



Proteomics Analysis of Lymphoblastoid Cell Lines from Patients with Amyotrophic Lateral Sclerosis




Abstract
1. Introduction
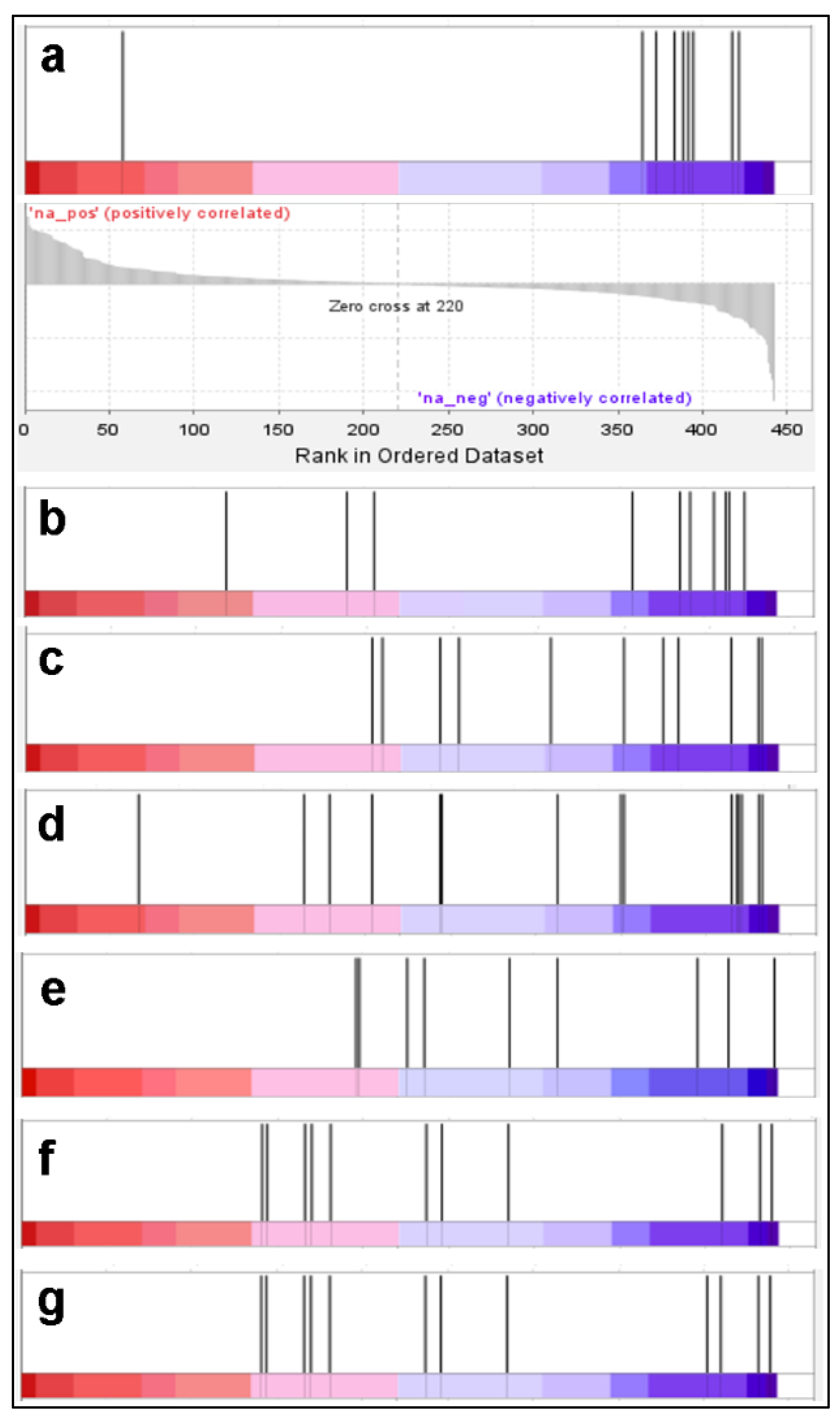
2. Results and Discussion
3. Materials and Methods
Cell Lines
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; Van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2016, 138, 225–238. [Google Scholar]
- Iacoangeli, A.; Al Khleifat, A.; Jones, A.R.; Sproviero, W.; Shatunov, A.; Opie-Martin, S.; Morrison, K.E.; Shaw, P.J.; Shaw, C.E.; Fogh, I. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol. Commun. 2019, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski Jr, T.; Bosco, D.; Leclerc, A.; Tamrazian, E.; Vanderburg, C.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.; Munsat, T. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Hulisz, D. Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar] [PubMed]
- Pansarasa, O.; Garofalo, M.; Scarian, E.; Dragoni, F.; Garau, J.; Di Gerlando, R.; Diamanti, L.; Bordoni, M.; Gagliardi, S. Biomarkers in Human Peripheral Blood Mononuclear Cells: The State of the Art in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 2580. [Google Scholar] [CrossRef]
- Riancho, J.; Arozamena, S.; De Munain, A. Dermic-derived fibroblasts for the study of amyotrophic lateral sclerosis. Neural Regen. Res. 2020, 15, 2043. [Google Scholar] [CrossRef]
- Hussain, T.; Mulherkar, R. Lymphoblastoid cell lines: A continuous in vitro source of cells to study carcinogen sensitivity and DNA repair. Int. J. Mol. Cell. Med. 2012, 1, 75. [Google Scholar]
- Hui-Yuen, J.; McAllister, S.; Koganti, S.; Hill, E.; Bhaduri-McIntosh, S. Establishment of Epstein-Barr virus growth-transformed lymphoblastoid cell lines. JoVE (J. Vis. Exp.) 2011, 57, e3321. [Google Scholar]
- Pansarasa, O.; Bordoni, M.; Drufuca, L.; Diamanti, L.; Sproviero, D.; Trotti, R.; Bernuzzi, S.; La Salvia, S.; Gagliardi, S.; Ceroni, M. Lymphoblastoid cell lines as a model to understand amyotrophic lateral sclerosis disease mechanisms. Dis. Model. Mech. 2018, 11, dmm031625. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P. Lymphoblastoid cell lines as models to study mitochondrial function in neurological disorders. Int. J. Mol. Sci. 2021, 22, 4536. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Bury, J.J.; Heath, P.R.; Wyles, M.; Higginbottom, A.; Gelsthorpe, C.; Highley, J.R.; Hautbergue, G.; Rattray, M.; Kirby, J. C9ORF72 GGGGCC expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosis. PLoS ONE 2015, 10, e0127376. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.; Carter, E.K.; Dammer, E.B.; Duong, D.M.; Gerasimov, E.S.; Liu, Y.; Liu, J.; Betarbet, R.; Ping, L.; Yin, L. Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 2022, 25, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Channaveerappa, D.; Lux, J.C.; Wormwood, K.L.; Heintz, T.A.; McLerie, M.; Treat, J.A.; King, H.; Alnasser, D.; Goodrow, R.J.; Ballard, G.; et al. Atrial electrophysiological and molecular remodelling induced by obstructive sleep apnoea. J. Cell Mol. Med. 2017, 21, 2223–2235. [Google Scholar] [CrossRef]
