

Synthesis and Antineoplastic Activity of a Dimer, Spiroindolinone Pyrrolidinecarboxamide

Abstract
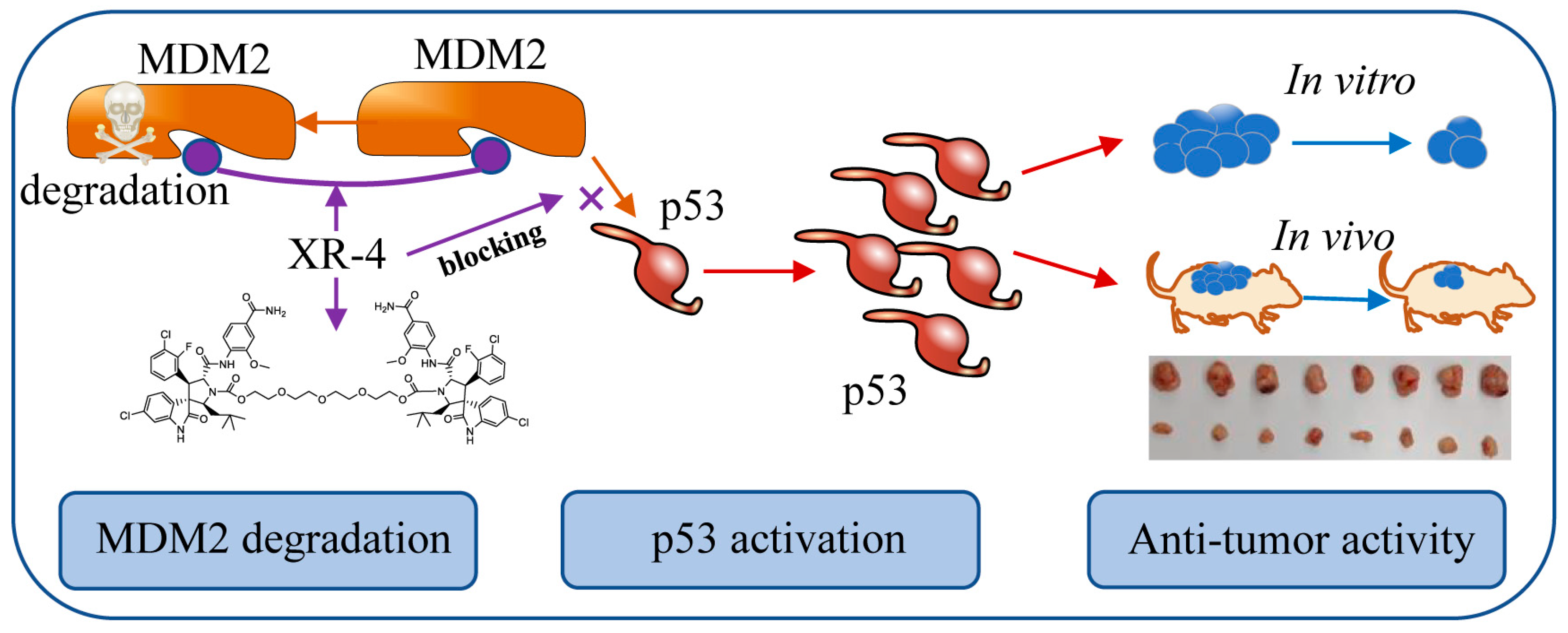
1. Introduction
2. Results
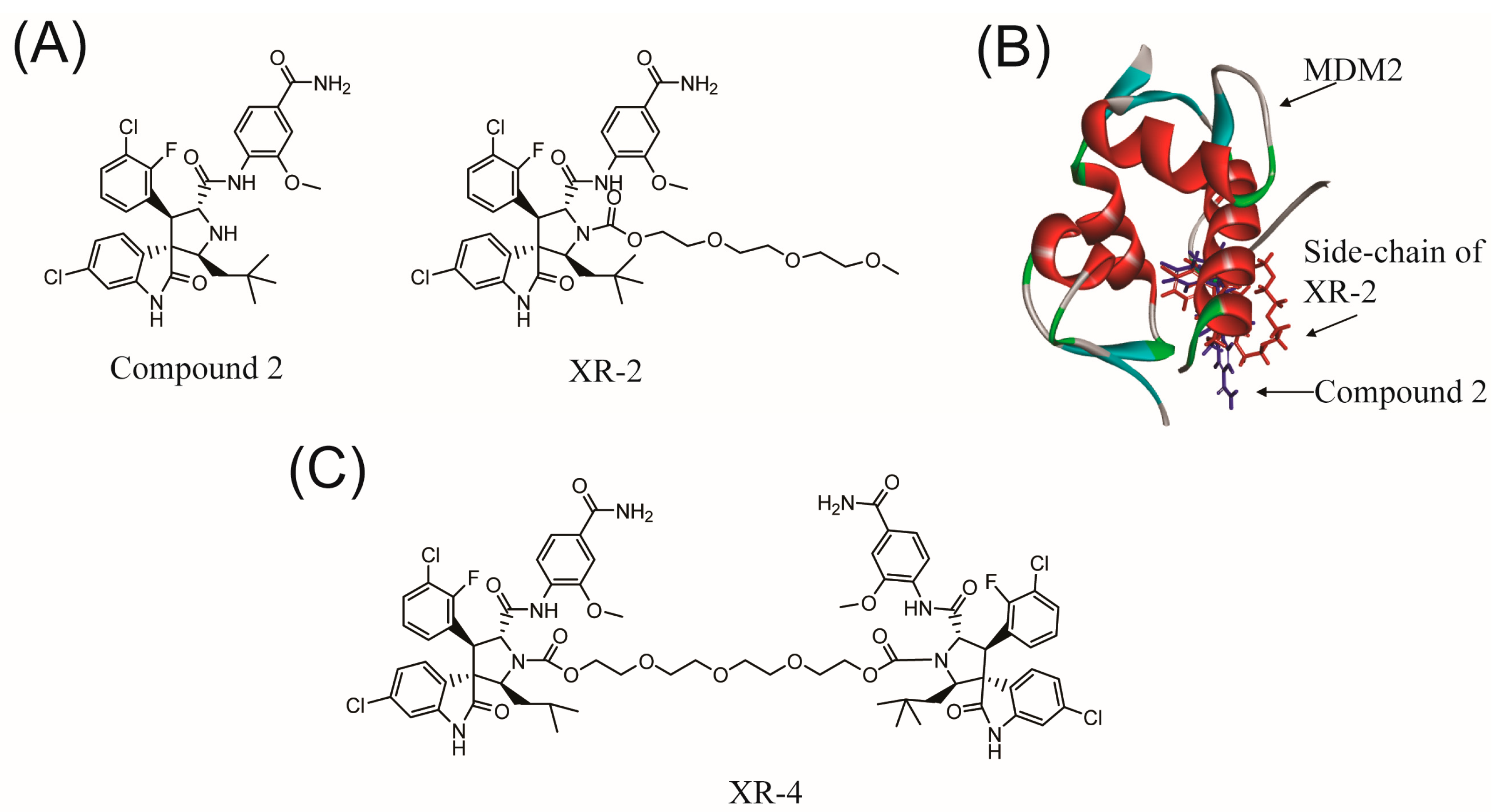
2.1. Rational Design of Dimer Spiroindolinone Pyrrolidinecarboxamide
2.2. Chemistry
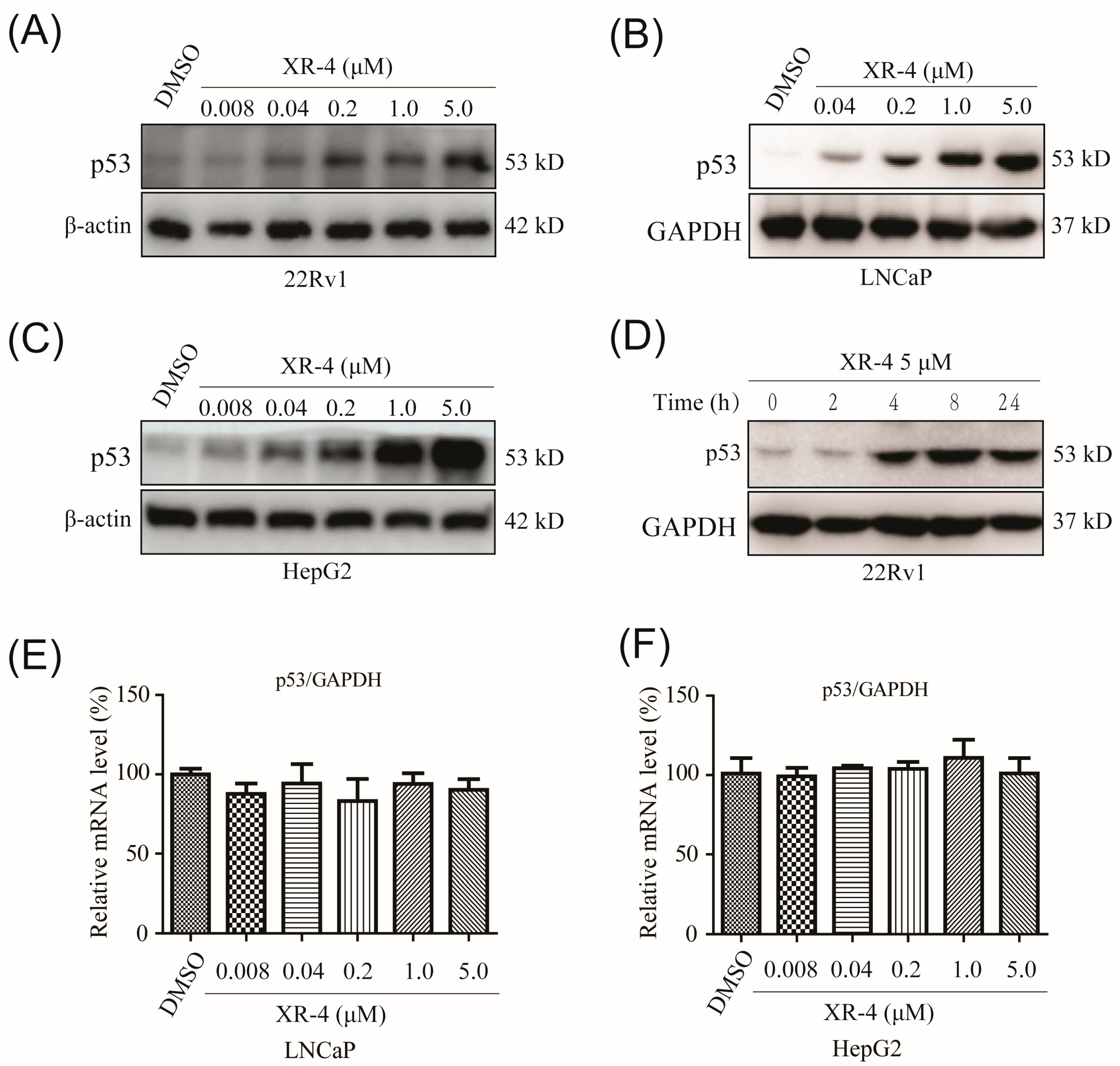
2.3. XR-4 Upregulates p53 Levels in Cancer Cells
2.4. XR-4 Activates p53 Downstream Target Genes
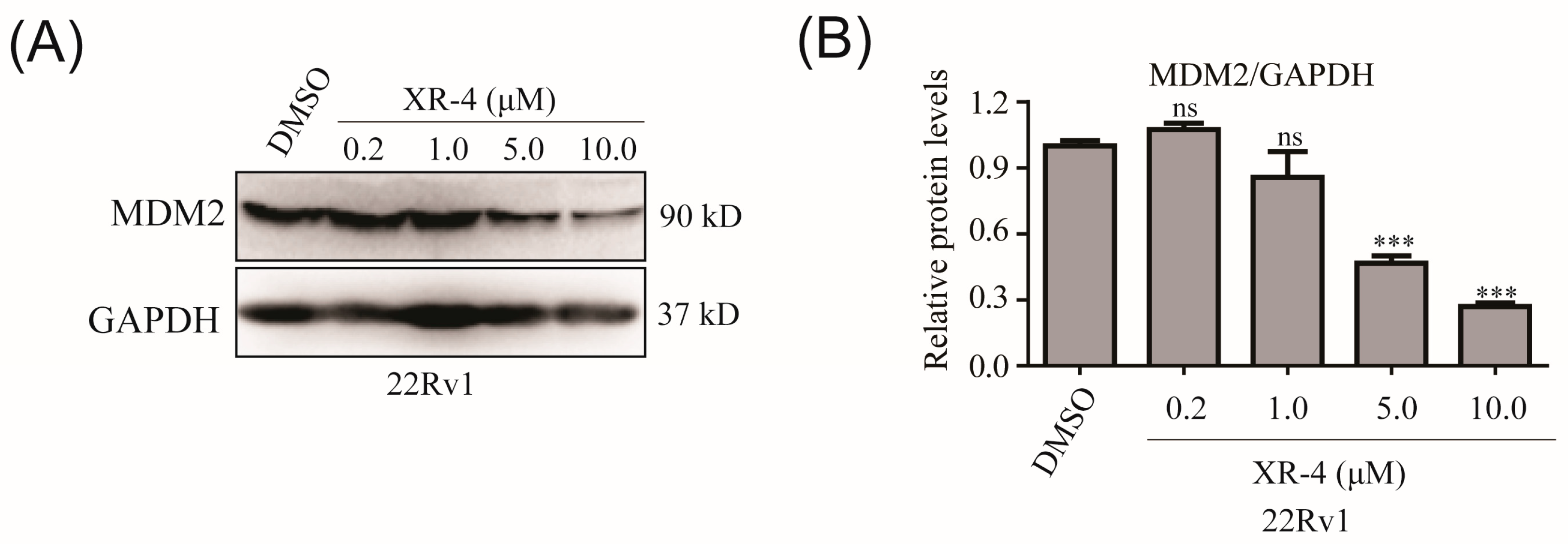
2.5. XR-4 Downregulates MDM2 Protein Levels
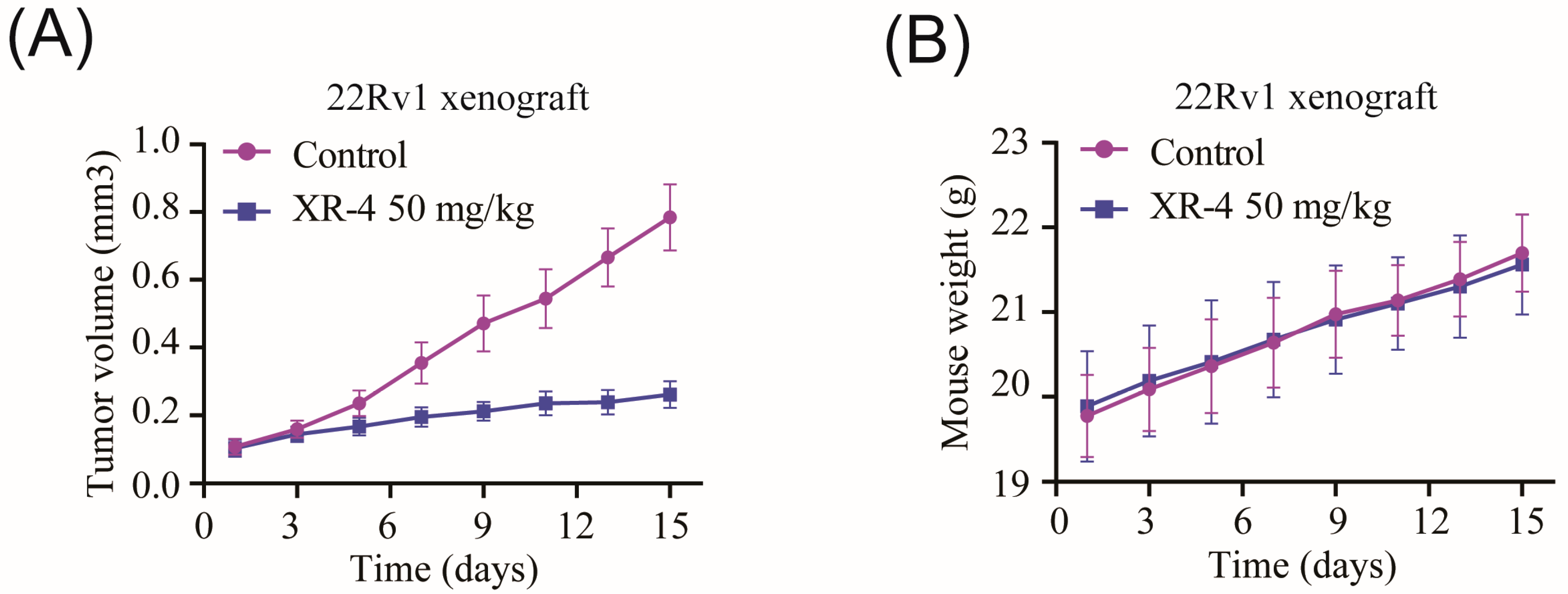
2.6. XR-4 Suppresses the Viability of Wild-Type p53 Cancer Cells Both In Vitro and In Vivo
