

Identification of Plant Peptides as Novel Inhibitors of Orthohepevirus A (HEV) Capsid Protein by Virtual Screening

 ,
,  , ,
, ,
Abstract
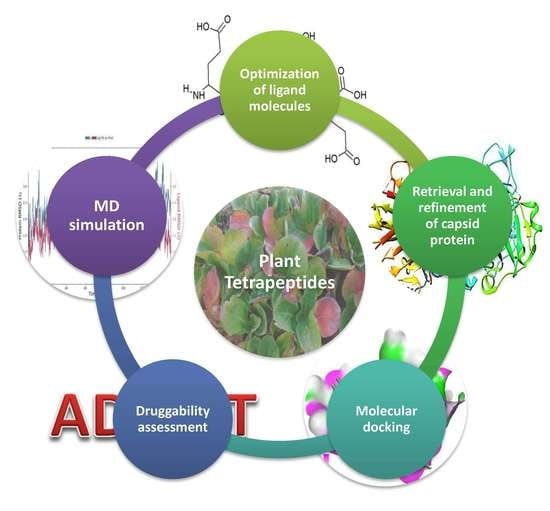
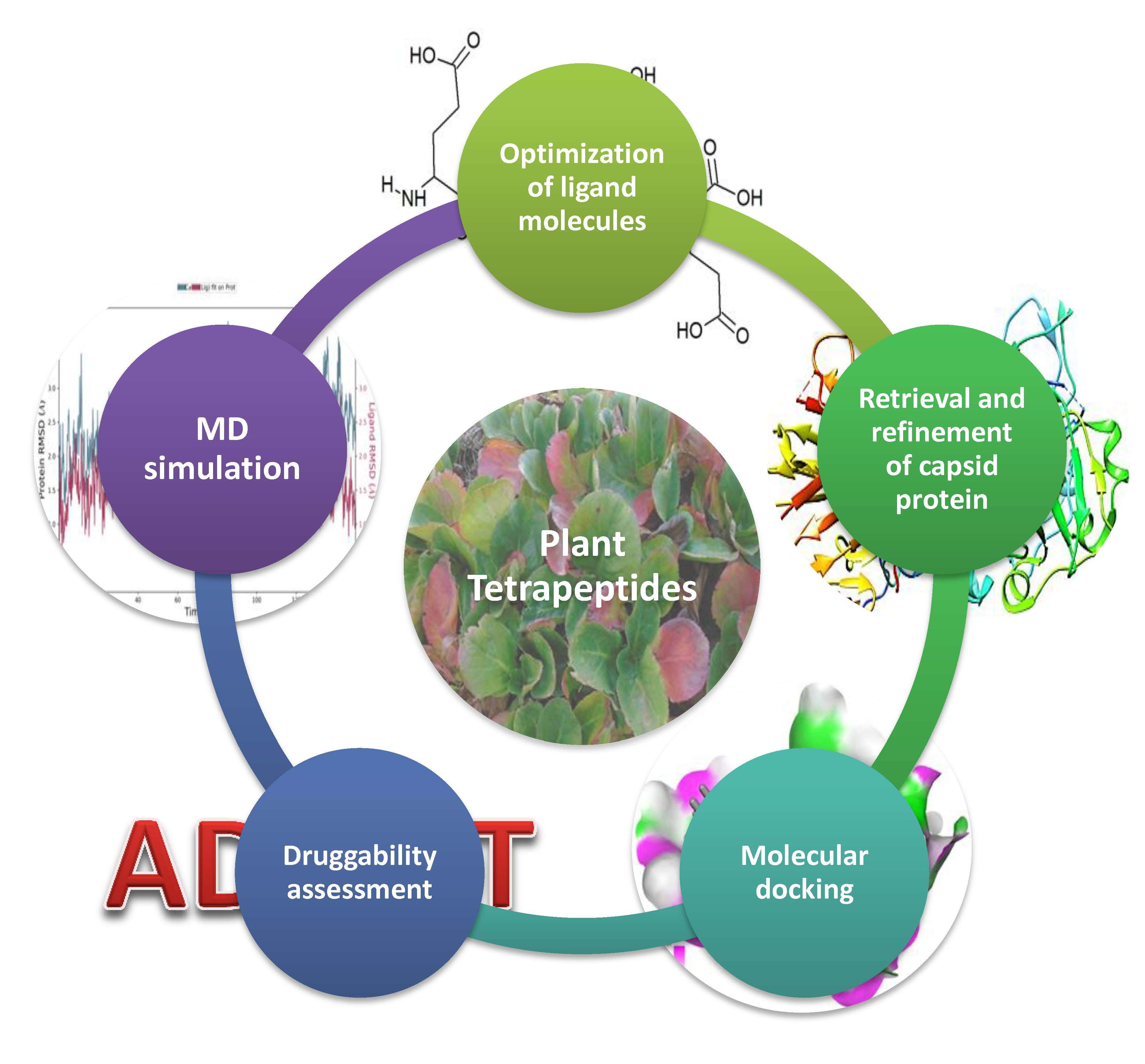
1. Introduction
2. Results
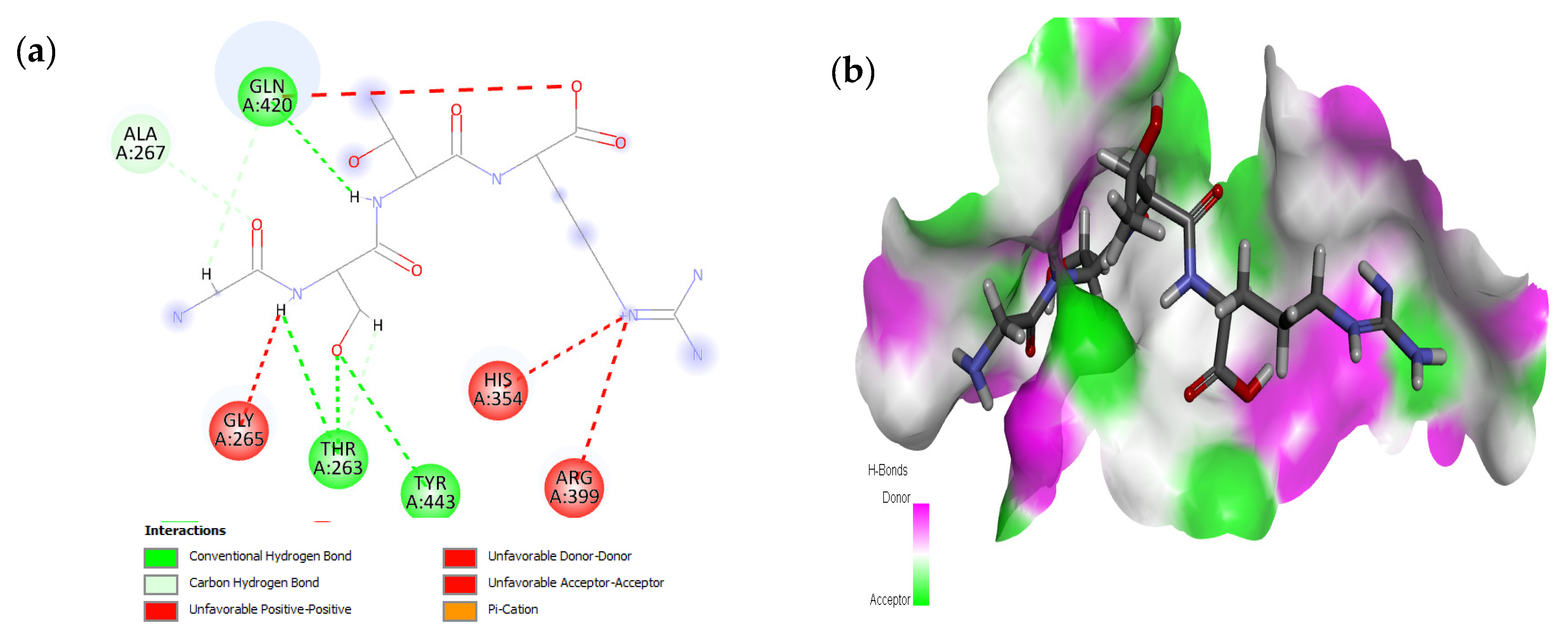
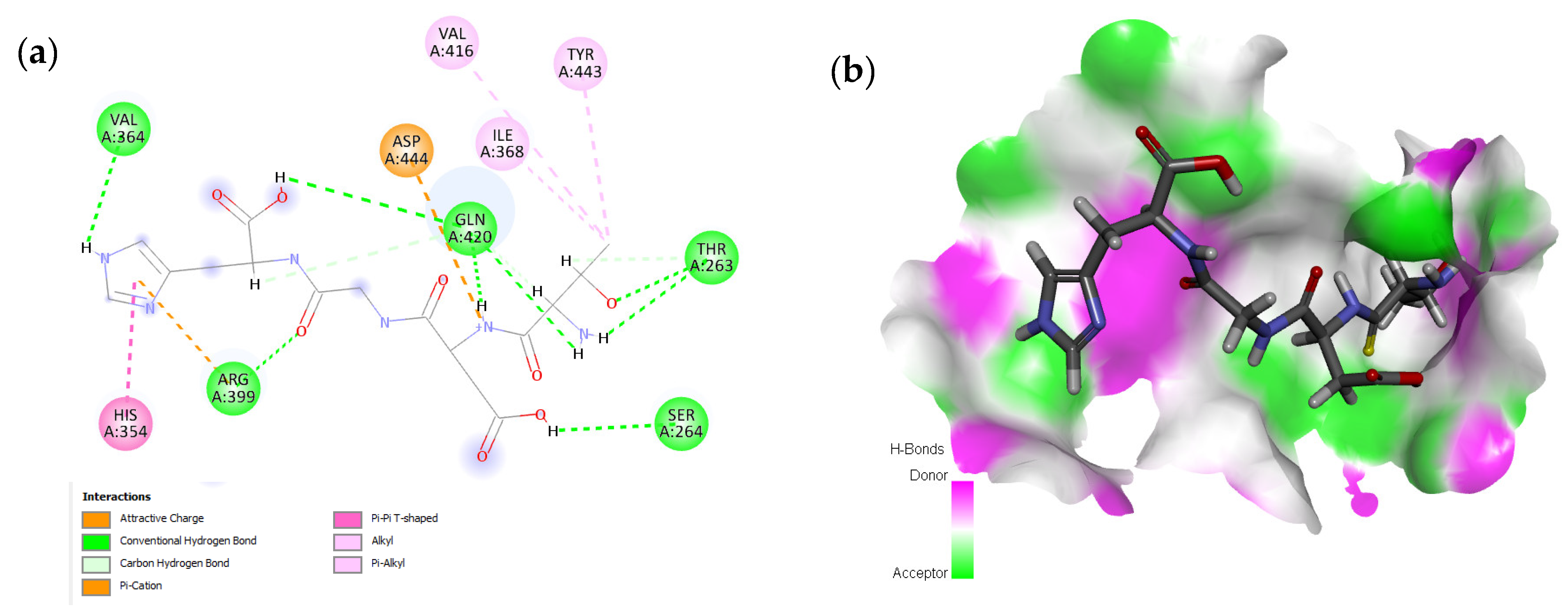
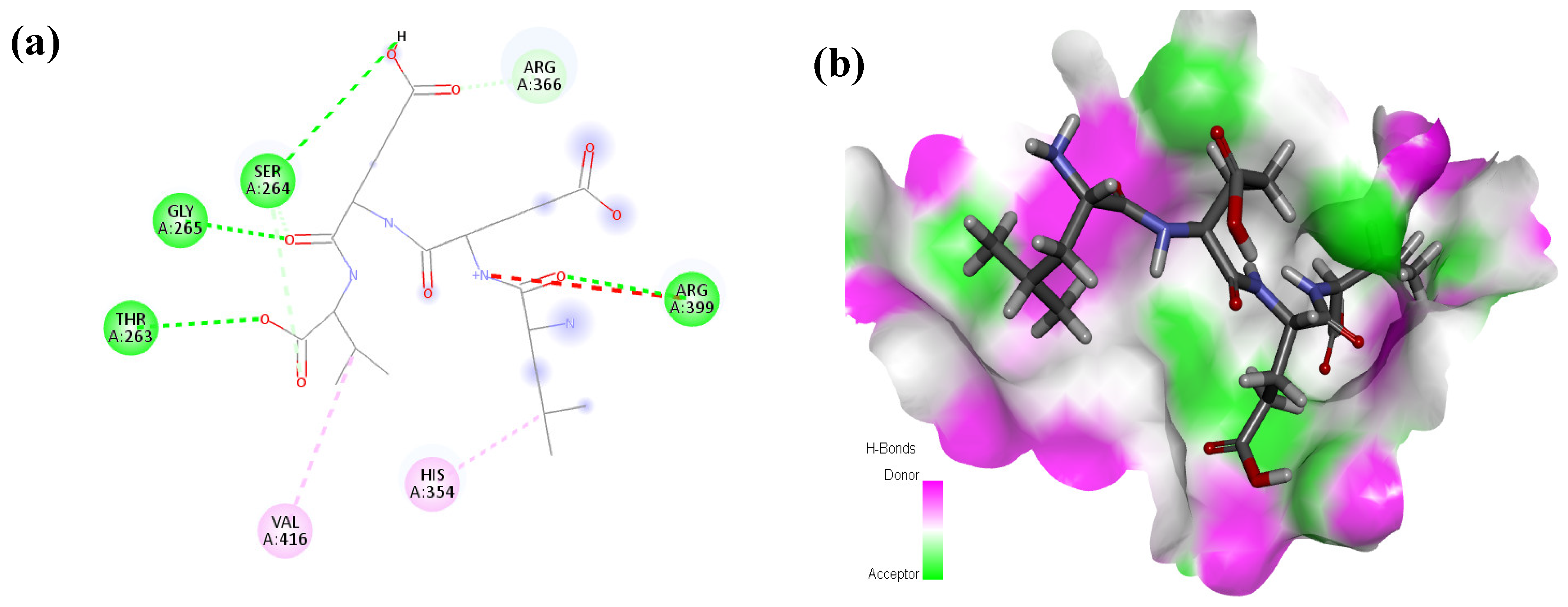
2.1. Molecular Docking and Interaction Analysis
2.2. Drug Scanning and ADMET Profiling
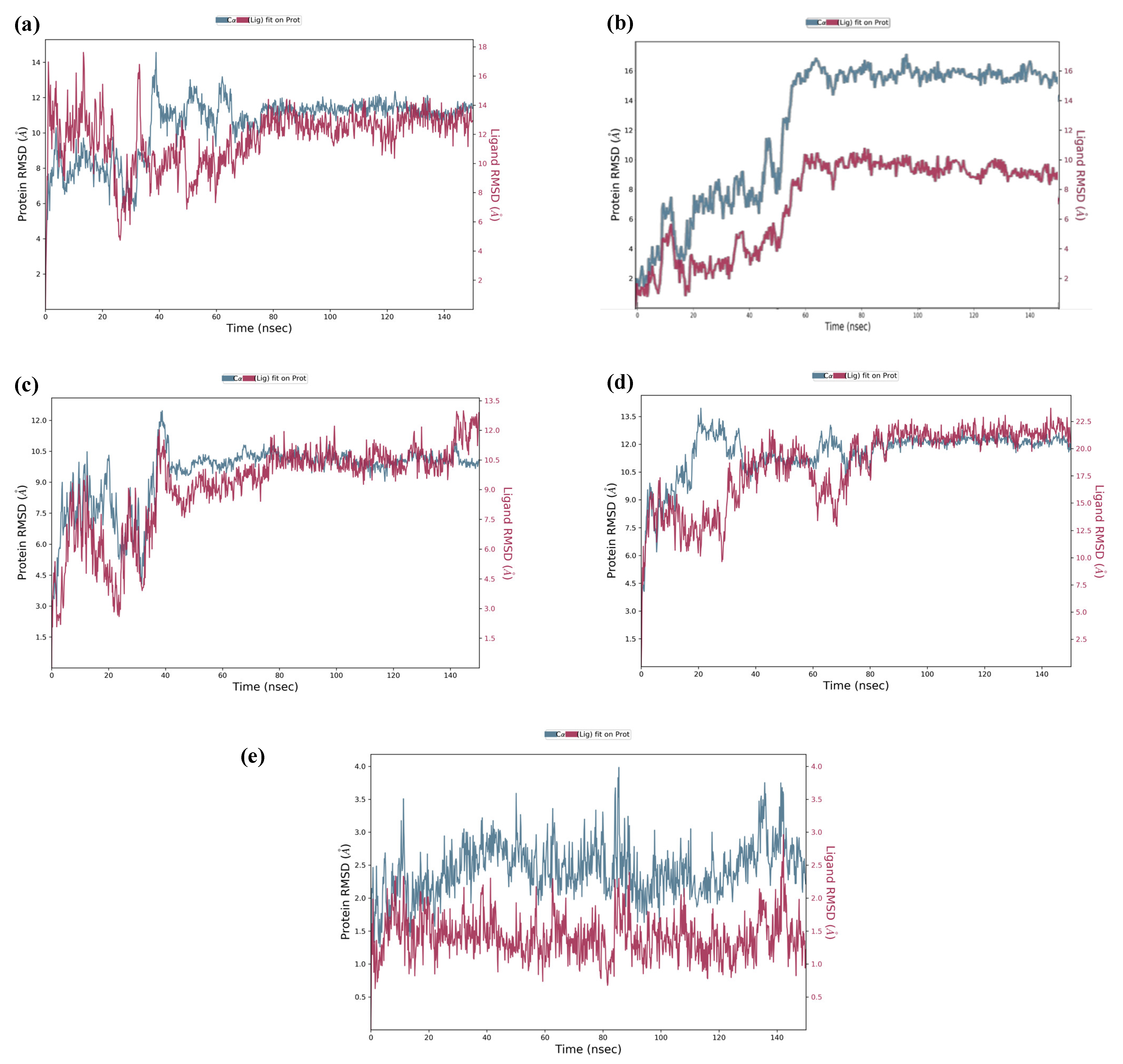
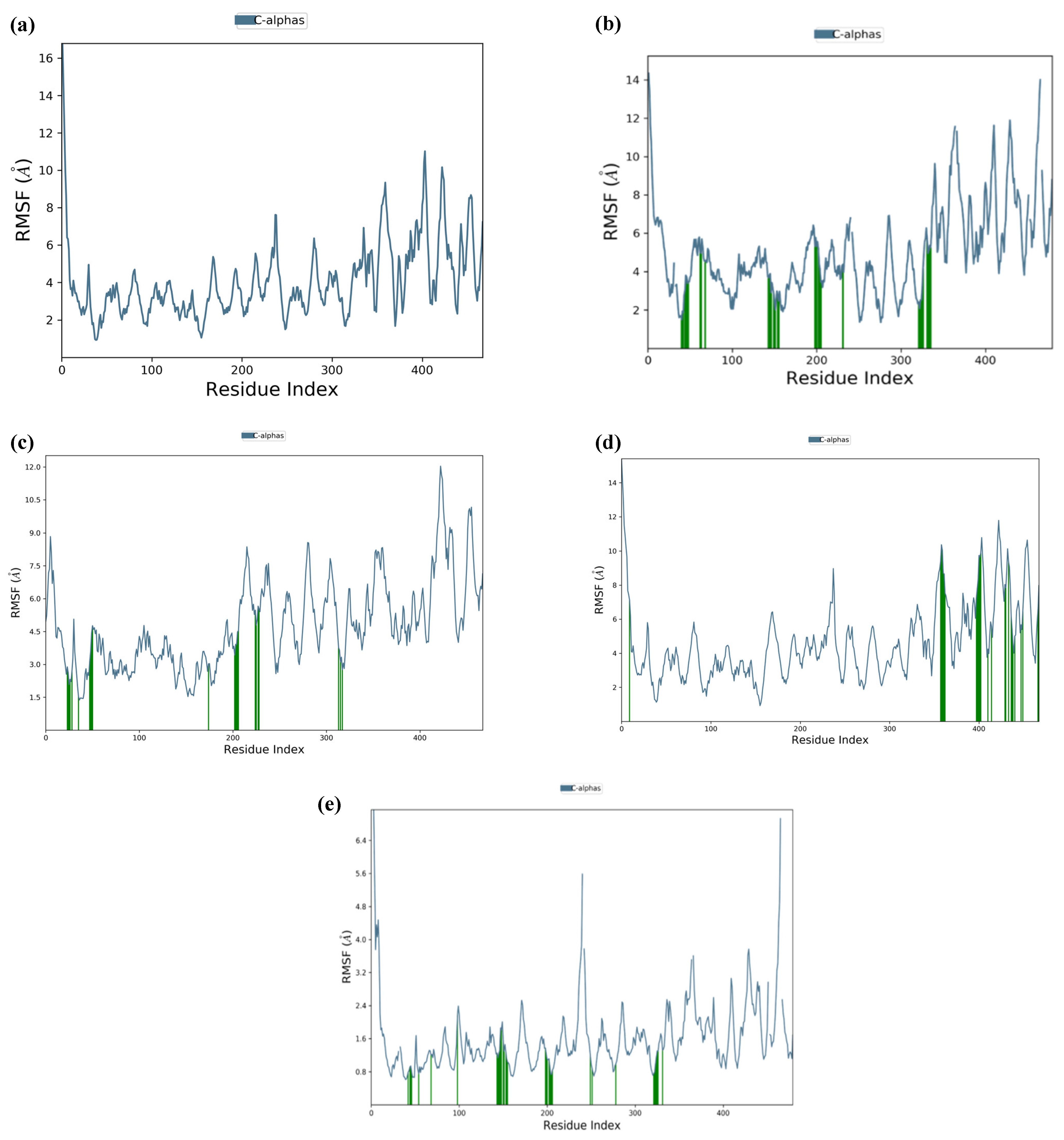
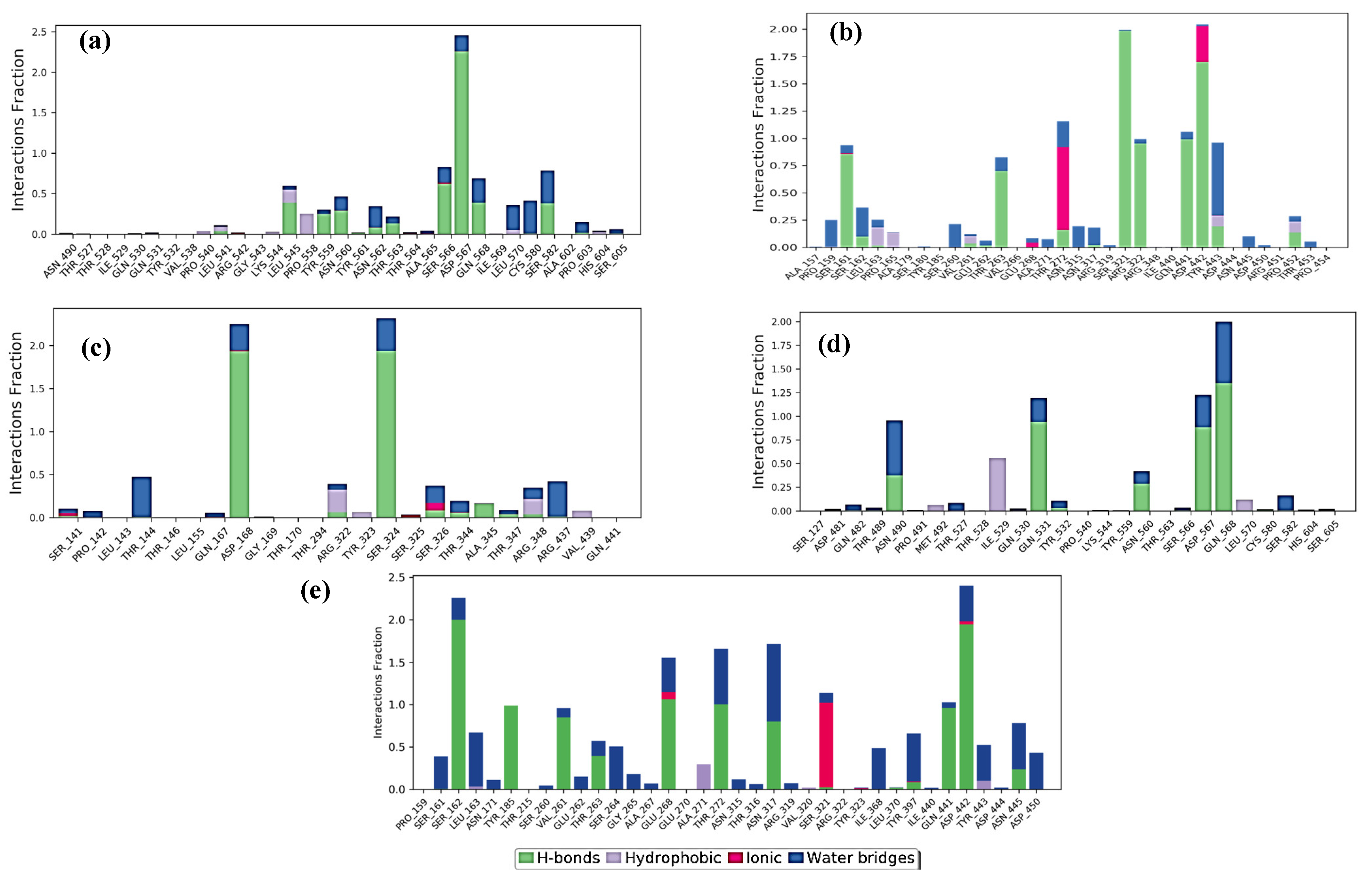
2.3. Molecular Dynamics Simulation
3. Discussion
4. Materials and Methods
4.1. Retrieval and Optimization of Ligand Molecules
4.2. Retrieval and Refinement of the Receptor Protein
4.3. Molecular Docking Analysis
4.4. Evaluation of Pharmacokinetics Parameters
4.5. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ma, Z.; Robert, A.; Kamar, N.; Pan, Q. Chronic hepatitis E: Advancing research and patient care. J. Hepatol. 2022, 77, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Izopet, J.; Pavio, N.; Aggarwal, R.; Labrique, A.; Wedemeyer, H.; Dalton, H.R. Hepatitis E virus infection. Nat. Rev. Dis. Prim. 2017, 3, 17086. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, G.; Sharma, S.; Kumar, A.; Prasad, S.; Agarwal, S.; Kar, P. Reduced glutathione in hepatitis E infection and pregnancy outcome. J. Obstet. Gynaecol. Res. 2016, 42, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Primadharsini, P.P.; Nagashima, S.; Okamoto, H. Genetic variability and evolution of hepatitis E virus. Viruses 2019, 11, 456. [Google Scholar] [CrossRef]
- Purdy, M.A.; Harrison, T.J.; Jameel, S.; Meng, X.-J.; Okamoto, H.; Van der Poel, W.; Smith, D.B.; Consortium, I.R. ICTV virus taxonomy profile: Hepeviridae. J. Gen. Virol. 2017, 98, 2645. [Google Scholar] [CrossRef]