- Dupree, E.J.; Crimmins, B.S.; Holsen, T.M.; Darie, C.C. Proteomic Analysis of the Lake Trout (Salvelinus namaycush) Liver Identifies Proteins from Evolutionarily Close and -Distant Fish Relatives. Proteomics 2019, 19, e1800429. [Google Scholar] [CrossRef]
- Dupree, E.J.; Crimmins, B.S.; Holsen, T.M.; Darie, C.C. Developing Well-Annotated Species-Specific Protein Databases Using Comparative Proteogenomics. Adv. Exp. Med. Biol. 2019, 1140, 389–400. [Google Scholar] [PubMed]
- Neagu, A.-N.; Jayathirtha, M.; Baxter, E.; Donnelly, M.; Petre, B.A.; Darie, C.C. Applications of Tandem Mass Spectrometry (MS/MS) in Protein Analysis for Biomedical Research. Molecules 2022, 27, 2411. [Google Scholar] [CrossRef] [PubMed]
- Neagu, A.-N.; Whitham, D.; Buonanno, E.; Jenkins, A.; Alexa-Stratulat, T.; Tamba, B.I.; Darie, C.C. Proteomics and its applications in breast cancer. Am. J. Cancer Res. 2021, 11, 4006–4049. [Google Scholar]
- Darie, C.C.; Deinhardt, K.; Zhang, G.; Cardasis, H.S.; Chao, M.V.; Neubert, T.A. Identifying transient protein-protein interactions in EphB2 signaling by blue native PAGE and mass spectrometry. Proteomics 2011, 11, 4514–4528. [Google Scholar] [CrossRef]
- Ngounou Wetie, A.G.; Sokolowska, I.; Channaveerappa, D.; Dupree, E.J.; Jayathirtha, M.; Woods, A.G.; Darie, C.C. Proteomics and Non-proteomics Approaches to Study Stable and Transient Protein-Protein Interactions. Adv. Exp. Med. Biol. 2019, 1140, 121–142. [Google Scholar] [PubMed]
- Ngounou Wetie, A.G.; Sokolowska, I.; Woods, A.G.; Roy, U.; Deinhardt, K.; Darie, C.C. Protein-protein interactions: Switch from classical methods to proteomics and bioinformatics-based approaches. Cell. Mol. Life Sci. 2014, 71, 205–228. [Google Scholar] [CrossRef]
- Jayathirtha, M.; Neagu, A.N.; Whitham, D.; Alwine, S.; Darie, C.C. Investigation of the effects of downregulation of jumping translocation breakpoint (JTB) protein expression in MCF7 cells for potential use as a biomarker in breast cancer. Am. J. Cancer Res. 2022, 12, 4373–4398. [Google Scholar] [PubMed]
- Jayathirtha, M.; Neagu, A.N.; Whitham, D.; Alwine, S.; Darie, C.C. Investigation of the effects of overexpression of jumping translocation breakpoint (JTB) protein in MCF7 cells for potential use as a biomarker in breast cancer. Am. J. Cancer Res. 2022, 12, 1784–1823. [Google Scholar]