3. Conclusions
4. Materials and Methods
4.1. Chemistry
- Bis(1-chloroethyl) (((oxybis(ethane-2,1-diyl))bis(oxy))bis(ethane-2,1-diyl)) bis(carbonate) (linker intermediate): The solution of 2,2’-((Oxybis(ethane-2,1-diyl)) bis(oxy)) diethanol (1.94 g, 10 mmol, purchased from Sigma-Aldrich, Darmstadt, Germany) and 1-chloroethyl chloroformate (4.29 g, 30 mmol, purchased from Sigma-Aldrich) was stirred in CH2Cl2 (100 mL, purchased from Bidepharm, Shanghai, China) at room temperature for 6 h in the presence of Et3N (4.04 g, 52.5 mmol, purchased from Bidepharm, Shanghai, China). Then, 200 mL of water was added, and the resulting mixture was extracted with CH2Cl2 (2 × 50 mL). The organic phase was separated, and subsequently, washed with brine, 1M hydrochloric acid, and water. After being concentrated through a vacuum, the target product was obtained as an oil, which was directly used in the next step.
- 1H NMR (400 MHz, CDCl3): 6.30 (q, J = 5.8 Hz, 1H), 4.29 (dd, J = 3.12 Hz, 2H), 3.68 (dd, J = 4.6 Hz, 2H), 3.59 (s, 4H), 1.76 (d, J = 5.8 Hz, 3H).
- ((Oxybis(ethane-2,1-diyl))bis(oxy))bis(ethane-2,1-diyl) (2′S,2′′′S,3R,3″R,4′S,4′′′S,5′R,5′′′R)-bis(5′-((4-carbamoyl-2-methoxyphenyl)carbamoyl)-6-chloro-4′-(3-chloro-2-fluorophenyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-1′-carboxylate) (XR-4): Compound 