 reacted with hydrazine without preference, resulting in a ~50:50 ratio of 6 and 12 (Table 2, entries 1–2). The ratio of 6:12 was improved to 65:35 when switching the solvent to ethanol and further improved to 70:30 with IPA as a solvent (Table 2, entries 3–4). In order to achieve >95% conversion of 8, an elevated reaction temperature (95 °C) and excess hydrazine was needed. However, these promising results inspired our further solvent screening for better regioselectivity. Among all other solvents screened, DIPEA and 2-MeTHF were identified to afford a higher ratio of the desired regioisomer (Table 2, entries 5–9). For further optimization, 2-MeTHF was selected because it would be easily recycled, as it is water-immiscible. The equivalents of hydrazine were investigated to obtain optimal conditions for this transformation. It was found that 4 equivalents of hydrazine hydrate afforded >98% conversion of compound 8 (Table 2, entries 10–11). In addition, a solvent volume screen showed that 5 V of 2-MeTHF afforded >99% conversion with a 70:30 ratio of 6/12 (Table 2, entry 12). As a result, the optimized conditions for cyclization were identified for scale-up (hydrazine hydrate (4 eq.), NaOAc (1.2 eq.), 2-MeTHF (5 V), 95 °C).
Table 2.
Optimization of cyclization of 8.
Parenthetically, the transformation in 2-MeTHF needs an internal temperature of >90 °C to obtain full conversion; thus, a pressure reactor was utilized for scale-up. A mixture of 8 (20 g), NaOAc (1.2 eq.), and 4 eq. of hydrazine hydrate in 2-MeTHF (5 V) was stirred in a Parr reactor at 95 °C (internal temperature), affording >95% conversion after 18 h. It is worth mentioning that Kruger and coworkers found that the addition of sodium acetate is needed to mitigate the safety concerns regarding the utilization of hydrazine hydrate in scale [15]. The sodium acetate may quench the resulting HCl during cyclization, which suppresses the possible formation of high-energetic hydrazine HCl conjugates. Although the two isomers could be separated by column chromatography, a more scalable purification method by recrystallization to afford the desired isomer was identified. After screening a variety of solvents, a binary solvent of MeOH/H2O (80/20, v/v) was found as optimal for the recrystallization. For instance, a mixture of regioisomers (molar ratio of 6/12: 70/30) was dissolved in MeOH/H2O (80/20, v/v) at 80 °C; after cooling to room temperature, the desired isomer 6 