- Willforss, J.; Chawade, A.; Levander, F. NormalyzerDE: Online tool for improved normalization of omics expression data and high-sensitivity differential expression analysis. J. Proteome Res. 2018, 18, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Ahlmann-Eltze, C.; Anders, S. proDA: Probabilistic dropout analysis for identifying differentially abundant proteins in label-free mass spectrometry. Biorxiv 2020, 661496. [Google Scholar]
- Suomi, T.; Seyednasrollah, F.; Jaakkola, M.K.; Faux, T.; Elo, L.L. ROTS: An R package for reproducibility-optimized statistical testing. PLoS Comput. Biol. 2017, 13, e1005562. [Google Scholar] [CrossRef]
- Cacabelos, D.; Ayala, V.; Granado-Serrano, A.B.; Jove, M.; Torres, P.; Boada, J.; Cabre, R.; Ramirez-Nunez, O.; Gonzalo, H.; Soler-Cantero, A.; et al. Interplay between TDP-43 and docosahexaenoic acid-related processes in amyotrophic lateral sclerosis. Neurobiol. Dis. 2016, 88, 148–160. [Google Scholar] [CrossRef]
- Iridoy, M.O.; Zubiri, I.; Zelaya, M.V.; Martinez, L.; Ausín, K.; Lachen-Montes, M.; Santamaría, E.; Fernandez-Irigoyen, J.; Jericó, I. Neuroanatomical Quantitative Proteomics Reveals Common Pathogenic Biological Routes between Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). Int. J. Mol. Sci. 2019, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rangaswamy, S.; Kodavati, M.; Mitra, J.; Guo, W.; Guerrero, E.N.; Van Den Bosch, L.; Hegde, M.L. RT2 PCR array screening reveals distinct perturbations in DNA damage response signaling in FUS-associated motor neuron disease. Mol. Brain 2019, 12, 1–5. [Google Scholar] [CrossRef]
- Leoni, E.; Bremang, M.; Mitra, V.; Zubiri, I.; Jung, S.; Lu, C.H.; Adiutori, R.; Lombardi, V.; Russell, C.; Koncarevic, S.; et al. Combined tissue-fluid proteomics to unravel phenotypic variability in amyotrophic lateral sclerosis. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Mech, A.M.; Aktar, T.; Zhang, Y.; Schiavo, G. Altered sensory neuron development in CMT2D mice is site-specific and linked to increased GlyRS levels. Front. Cell. Neurosci. 2020, 14, 232. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.I.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Dowell, J.A.; Wright, L.J.; Armstrong, E.A.; Denu, J.M. Benchmarking quantitative performance in label-free proteomics. ACS Omega 2021, 6, 2494–2504. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Lopez-Gonzalez, R.; Krishnan, G.; Phillips, H.L.; Li, A.N.; Seeley, W.W.; Yao, W.D.; Almeida, S.; Gao, F.B. C9ORF72-ALS/FTD-associated poly (GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 2019, 22, 851–862. [Google Scholar] [CrossRef]
- Tsukahara, A.; Hosokawa, T.; Nishioka, D.; Kotani, T.; Ishida, S.; Takeuchi, T.; Kimura, F.; Arawaka, S. Neuron-specific enolase level is a useful biomarker for distinguishing amyotrophic lateral sclerosis from cervical spondylotic myelopathy. Sci. Rep. 2021, 11, 1–9. [Google Scholar]