2 was synthesized according to the literature [28]. Compound 2 (612 mg, 1.0 mmol) was dissolved in acetone (100 mL, purchased from Bidepharm, Shanghai, China), and then anhydrous Cs2CO3 (2.0 mmol, purchased from Sigma-Aldrich, Darmstadt, Germany) and bis(1-chloroethyl) (((oxybis(ethane-2,1-diyl))bis(oxy))bis(ethane-2,1-diyl)) bis(carbonate) (204 mg, 0.5 mmol) were added, and the resulting mixture was stirred overnight at room temperature. The reaction mixture was then diluted with water (20 mL) and extracted with ethyl acetate (2 × 50 mL, purchased from Bidepharm, Shanghai, China) and washed with brine, hydrochloric acid, and water, before being concentrated through a vacuum. After purification via silica gel column chromatography, the target product, XR-4, was obtained (405 mg, yield: 55%; purity: 97%) as a white solid. The melting point of XR-4 was about 155–156 °C. 1H NMR (CDCl3, 400 MHz): 10.59 (s, 1H), 8.44 (d, J = 8.24 Hz, 1H), 7.79 (s, 1H), 7.54 (s, 1H), 7.48 (t, J = 6.90 Hz, 1H), 7.28–7.39 (m, 2H), 7.17–7.25 (m, 2H), 7.04 (t, J = 7.96 Hz, 1H), 6.20–6.55 (bs, 1H), 5.35–5.65 (bs, 1H), 4.69 (t, J = 8.84 Hz, 1H), 4.35–4.55 (m, 3H), 3.92 (s, 3H), 3.81 (t, J = 4.46 Hz, 2H), 3.60–3.75 (m, 5H), 3.24 (t, J = 10.7 Hz, 1H), 1.78 (s, 1H), 0.85–1.00 (m, 10H).