was obtained as a solid in an ~80% recovery yield with ~97% purity (qNMR). The protocol was successfully demonstrated with three batches in 20–80 g scale reactions. As shown in Table 3, in a Parr reactor, with 2-MeTHF as a solvent, the reaction of 8 and hydrazine hydrate at 95 °C afforded the crudes of the regioisomers (molar ratio of 6/12: 70/30) in a quantitative yield. After treating the regioisomers with MeOH/H2O (80/20, v/v), the desired isomer 6 was obtained in a 50–56% isolated yield with 96–98 A% purity (GCMS) (Table 3, entries 1–3).
Table 3.
Synthesis and purification of 7-bromo-4-chloro-1H-indazol-3-amine.
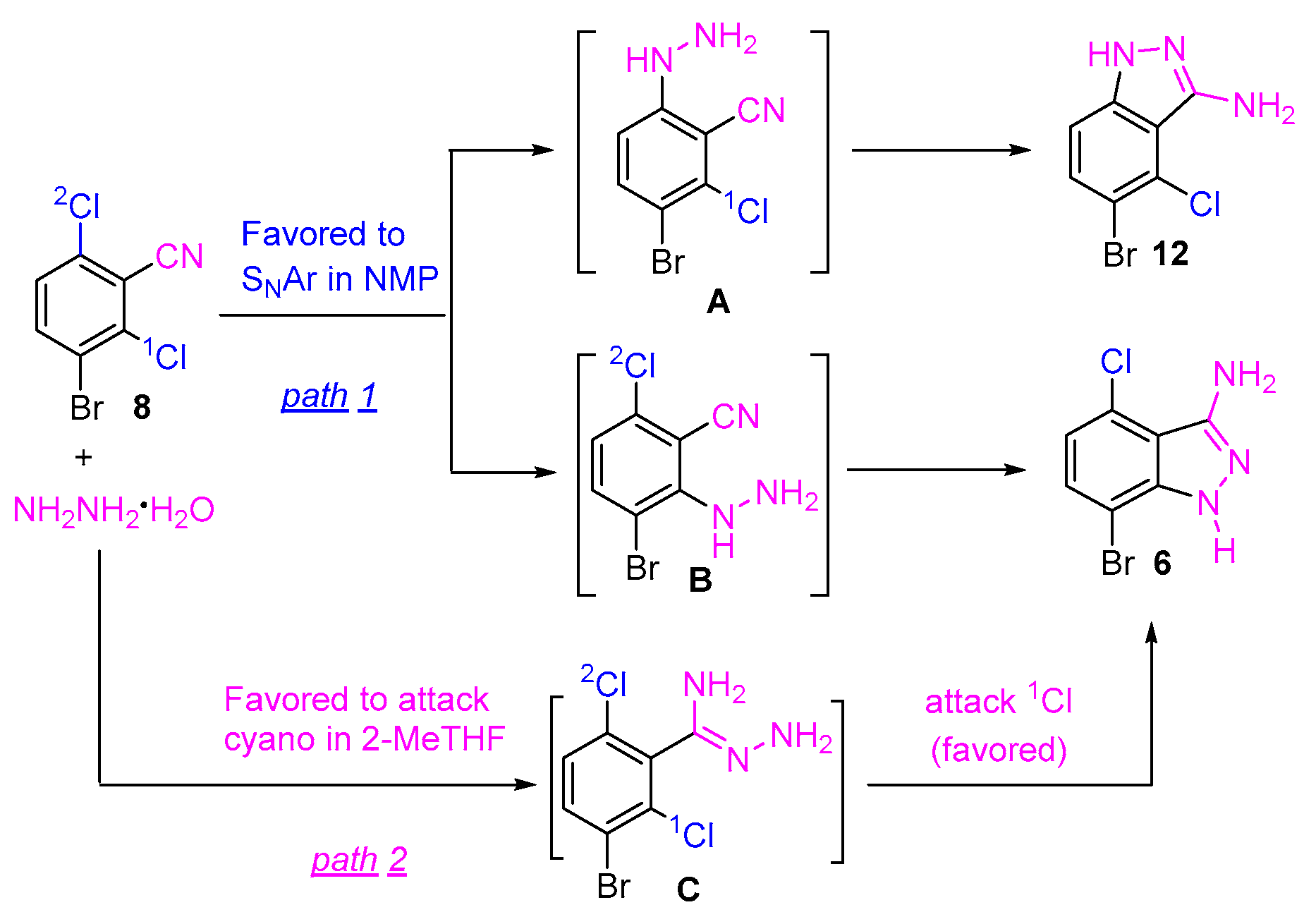
The subtle steric and electronic distinction of the two chlorine atoms might play an important role in the regioselective cyclization of 8 with hydrazine hydrate. However, the detailed mechanistic basis of the regioselectivity is unclear. It is known that the condensation reaction between 2,6-dichlorobenzonitrile and hydrazine hydrate can proceed with two possible pathways: (1) SNAr occurs first, followed by an intramolecular cyano attack; or (2) the cyano attack occurs first, followed by an intramolecular SNAr reaction [6,15]. The reaction of 8 and hydrazine might proceed with a similar pathway. As shown in Scheme 3, the indazole 6 could be formed through either hydrazine attacking the chloro first (SNAr reaction), followed by intramolecular cyclization (path 1), or the hydrazine attacking the cyano group first, followed by intramolecular SNAr cyclization (path 2). Both pathways afforded the indazole 6. It is believed that pathway 1 might need less energy in the transformation of the intermediates to the indazole 6 (fast reaction rate, less hydrazine, and low reaction temperature), while pathway 2 might need higher energy to convert the intermediates to the indazole compound (long reaction time, more hydrazine, higher reaction temperature). Our experimental results indicated that the regioselective cyclization was highly solvent-dependent. We assumed that, in an aprotic polar solvent, such as NMP, pathway 1 was dominant and the fast reaction rate resulted in no selectivity. On the other hand, pathway 2 was dominant in 2-MeTHF. Presumably, the resulting intermediate C favors the intramolecular SNAr reaction on 1Cl to afford the compound 6, in which the inductive effect of the ortho bromine atom might play the role.
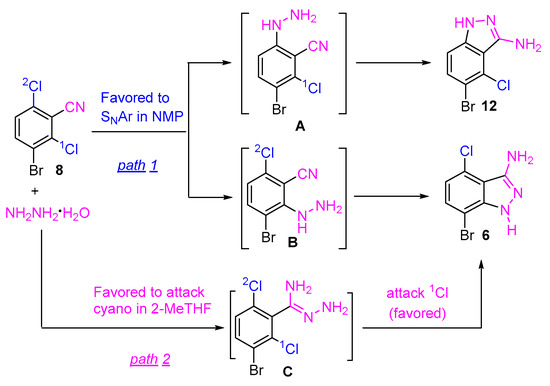
Scheme 3.