- Rushkevich, Y.N.; Pashkouskaya, I.; Likhachev, S. Neurospecific proteins in cerebrospinal fluid and in the bloodserum of patients with amyotrophic lateral sclerosis. Zhurnal Nevrol. I Psikhiatrii Im. SS Korsakova 2018, 118, 75–80. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard Jr, J.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Dalla Bella, E.; Tramacere, I.; Antonini, G.; Borghero, G.; Capasso, M.; Caponnetto, C.; Chiò, A.; Corbo, M.; Eleopra, R.; Filosto, M.; et al. Protein misfolding, amyotrophic lateral sclerosis and guanabenz: Protocol for a phase II RCT with futility design (ProMISe trial). BMJ Open 2017, 7, e015434. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, J.; Van Den Bosch, L. The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 12175. [Google Scholar] [CrossRef]
- Ferreira, P.A. The coming-of-age of nucleocytoplasmic transport in motor neuron disease and neurodegeneration. Cell. Mol. Life Sci. 2019, 76, 2247–2273. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Bogaert, E.; Van Damme, P.; Van Den Bosch, L. Inside out: The role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016, 132, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, C.G.; Lum, J.S.; Farrawell, N.E.; Yerbury, J.J. Ubiquitin homeostasis disruption, a common cause of proteostasis collapse in amyotrophic lateral sclerosis? Neural Regen. Res. 2022, 17, 2218. [Google Scholar] [PubMed]
- Zhang, M.; Xi, Z.; Saez-Atienzar, S.; Chia, R.; Moreno, D.; Sato, C.; Montazer Haghighi, M.; Traynor, B.J.; Zinman, L.; Rogaeva, E. Combined epigenetic/genetic study identified an ALS age of onset modifier. Acta Neuropathol. Commun. 2021, 9, 1–9. [Google Scholar] [CrossRef]
- Polverino, A.; Rucco, R.; Stillitano, I.; Bonavita, S.; Grimaldi, M.; Minino, R.; Pesoli, M.; Trojsi, F.; d’Ursi, A.M.; Sorrentino, G.; et al. In amyotrophic lateral sclerosis blood cytokines are altered, but do not correlate with changes in brain topology. Brain Connect. 2020, 10, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Hensel, N.; Claus, P. The actin cytoskeleton in SMA and ALS: How does it contribute to motoneuron degeneration? Neuroscientist 2018, 24, 54–72. [Google Scholar] [CrossRef]
- Giampetruzzi, A.; Danielson, E.W.; Gumina, V.; Jeon, M.; Boopathy, S.; Brown, R.H.; Ratti, A.; Landers, J.E.; Fallini, C. Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Ulgen, E.; Ozisik, O.; Sezerman, O. pathfindR: An R package for comprehensive identification of enriched pathways in omics data through active subnetworks. Front. Genet. 2019, 10, 858. [Google Scholar] [CrossRef]