- Wang, B.; Yang, X.-L. Chirohepevirus from bats: Insights into hepatitis E virus diversity and evolution. Viruses 2022, 14, 905. [Google Scholar] [CrossRef]
- Nan, Y.; Zhang, Y.-J. Molecular biology and infection of hepatitis E virus. Front. Microbiol. 2016, 7, 1419. [Google Scholar] [CrossRef]
- Sridhar, S.; Teng, J.L.; Chiu, T.-H.; Lau, S.K.; Woo, P.C. Hepatitis E virus genotypes and evolution: Emergence of camel hepatitis E variants. Int. J. Mol. Sci. 2017, 18, 869. [Google Scholar] [CrossRef]
- Xing, L.; Wang, J.C.; Li, T.-C.; Yasutomi, Y.; Lara, J.; Khudyakov, Y.; Schofield, D.; Emerson, S.U.; Purcell, R.H.; Takeda, N. Spatial configuration of hepatitis E virus antigenic domain. J. Virol. 2011, 85, 1117–1124. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Tanaka, T.; Yamada, K.; Nishizawa, T.; Okamoto, H. A PSAP motif in the ORF3 protein of hepatitis E virus is necessary for virion release from infected cells. J. Gen. Virol. 2011, 92, 269–278. [Google Scholar] [CrossRef]
- Ahmad, I.; Holla, R.P.; Jameel, S. Molecular virology of hepatitis E virus. Virus Res. 2011, 161, 47–58. [Google Scholar] [CrossRef]
- Paliwal, D.; Panda, S.K.; Kapur, N.; Varma, S.P.K.; Durgapal, H. Hepatitis E virus (HEV) protease: A chymotrypsin-like enzyme that processes both non-structural (pORF1) and capsid (pORF2) protein. J. Gen. Virol. 2014, 95, 1689–1700. [Google Scholar] [CrossRef]
- Kumar, A.; Sahu, U.; Agnihotri, G.; Dixit, A.; Khare, P. A novel multi-epitope peptide vaccine candidate targeting Hepatitis E virus: An in-silico approach. bioRxiv 2022. [Google Scholar] [CrossRef]
- Quintero-Gil, C.; Parra-Suescún, J.; Lopez-Herrera, A.; Orduz, S. In-Silico design and molecular docking evaluation of peptides derivatives from bacteriocins and porcine beta defensin-2 as inhibitors of Hepatitis E virus capsid protein. Virusdisease 2017, 28, 281–288. [Google Scholar] [CrossRef]