A plausible pathway for the formation of indazoles.
3. Experimental Section
3.1. General Method
Reagents and solvents were obtained from commercial suppliers and used as received, unless otherwise indicated. Where applicable, reactions were conducted in oven-dried (120 °C) glassware, which was assembled while hot and cooled to ambient temperature under an inert atmosphere. Reactors were pre-rinsed with reaction solvent and subjected to evacuation/back-fill cycles (3×) as necessary. Reactions were monitored by TLC (precoated silica gel 60 F254 plates, EMD Chemicals, Burlington, MA, USA), Agilent GCMS (Santa Clara, CA, USA), or crude 1H NMR. HRMS was recorded using a Perkin Elmer Axion 2 ToF MS (Waltham, MA, USA) in positive ionization mode with a scan range of 100–1000 m/z, flight tube voltage of 8 kV, spray voltage of 3.5 kV, and methanol as a solvent. TLC was visualized with UV light. The proton (1H NMR), carbon (13C NMR), and 2-DNMR spectra of the compounds were recorded on a Bruker Avance III HD Ascend 600 MHz spectrometer or 400 MHz spectrometer (Billerica, MA, USA). The NMR solvent used was DMSO-d6. The chemical shifts were reported in parts per million (ppm). Coupling constants J are reported in hertz (Hz). The abbreviations used to designate signal multiplicity were s, singlet; d, doublet.
3.2. Chemistry
3.2.1. Preparation of 3-Bromo-2,6-dichlorobenzonitrile (8)
To a 5 L ChemRxnHub reactor (Chemglass Life Sciences LLC, Vineland, NJ, USA) at room temperature, 2,6-dichlorobenzonitrile (290.0 g, 1.68 mol) was added, followed by the addition of 96% sulfuric acid (10 eq., 0.92 L, 16.8 mol) with stirring at 0 °C. After the completion of the addition of sulfuric acid, the reaction mixture was stirred for 15 min to obtain a clear yellowish solution. N-bromosuccinimide (321 g, 1.07 eq., 1.8 mol) was added in portions over the course of 10 min at 0 °C. The reaction mixture was stirred at 25 °C for 18 h to afford a thick, pale-yellowish orange slurry. After the completion of the reaction (monitored by 1HNMR), the crude mixture was slowly transferred to ice water (2.9 L, 10 V). The slurry was stirred for 45 min and the resulting precipitates were collected by filtration. The solid cake was washed with water (500 mL × 5), dried under a house vacuum, and then washed with ethyl acetate (300 mL × 3). The solid was dried under a vacuum to obtain the product 8 (355 g, yield: 80%; purity by qNMR: 95%; purity by GCMS: 97%, containing 2% of dibromobenzonitrile 8a).
1H NMR (600 MHz, DMSO-d6) δ 8.11 (d, J = 8.8 Hz, 1H), 7.66 (d, J = 8.8 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 138.9, 137.3, 136.5, 129.8, 121.7, 114.5, 113.3. 13C NMR DEPT 135 (151 MHz, DMSO-d6) δ 138.9, 129.8. MS-EI (m/z): 251.
3.2.2. Preparation of 7-Bromo-4-chloro-1H-indol-3-amine (6)
To a degassed 1 L Parr reactor was charged 3-bromo-2,6-dichlorobenzonitrile (80.0 g, 1 eq., 296 mmol), hydrazine hydrate (76 mL, 4 eq., 1.2 mol), and 2-MeTHF (5 V, 400 mL) at room temperature. The reaction mixture was heated at 105 °C and stirred for 18 h. After completion, the mixture was cooled to 25 °C. Water (3 V, 240 mL) was added and the mixture was extracted with ethyl acetate (300 mL × 3). The organic layer was combined and washed with brine (300 mL). The organic layer was separated and evaporated to dryness to afford a mixture of 6 and 12 (ratio: 70:29) with a quantitative mass (73 g crude). Methanol/water (4/1, v/v, ~20 V, 1.4 L) was added to the crude solid. The mixture was refluxed to afford a clear solution. The resulting solution was stirred at room temperature overnight. The white precipitates were filtered and washed with MTBE (20 mL × 4) to obtain the compound 6 (38 g, yield: 53%; purity by qNMR: 96%; purity by GCMS: 97%, containing 2% of dibromochloroindazole (m/z: 323)).