- 13C NMR (CDCl3, 101 MHz): 174.09, 171.38, 168.83, 157.38, 154.90, 149.29, 148.00, 139.75, 134.55, 130.00, 129.75, 128.43, 127.15, 125.26, 124.62, 124.49, 123.54, 123.10, 121.19, 119.97, 117.76, 115.80, 109.62, 70.48, 68.35, 67.66, 66.32, 66.28, 65.16, 55.39, 50.54, 42.50, 30.15, 29.61, 29.50. HRMS (ESI-TOF): m/z calculated for C72H78Cl4F2N8NaO15+ [M + 2H + Na]+: 1495.4207, found: 1495.4204. The NMR and HRMS data of XR-4 were showed in supplementary materials.
4.2. Cell Culture
4.3. RNA Extraction, Reverse Transcription, and Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.4. Western Blot
4.5. Cell Viability Assay
4.6. Colony Formation Assay
4.7. Flow Cytometry Assay
4.8. In Vivo Studies
4.9. In Silico Docking
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Pearson, P.L.; Van der Luijt, R.B. The genetic analysis of cancer. J. Intern. Med. 1998, 243, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wu, Z.X.; Assaraf, Y.G.; Chen, Z.S.; Wang, L. Overcoming anti-cancer drug resistance via restoration of tumor suppressor gene function. Drug Resist. Updat. 2021, 57, 100770. [Google Scholar] [CrossRef]
- Majors, B.S.; Betenbaugh, M.J.; Chiang, G.G. Links between metabolism and apoptosis in mammalian cells: Applications for anti-apoptosis engineering. Metab. Eng. 2007, 9, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Daniels, G.A.; Piccioni, D.E.; Kesari, S.; Fanta, P.T.; Schwab, R.B.; Kurzrock, R. Next generation sequencing demonstrates association between tumor suppressor gene aberrations and poor outcome in patients with cancer. Cell Cycle 2015, 14, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Ertel, A.; Casimiro, M.C.; Yu, Z.; Meng, H.; McCue, P.A.; Walters, R.; Fortina, P.; Lisanti, M.P.; Pestell, R.G. Novel oncogene-induced metastatic prostate cancer cell lines define human prostate cancer progression signatures. Cancer Res. 2013, 73, 978–989. [Google Scholar] [CrossRef]
- Bourdon, J.C. p53 and its isoforms in cancer. Br. J. Cancer 2007, 97, 277–282. [Google Scholar] [CrossRef]
- Vaddavalli, P.L.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 2022, 38, 598–612. [Google Scholar] [CrossRef]
- Romer, L.; Klein, C.; Dehner, A.; Kessler, H.; Buchner, J. p53--a natural cancer killer: Structural insights and therapeutic concepts. Angew. Chem. Int. Ed. Engl. 2006, 45, 6440–6460. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin Cancer Biol. 2022, 79, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef]
- Piette, J.; Neel, H.; Marechal, V. Mdm2: Keeping p53 under control. Oncogene 1997, 15, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gao, H.; Ji, Y.; Zhou, Q.; Du, Z.; Tian, L.; Jiang, Y.; Yao, K.; Zhou, Z. Targeting p53-MDM2 interaction by small-molecule inhibitors: Learning from MDM2 inhibitors in clinical trials. J. Hematol. Oncol. 2022, 15, 91. [Google Scholar] [CrossRef]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Graves, B. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, A.; Lu, J.; Liu, L.; Du, D.; Bernard, D.; McEachern, D.; Wang, S. Discovery of 4-((3’R,4’S,5’R)-6’’-Chloro-4’-(3-chloro-2-fluorophenyl)-1’-ethyl-2’’-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3’’-indoline]-5’-carboxamido)bicyclo[2.2.2]octane-1-carboxylic Acid (AA-115/APG-115): A Potent and Orally Active Murine Double Minute 2 (MDM2) Inhibitor in Clinical Development. J. Med. Chem. 2017, 60, 2819–2839. [Google Scholar] [PubMed]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.Y. Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a026245. [Google Scholar] [CrossRef] [PubMed]