- Zhou, Z.; Xie, Y.; Wu, C.; Nan, Y. The hepatitis E virus open reading frame 2 protein: Beyond viral capsid. Front. Microbiol. 2021, 12, 739124. [Google Scholar] [CrossRef]
- Kamar, N.; Abravanel, F.; Behrendt, P.; Hofmann, J.; Pageaux, G.P.; Barbet, C.; Moal, V.; Couzi, L.; Horvatits, T.; De Man, R.A. Ribavirin for hepatitis E virus infection after organ transplantation: A large European retrospective multicenter study. Clin. Infect. Dis. 2020, 71, 1204–1211. [Google Scholar] [CrossRef]
- Gorris, M.; van Der Lecq, B.M.; van Erpecum, K.J.; de Bruijne, J. Treatment for chronic hepatitis E virus infection: A systematic review and meta-analysis. J. Viral Hepat. 2021, 28, 454–463. [Google Scholar] [CrossRef]
- Hui, W.; Wei, L.; Li, Z.; Guo, X. Treatment of hepatitis E. Hepat. E Virus 2016, 948, 211–221. [Google Scholar]
- Sayed, I.M.; Karam-Allah Ramadan, H.; Hafez, M.H.; Elkhawaga, A.A.; El-Mokhtar, M.A. Hepatitis E virus (HEV) open reading frame 2: Role in pathogenesis and diagnosis in HEV infections. Rev. Med. Virol. 2022, 32, e2401. [Google Scholar] [CrossRef]
- Mustafa, G.; Arif, R.; Atta, A.; Sharif, S.; Jamil, A. Bioactive compounds from medicinal plants and their importance in drug discovery in Pakistan. Matrix Sci. Pharma 2017, 1, 17–26. [Google Scholar] [CrossRef]
- Mahrosh, H.S.; Tanveer, M.; Arif, R.; Mustafa, G. Computer-Aided prediction and identification of phytochemicals as potential drug candidates against MERS-CoV. BioMed Res. Int. 2021, 2021, 5578689. [Google Scholar] [CrossRef]