1H NMR (600 MHz, DMSO-d6) δ 12.23 (s, 1H), 7.41 (d, J = 7.9 Hz, 1H), 6.85 (d, J = 7.9 Hz, 1H), 5.33 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 149.1, 141.1, 129.5, 125.2, 119.1, 111.9, 101.0. 13C NMR DEPT 135 (151 MHz, DMSO-d6) δ 129.5, 119.1. HRMS (m/z): [M + H]+ calcd for C7H5BrClN3·H+: 247.9413 amu; found: 247.9412 amu.
For comparison, the undesired isomer 12 was purified by column chromatography (SiO2, ethyl acetate/heptanes = 10/90) to obtain the characterization data.
Compound 12: 1H NMR (600 MHz, DMSO-d6) δ 12.0 (s, 1H), 7.45 (d, J = 8.8 Hz, 1H), 7.19 (d, J = 8.8 Hz, 1H), 5.27 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 148.5, 141.8, 131.1, 125.7, 112.3, 110.9, 110.3. HRMS (m/z): [M + H]+ calcd for C7H5BrClN3·H+: 247.9413 amu; found: 247.9400 amu.
4. Conclusions
The practical synthesis of the heterocyclic compound 7-bromo-4-chloro-1H-indazol-3-amine has been developed from the readily available and inexpensive raw material 2,6-dichlorobenzonitrile. The two-step synthetic sequence utilizes a highly regioselective bromination and cyclization step to give an overall yield of 38–45% for the functionalized 3-aminoindazole (6). Mild bromination conditions (NBS/H2SO4) were identified, affording bromide 8 in a 76–81% yield from 2,6-dichlorobenzonitrile. The subsequent regioselective cyclization in 2-MeTHF afforded the desired 3-aminoindazole regioisomer in a 50–56% isolated yield with purity of up to 98%. The two-step protocol was demonstrated on hundred-gram scales and eliminates the need for column chromatography purification. This new chemistry provides a practical synthetic route to 7-bromo-4-chloro-1H-indazol-3-amine (6), a heterocyclic fragment used in the synthesis of the potent anti-HIV therapeutic, Lenacapavir.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29122705/s1, GCMS method. Detailed procedure of synthesis of 6 from 3-bromo-6-chloro-2-fluorobenzonitrile; Detailed procedure of synthesis of 9 and 10. Table S1: Recrystallization of 7-bromo-4-chloro-1H-indazol-3-amine 6. Figure S1: 1HNMR of 3-bromo-2,6-dichlorobenzonitrile (8) in DMSO-d6 (600 MHz). Figure S2. 13CNMR of 3-bromo-2,6-dichlorobenzonitrile (8) in DMSO-d6. Figure S3. DEPT-135 of 3-bromo-2,6-dichlorobenzonitrile (8) in DMSO-d6. Figure S4. 1HNMR of 7-bromo-4-chloro-1H-indazole-3-amine (6) in DMSO-d6. Figure S5. 13CNMR of 7-bromo-4-chloro-1H-indazole-3-amine (6) in DMSO-d6. Figure S6. DEPT-135 of 7-bromo-4-chloro-1H-indazole-3-amine (6) in DMSO-d6. Figure S7. 1H-1H COSY of 7-bromo-4-chloro-1H-indazole-3-amine (6) in DMSO-d6. Figure S8. HMBC of 7-bromo-4-chloro-1H-indazole-3-amine (6) in DMSO-d6. Figure S9. 1HNMR of 5-bromo-4-chloro-1H-indazole-3-amine (12) in DMSO-d6. Figure S10. 13CNMR of 5-bromo-4-chloro-1H-indazole-3-amine (12) in DMSO-d6. Figure S11. 1H-1H COSY of 5-bromo-4-chloro-1H-indazole-3-amine (12) in DMSO-d6. Figure S12. HSQC of 5-bromo-4-chloro-1H-indazole-3-amine (12) in DMSO-d6. Figure S13. 1HNMR of 4-chloro-1H-indazole-3-amine (9) in DMSO-d6. Figure S14. 1HNMR of 10a in DMSO-d6. Figure S15. 1HNMR of 10 in DMSO-d6.
Author Contributions
N.A., M.L. and S.M.M.R. conducted the experiments and manuscript writing. J.M.B. performed the analytical measurements of all products and manuscript writing. T.D.R., G.M.L., S.A., B.F.G. and D.K. designed and organized this work and conducted manuscript writing. L.J. planned, designed, and organized all of this study’s research and the manuscript’s preparation. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by funding from the Bill & Melinda Gates Foundation (BMGF) (OPP1176590).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the main text and the Supplementary Materials of this article.