- Tank, E.M.; Figueroa-Romero, C.; Hinder, L.M.; Bedi, K.; Archbold, H.C.; Li, X.; Weskamp, K.; Safren, N.; Paez-Colasante, X.; Pacut, C.; et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Ramaswami, G.; Geschwind, D. Gene co-expression network analysis in human spinal cord highlights mechanisms underlying amyotrophic lateral sclerosis susceptibility. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Verheijen, M.H.; Peviani, M.; Hendricusdottir, R.; Bell, E.M.; Lammens, M.; Smit, A.B.; Bendotti, C.; Van Minnen, J. Increased axonal ribosome numbers is an early event in the pathogenesis of amyotrophic lateral sclerosis. PLoS ONE 2014, 9, e87255. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mandrioli, J.; D’Amico, R.; Zucchi, E.; Gessani, A.; Fini, N.; Fasano, A.; Caponnetto, C.; Chiò, A.; Dalla Bella, E.; Lunetta, C.; et al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine 2018, 97, e11119. [Google Scholar] [CrossRef]
- Warita, H.; Kato, M.; Asada, R.; Yamashita, A.; Hayata, D.; Adachi, K.; Aoki, M. Safety, tolerability, and pharmacodynamics of intrathecal injection of recombinant human HGF (KP-100) in subjects with amyotrophic lateral sclerosis: A phase I trial. J. Clin. Pharmacol. 2019, 59, 677–687. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; Gentile, G.; Aronica, E.; Cavallaro, S. Splicing Players Are Differently Expressed in Sporadic Amyotrophic Lateral Sclerosis Molecular Clusters and Brain Regions. Cells 2020, 9, 159. [Google Scholar] [CrossRef]
- Nagara, Y.; Tateishi, T.; Yamasaki, R.; Hayashi, S.; Kawamura, M.; Kikuchi, H.; Iinuma, K.M.; Tanaka, M.; Iwaki, T.; Matsushita, T.; et al. Impaired Cytoplasmic–Nuclear Transport of Hypoxia-Inducible Factor-1α in Amyotrophic Lateral Sclerosis. Brain Pathol. 2013, 23, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Gosset, P.; Kluza, J.; Brunaud-Danel, V.; Lassalle, P.; Marchetti, P.; Defebvre, L.; Destée, A.; Devos, D. Deregulation of the hypoxia inducible factor-1α pathway in monocytes from sporadic amyotrophic lateral sclerosis patients. Neuroscience 2011, 172, 110–117. [Google Scholar] [CrossRef]
- Dupree, E.J.; Goodwin, A.; Darie, C.C.; Boolani, A. A Pilot Exploratory Proteomics Investigation of Mental Fatigue and Mental Energy. Adv. Exp. Med. Biol. 2019, 1140, 601–611. [Google Scholar]


{kind=link}
| Protein Names | Accession Number | Gene Symbol | p-Value | Av Control | Av ALS | Av Control/AV ALS | |
|---|---|---|---|---|---|---|---|
| 1 | Beta actin variant | 62897625 | <0.00010 | 187.8 | 0.0 | inf | |
| 2 | Fructose-bisphosphate aldolase A (EC 4.1.2.13) (lung cancer antigen NY-LU-1) (muscle-type aldolase) | 16740581 | ALDOA ALDA | <0.00010 | 45.8 | 0.0 | inf |
| 3 | MHC class I antigen | 302144416 | HLA-A | <0.00010 | 30.0 | 0.0 | inf |
| 4 | T-complex protein 1 subunit theta (CCT-theta) | 544711070 | CCT8 hCG_1810843 | <0.00010 | 15.5 | 0.0 | inf |
| 5 | MHC class II antigen | 193227846 | HLA-DRB1 | <0.00010 | 4.8 | 0.0 | inf |