- Tietcheu Galani, B.R.; Ayissi Owona, V.B.; Guemmogne Temdie, R.J.; Metzger, K.; Atsama Amougou, M.; Djamen Chuisseu, P.D.; Fondjo Kouam, A.; Ngounoue Djuidje, M.; Aliouat-Denis, C.-M.; Cocquerel, L. In Silico and in vitro screening of licensed antimalarial drugs for repurposing as inhibitors of hepatitis E virus. In Silico Pharmacol. 2021, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, G.; Mahrosh, H.S.; Zafar, M.; Attique, S.A.; Arif, R. Exploring the antihyperglycemic potential of tetrapeptides devised from AdMc1 via different receptor proteins inhibition using in silico approaches. Int. J. Immunopathol. Pharmacol. 2022, 36, 03946320221103120. [Google Scholar] [CrossRef] [PubMed]
- Mahrosh, H.S.; Mustafa, G. An in silico approach to target RNA-dependent RNA polymerase of COVID-19 with naturally occurring phytochemicals. Environ. Dev. Sustain. 2021, 23, 16674–16687. [Google Scholar] [CrossRef]
- Pischke, S.; Schulze-zur-Wiesch, J.; Lütgehetmann, M.; Kreuels, B.; Lueth, S.; Kapaun, P.; Benten, D.; Schmiedel, S.; Sterneck, M.; Lohse, A.W. High clinical manifestation rate in an imported outbreak of hepatitis E genotype 1 infection in a German group of travellers returning from India. Ann. Hepatol. 2017, 16, 57–62. [Google Scholar] [CrossRef]
- Bi, H.; Yang, R.; Wu, C.; Xia, J. Hepatitis E virus and blood transfusion safety. Epidemiol. Infect. 2020, 148, E158. [Google Scholar] [CrossRef]
- Hughes, J.M.; Wilson, M.E.; Teshale, E.H.; Hu, D.J.; Holmberg, S.D. The two faces of hepatitis E virus. Clin. Infect. Dis. 2010, 51, 328–334. [Google Scholar]
- Nguyen, H.; Shukla, P.; Torian, U.; Faulk, K.; Emerson, S. Hepatitis E virus genotype 1 infection of swine kidney cells in vitro is inhibited at multiple levels. J. Virol. 2014, 88, 868–877. [Google Scholar] [CrossRef]
- Montpellier, C.; Wychowski, C.; Sayed, I.M.; Meunier, J.-C.; Saliou, J.-M.; Ankavay, M.; Bull, A.; Pillez, A.; Abravanel, F.; Helle, F. Hepatitis E virus lifecycle and identification of 3 forms of the ORF2 capsid protein. Gastroenterology 2018, 154, 211–223.e8. [Google Scholar] [CrossRef]