Acknowledgments
The Medicines for All Institute (M4ALL) would like to express its gratitude to Trevor Laird, John Dillon, and Ryan Nelson (BMGF) for their helpful technical guidance throughout this project, as well as Silpa Sundaram (BMGF), Susan Hershenson (BMGF), John Walker (BMGF), and Scott Rosenblum (BMGF) for their ongoing collaboration and support of the M4ALL mission. The authors are also grateful to Janie Wierzbicki, Sarah Cox, and Michael Osberg for their inputs in this work.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lesuisse, D.; Dutruc-Rosset, G.; Tiraboschi, G.; Dreyer, M.K.; Maignan, S.; Chevalier, A.; Halley, F.; Bertrand, P.; Burgevin, M.-C.; Quarteronet, D.; et al. Rational Design of Potent GSK3β Inhibitors with Selectivity for Cdk1 and Cdk2. Bioorg. Med. Chem. Lett. 2010, 20, 1985–1989. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Ueda, K.; Ishiyama, T.; Goto, R.; Muramatsu, S.; Hashimoto, M.; Watanabe, K.; Tanaka, N. Synthesis and SAR Studies of 3,6-Disubstituted Indazole Derivatives as Potent Hepcidin Production Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2148–2152. [Google Scholar] [CrossRef] [PubMed]
- Kansal, N.; Silakari, O.; Ravikumar, M. Three Dimensional Pharmacophore Modelling for C-Kit Receptor Tyrosine Kinase Inhibitors. Eur. J. Med. Chem. 2010, 45, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical Targeting of HIV Capsid Protein with a Long-Acting Small Molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Bester, S.M.; Wei, G.; Zhao, H.; Adu-Ampratwum, D.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Annamalai, A.S.; Singh, P.K.; Shkriabai, N.; et al. Structural and Mechanistic Bases for a Potent HIV-1 Capsid Inhibitor. Science 2020, 370, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Kruger, A.W.; Rozema, M.J.; Chu-Kung, A.; Gandarilla, J.; Haight, A.R.; Kotecki, B.J.; Richter, S.M.; Schwartz, A.M.; Wang, Z. The Discovery and Development of a Safe, Practical Synthesis of ABT-869. Org. Process Res. Dev. 2009, 13, 1419–1425. [Google Scholar] [CrossRef]
- Lefebvre, V.; Cailly, T.; Fabis, F.; Rault, S. Two-Step Synthesis of Substituted 3-Aminoindazoles from 2-Bromobenzonitriles. J. Org. Chem. 2010, 75, 2730–2732. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, T.; Lombardi, B.A.; Nesi, M.; Perrone, E.; Bossi, R.; Polucci, P. Indazole Derivatives as Kinase Inhibitors for the Treatment of Cancer. WO2008074749A1, 26 June 2008. [Google Scholar]
- Burke, M.J.; Trantow, B.M. An Efficient Route to 3-Aminoindazoles and 3-Amino-7-Azaindazoles. Tetrahedron Lett. 2008, 49, 4579–4581. [Google Scholar] [CrossRef]
- Suryakiran, N.; Prabhakar, P.; Venkateswarlu, Y. Synthesis of 3-Amino-Substituted N -Alkylindazoles via Palladium(II)-Catalyzed Intramolecular N-Arylation of Tosylhydrazines. Chem. Lett. 2007, 36, 1370–1371. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, H.; Li, Z.; Liang, Z.; Qi, S.; Cai, M.; Zhang, S.; Jia, X.; Zhang, G.; Hu, M.-L. Rapid Access to 3-Aminoindazoles from Nitriles with Hydrazines: A Strategy to Overcome the Basicity Barrier Imparted by Hydrazines. Chem. Commun. 2020, 56, 9521–9524. [Google Scholar] [CrossRef] [PubMed]
- Albanesi, S.; Koren, D. An Update on Lenacapavir. Contagion 2023, 8, 10–11. [Google Scholar]
- Price from Sigmaaldrich: 3-Bromo-6-Chloro-2-Fluorobenzonitrile: 1g/$207; 2,6-Dichlorobenzonitrileprice: 1g/$2.6. Available online: https://www.sigmaaldrich.com/US/en (accessed on 2 June 2024).
- Eckhardt, M.; Giroud, M.; Langkopf, E.; Mayer, C.; Wagner, H.; Wiedenmayer, D. Heteroaromatic Carboxamide Derivatives as Plasma Kallikrein Inhibitors. WO2021160718A1, 19 August 2021. [Google Scholar]
- Wang, Z.; Richter, S.M.; Gandarilla, J.; Kruger, A.W.; Rozema, M.J. Safe Scale-Up of a Hydrazine Condensation by the Addition of a Base. Org. Process Res. Dev. 2013, 17, 1603–1610. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).