| 6 | HLA class II histocompatibility antigen, DR alpha chain (MHC class II antigen DRA) | 52426774 | HLA-DRA HLA-DRA1 | <0.00010 | 3.5 | 0.0 | inf |
| 7 | phosphoribosylaminoimidazole carboxylase, phosphoribosylaminoimidazole succinocarboxamide synthetase, isoform CRA_a | 119625894 | PAICS | 0.00036 | 2.8 | 0.0 | inf |
| 8 | cDNA FLJ54071, highly similar to drebrin-like protein | 194383996 | 0.00075 | 2.5 | 0.0 | inf | |
| 9 | Phosphopyruvate hydratase (EC 4.2.1.11) (2-phospho-D-glycerate hydro-lyase) | 119590453 | EDARADD hCG_1640319 | 0.00075 | 2.5 | 0.0 | inf |
| 10 | Serine/arginine-rich splicing factor 2 (Splicing component, 35 kDa) (splicing factor SC35) (splicing factor, arginine/serine-rich 2) | 119609849 | SFRS2 hCG_27842 | 0.0015 | 2.3 | 0.0 | inf |
| 11 | Ubiquitin-conjugating enzyme E2 variant 1 (UEV-1) (CROC-1) (TRAF6-regulated IKK activator 1 beta Uev1A) | 73536113 | UBE2V1 CROC1 UBE2V UEV1 P/OKcl.19 | 0.0032 | 2.0 | 0.0 | inf |
| 12 | Hematopoietic cell-specific Lyn substrate 1, isoform CRA_a | 119599911 | HCSL1 | 0.0032 | 2.0 | 0.0 | inf |
| 13 | Small nuclear ribonucleoprotein Sm D2 (Sm-D2) (snRNP core protein D2) | 4759158 | SNRPD2 SNRPD1 | 0.0065 | 1.8 | 0.0 | inf |
| 14 | Transcription factor BTF3 (nascent polypeptide-associated complex subunit beta) (NAC-beta) (RNA polymerase B transcription factor 3) | 20070130 | BTF3 NACB OK/SW-cl.8 | 0.013 | 1.5 | 0.0 | inf |
| 15 | Calcyclin-binding protein (CacyBP) (hCacyBP) (S100A6-binding protein) (Siah-interacting protein) | 193787507 | CACYBP S100A6BP SIP PNAS-107 | 0.013 | 1.5 | 0.0 | inf |
| 16 | Vasodilator-stimulated phosphoprotein isoform X1 | 530417131 | VASP | 0.013 | 1.5 | 0.0 | inf |
| 17 | UV excision repair protein RAD23 homolog B (HR23B) (hHR23B) (XP-C repair-complementing complex 58 kDa protein) (p58) | 18089249 | RAD23B | 0.0046 | 2.5 | 0.3 | 10.00 |
| 18 | MHC class II antigen | 32401120 | HLA-DQA1/DRA | <0.00010 | 4.8 | 0.5 | 9.50 |
| 19 | cDNA FLJ77316, highly similar to Homo sapiens interferon, gamma-inducible protein 30 (IFI30), mRNA | 158256534 | 0.00013 | 4.5 | 0.5 | 9.00 | |
| 20 | Proteasome subunit alpha type-2 | 21465643 | PSMA2 | 0.00024 | 4.3 | 0.5 | 8.50 |
| 21 | Suppression of tumorigenicity 13 (colon carcinoma) (Hsp70 interacting protein), isoform CRA_b | 119580799 | ST13 | <0.00010 | 5.3 | 0.8 | 7.00 |
| 22 | Heterogeneous nuclear ribonucleoprotein A1 (Helix-destabilizing protein) (hnRNP core protein A1) | 194389862 | 0.0015 | 3.5 | 0.5 | 7.00 | |
| 23 | Stomatin-like protein 2, mitochondrial (SLP-2) (EPB72-like protein 2) (paraprotein target 7) (paratarg-7) | 6841440 | STOML2 SLP2 HSPC108 | 0.0049 | 3.0 | 0.5 | 6.00 |
| 24 | Transferrin receptor (p90, CD71), isoform CRA_b | 119574056 | TFRC | 0.0087 | 2.8 | 0.5 | 5.50 |