- Mushtaq, A.; Mustafa, G.; Ansari, T.M.; Shad, M.A.; Cruz-Reyes, J.; Jamil, A. Antiviral activity of hexapeptides derived from conserved regions of bacterial proteases against HCV NS3 protease. Pak. J. Pharm. Sci. 2021, 34, 217–223. [Google Scholar]
- Glitscher, M.; Himmelsbach, K.; Woytinek, K.; Johne, R.; Reuter, A.; Spiric, J.; Schwaben, L.; Grünweller, A.; Hildt, E. Inhibition of hepatitis E virus spread by the natural compound silvestrol. Viruses 2018, 10, 301. [Google Scholar] [CrossRef]
- Das, N.; Kalita, P.P.; Sarma, M.P.; Bhattacharjee, M. Molecular Modeling of HEV Core Protein and Active Compounds from Northeast Folk Medicine. J. Biochem. Technol. 2021, 12, 12–18. [Google Scholar] [CrossRef]
- Zhang, C.; Freistaedter, A.; Schmelas, C.; Gunkel, M.; Dao Thi, V.L.; Grimm, D. An RNA Interference/Adeno-Associated Virus Vector–Based Combinatorial Gene Therapy Approach Against Hepatitis E Virus. Hepatol. Commun. 2022, 6, 878–888. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Li, Z.; Wan, H.; Shi, Y.; Ouyang, P. Personal experience with four kinds of chemical structure drawing software: Review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J. Chem. Inf. Comput. Sci. 2004, 44, 1886–1890. [Google Scholar] [CrossRef]
- Mustafa, G.; Mahrosh, H.S.; Salman, M.; Sharif, S.; Jabeen, R.; Majeed, T.; Tahir, H. Identification of peptides as novel inhibitors to target IFN-γ, IL-3, and TNF-α in systemic lupus erythematosus. BioMed Res. Int. 2021, 2021, 1124055. [Google Scholar] [CrossRef]
- Arif, R.; Ahmad, S.; Mustafa, G.; Mahrosh, H.S.; Ali, M.; ul Qamar, M.T.; Dar, H.R. Molecular docking and simulation studies of antidiabetic agents devised from hypoglycemic polypeptide-P of Momordica charantia. BioMed Res. Int. 2021, 2021, 5561129. [Google Scholar] [CrossRef]