- Zanjirband, M.; Rahgozar, S. Targeting p53-MDM2 Interaction Using Small Molecule Inhibitors and the Challenges Needed to be Addressed. Curr. Drug Targets 2019, 20, 1091–1111. [Google Scholar] [CrossRef]
- Wang, B.; Wu, S.; Liu, J.; Yang, K.; Xie, H.; Tang, W. Development of selective small molecule MDM2 degraders based on nutlin. Eur. J. Med. Chem. 2019, 176, 476–491. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, J.; Aguilar, A.; McEachern, D.; Przybranowski, S.; Liu, L.; Yang, C.-Y.; Wang, M.; Han, X.; Wang, S. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem. 2019, 62, 448–466. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Ma, J.; Fang, Y.; Liu, Y.; Wu, S.; Dong, G.; Wang, W.; Sheng, C. Homo-PROTAC mediated suicide of MDM2 to treat non-small cell lung cancer. Acta Pharm. Sin. B 2021, 11, 1617–1628. [Google Scholar] [CrossRef]
- Wu, M.; Cui, J.; Hou, H.; Li, Y.; Liu, S.; Wan, L.; Zhang, L.; Huang, W.; Sun, G.; Liu, J.; et al. Novel MDM2 Inhibitor XR-2 Exerts Potent Anti-Tumor Efficacy and Overcomes Enzalutamide Resistance in Prostate Cancer. Front. Pharmacol. 2022, 13, 871259. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, M.; Zhang, L.; Wan, L.; Li, H.; Zhang, L.; Xiao, F. Nonsense-mediated mRNA decay inhibition synergizes with MDM2 inhibition to suppress TP53 wild-type cancer cells in p53 isoform-dependent manner. Cell Death Discov. 2022, 8, 402. [Google Scholar] [CrossRef]
- Shu, L.; Li, Z.; Gu, C.; Fishlock, D. Synthesis of a Spiroindolinone Pyrrolidinecarboxamide MDM2 Antagonist. Org. Process Res. Dev. 2013, 17, 247–256. [Google Scholar] [CrossRef]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef]
- Shamloo, B.; Usluer, S. p21 in Cancer Research. Cancers 2019, 11, 1178. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | P53 Type | XR-4 (μM) | RG7388 (μM) |
|---|---|---|---|
| LNCaP | wild | 0.043 | 0.068 |
| 22Rv1 | wild | 0.66 | 0.67 |
| HepG2 | wild | 0.55 | 0.46 |
| HCT116 | wild | 0.84 | 0.75 |
| MCF7 | wild | 0.31 | 0.26 |
| T24 | wild | 0.39 | 0.51 |
| DU145 | mutated | >50 | 12.6 |
| PC-3 | null | >50 | 21.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Wang, Y.; Li, X.; Xiao, F.; Ren, H.; Wu, M. Synthesis and Antineoplastic Activity of a Dimer, Spiroindolinone Pyrrolidinecarboxamide. Molecules 2023, 28, 3912. https://doi.org/10.3390/molecules28093912
Cui J, Wang Y, Li X, Xiao F, Ren H, Wu M. Synthesis and Antineoplastic Activity of a Dimer, Spiroindolinone Pyrrolidinecarboxamide. Molecules. 2023; 28(9):3912. https://doi.org/10.3390/molecules28093912
Chicago/Turabian StyleCui, Jingyi, Yujie Wang, Xiaoxin Li, Fei Xiao, Hongjun Ren, and Meng Wu. 2023. "Synthesis and Antineoplastic Activity of a Dimer, Spiroindolinone Pyrrolidinecarboxamide" Molecules 28, no. 9: 3912. https://doi.org/10.3390/molecules28093912
APA StyleCui, J., Wang, Y., Li, X., Xiao, F., Ren, H., & Wu, M. (2023). Synthesis and Antineoplastic Activity of a Dimer, Spiroindolinone Pyrrolidinecarboxamide. Molecules, 28(9), 3912. https://doi.org/10.3390/molecules28093912