- Althagafi, I.; El-Metwaly, N.; Farghaly, T.A. New series of thiazole derivatives: Synthesis, structural elucidation, antimicrobial activity, molecular modeling and MOE docking. Molecules 2019, 24, 1741. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule library screening by docking with PyRx. In Chemical Biology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 243–250. [Google Scholar]
- Mustafa, G.; Mahrosh, H.S.; Arif, R. In Silico characterization of growth differentiation factors as inhibitors of TNF-alpha and IL-6 in immune-mediated inflammatory disease rheumatoid arthritis. BioMed Res. Int. 2021, 2021, 5538535. [Google Scholar] [CrossRef]
- Sharma, S.; Sharma, A.; Gupta, U. Molecular Docking studies on the Anti-fungal activity of Allium sativum (Garlic) against Mucormycosis (black fungus) by BIOVIA discovery studio visualizer 21.1. 0.0. Ann. Antivir. Antiretrovir. 2021, 5, 028–032. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-Service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.; Chow, E.X.; Dror, H.; Eastwood, R.; Gregersen, M.; Klepeis, B.; Kolossvary, J.; Moraes, I.; Sacerdoti, M.; Salmon, F. Molecular dynamics—Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM, IEEE Conference on Supercomputing, New York, NY, USA, 22–26 June 2006; p. 84. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Ligand | Structure | S-Score | Interacting Amino Acids * |
|---|---|---|---|---|
| 1 | GSTR |  | −10.1 | Thr263, Gln420, Tyr443 |
| 2 | TDGH |  | −8.5 | Thr263, Ser264, Val364, Arg399, Gln420 |
| 3 | LEEV |  | −8.4 | Thr263, Ser264, Gly265, Arg399 |
| 4 | WDDG |  | −8.2 | Arg366, Arg399, Gln420 |
| 5 | EGDE |  | −8.0 | Gly265, His354, Val364, Arg399, Gln420, Asp444 |
| 6 | FTDG |  | −7.8 | Thr263, Val266, Val364, Arg366, Arg399, Asp444 |
| 7 | EPST |  | −7.5 | Thr263, Ser264, Gly265, Val266, Arg366, Asp444, Gln446 |
| 8 | SDAF |  | −6.5 | Val364, Arg366, Arg399, Glu417 |
| 9 | PRGS |  | −5.7 | Leu353, His354, Arg366, Gln446, Arg451, Thr453 |
| 10 | NTFP |  | −5.6 | Val364, Arg399, Glu417, Gln420, Gln421 |
| Peptides | Molecular Properties † | ||||||
|---|---|---|---|---|---|---|---|
| MW | HBD | HBA | nrotb | Log P | A | Violations | |
| GSTR | 419.44 | 10 | 8 | 17 | −3.58 | 99.13 | 1 |
| TDGH | 428.40 | 8 | 8 | 12 | −3.24 | 97.66 | 1 |
| LEEV | 488.54 | 7 | 7 | 16 | −0.72 | 120.09 | 1 |
| WDDG | 491.46 | 8 | 7 | 16 | −1.68 | 117.98 | 1 |
| EGDE | 448.39 | 8 | 8 | 15 | −3.01 | 97.83 | 1 |
| FTDG | 438.44 | 7 | 7 | 12 | −1.66 | 438.44 | 1 |
| EPST | 432.43 | 7 | 8 | 11 | −3.16 | 103.31 | 1 |
| SDAF | 438.44 | 7 | 7 | 12 | −1.69 | 105.51 | 1 |
| PRGS | 415.45 | 9 | 7 | 12 | −2.96 | 104.67 | 1 |
| NTFP | 477.52 | 6 | 7 | 11 | −1.68 | 122.96 | 1 |
| Peptides | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GSTR | TDGH | LEEV | WDDG | EGDE | FTDG | EPST | SDAF | PRGS | NTFP | |
| Absorption | ||||||||||
| BBB | – | – | – | – | – | – | – | – | – | – |
| HIA | – | + | – | – | – | – | – | – | – | + |
| Caco-2 Permeability | Caco-2- | Caco-2- | Caco-2- | Caco-2- | Caco-2- | Caco-2- | Caco-2- | Caco-2- | Caco-2- | Caco-2- |
| PGS | Substrate | NS | NS | NS | NS | Substrate | Substrate | NS | Substrate | Substrate |
| PGI | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI |
| ROCT | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI |
| Metabolism | ||||||||||
| CYP3A4 substrate | Substrate | Substrate | NS | Substrate | NS | NS | Substrate | NS | Substrate | Substrate |
| CYP2C9 substrate | NS | NS | Substrate | NS | NS | NS | NS | NS | NS | NS |
| CYP2D6 substrate | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| CYP3A4 inhibition | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI |
| CYP2C9 inhibition | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI |
| CYP2C19 inhibition | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI |
| CYP2D6 inhibition | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI |
| CYP1A2 inhibition | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI |
| Toxicity | ||||||||||
| AMES Toxicity | NAT | NAT | NAT | NAT | NAT | NAT | NAT | NAT | NAT | NAT |
| Carcinogens | NC | NC | NC | NC | NC | NC | NC | NC | NC | NC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustafa, G.; Mahrosh, H.S.; Attique, S.A.; Arif, R.; Farah, M.A.; Al-Anazi, K.M.; Ali, S. Identification of Plant Peptides as Novel Inhibitors of Orthohepevirus A (HEV) Capsid Protein by Virtual Screening. Molecules 2023, 28, 2675. https://doi.org/10.3390/molecules28062675
Mustafa G, Mahrosh HS, Attique SA, Arif R, Farah MA, Al-Anazi KM, Ali S. Identification of Plant Peptides as Novel Inhibitors of Orthohepevirus A (HEV) Capsid Protein by Virtual Screening. Molecules. 2023; 28(6):2675. https://doi.org/10.3390/molecules28062675
Chicago/Turabian StyleMustafa, Ghulam, Hafiza Salaha Mahrosh, Syed Awais Attique, Rawaba Arif, Mohammad Abul Farah, Khalid Mashay Al-Anazi, and Sajad Ali. 2023. "Identification of Plant Peptides as Novel Inhibitors of Orthohepevirus A (HEV) Capsid Protein by Virtual Screening" Molecules 28, no. 6: 2675. https://doi.org/10.3390/molecules28062675
APA StyleMustafa, G., Mahrosh, H. S., Attique, S. A., Arif, R., Farah, M. A., Al-Anazi, K. M., & Ali, S. (2023). Identification of Plant Peptides as Novel Inhibitors of Orthohepevirus A (HEV) Capsid Protein by Virtual Screening. Molecules, 28(6), 2675. https://doi.org/10.3390/molecules28062675