| 25 | MHC class I antigen | 161376703 | HLA-A | <0.00010 | 30.3 | 5.8 | 5.26 |
| 26 | Transaldolase (EC 2.2.1.2) | 48257056 | TALDO1 TAL TALDO TALDOR | 0.00016 | 6.0 | 1.3 | 4.80 |
| 27 | Transforming protein RhoA (EC 3.6.5.2) (Rho cDNA clone 12) (h12) | 21466025 | RHOA ARH12 ARHA RHO12 | 0.0013 | 5.0 | 1.3 | 4.00 |
| 28 | Eukaryotic translation initiation factor 4H (eIF-4H) (Williams–Beuren syndrome chromosomal region 1 protein) | 11559923 | EIF4H KIAA0038 WBSCR1 WSCR1 | 0.012 | 3.5 | 1.0 | 3.50 |
| 29 | Signal recognition particle 9 kDa protein (SRP9) | 11513832 | SRP9 | 0.0097 | 4.0 | 1.3 | 3.20 |
| 30 | Retinoic acid receptor alpha (RAR-alpha) (nuclear receptor subfamily 1 group B member 1) | 1314308 | RARA NR1B1 | <0.00010 | 27.8 | 8.8 | 3.17 |
| 31 | Acetyl-CoA acetyltransferase, cytosolic (EC 2.3.1.9) (acetyl-CoA transferase-like protein) (cytosolic acetoacetyl-CoA thiolase) | 546901 | ACAT2 ACTL | 0.0014 | 6.3 | 2.0 | 3.13 |
| 32 | SERPINE1 mRNA-binding protein 1, isoform CRA_ | 119626894 | SERBP1 | 0.015 | 3.8 | 1.3 | 3.00 |
| 33 | cDNA, FLJ94551 | 189054116 | 0.0066 | 5.0 | 1.8 | 2.86 | |
| 34 | cDNA FLJ53392, highly similar to ubiquitin-activating enzyme E1 | 194384538 | 0.00016 | 10.3 | 3.8 | 2.73 | |
| 35 | Peroxiredoxin-5, mitochondrial (EC 1.11.1.24) (Alu corepressor 1) (antioxidant enzyme B166) (AOEB166) (liver tissue 2D-page spot 71B) (PLP) (peroxiredoxin V) (Prx-V) (peroxisomal antioxidant enzyme) (TPx type VI) (thioredoxin peroxidase PMP20) (thioredoxin-dependent peroxiredoxin 5) | 15826629 | PRDX5 ACR1 SBBI10 | 0.00046 | 9.3 | 3.5 | 2.64 |
| 36 | MHC class I antigen | 572455386 | HLA-B | <0.00010 | 19.0 | 7.3 | 2.62 |
| 37 | PREDICTED: cofilin-1 isoform X2 [Nomascus leucogenys] | 332250232 | <0.00010 | 29.8 | 11.5 | 2.59 | |
| 38 | Endoplasmic reticulum resident protein 29 (ERp29) (endoplasmic reticulum resident protein 28) (ERp28) (endoplasmic reticulum resident protein 31) (ERp31) | 192987144 | ERP29 C12orf8 ERP28 | 0.0013 | 9.0 | 3.8 | 2.40 |
| 39 | ATP-binding cassette sub-family E member 1 (2′-5′-oligoadenylate-binding protein) (HuHP68) (RNase L inhibitor) (ribonuclease 4 inhibitor) (RNS4I) | 987870 | ABCE1 RLI RNASEL1 RNASELI RNS4I OK/SW-cl.40 | 0.0095 | 6.3 | 2.8 | 2.27 |
| 40 | Ubiquitin-like protein ISG15 (interferon-induced 15 kDa protein) (interferon-induced 17 kDa protein) (IP17) (ubiquitin cross-reactive protein) (hUCRP) | 4826774 | ISG15 G1P2 UCRP | 0.0095 | 6.3 | 2.8 | 2.27 |
| 41 | Quinone oxidoreductase (EC 1.6.5.5) (NADPH:quinone reductase) (zeta-crystallin) | 13236495 | CRYZ | 0.017 | 5.5 | 2.5 | 2.20 |
| 42 | Proteasome subunit alpha type-4 (macropain subunit C9) (multicatalytic endopeptidase complex subunit C9) (proteasome component C9) (proteasome subunit L) | 34783332 | PSMA4 HC9 PSC9 | 0.0031 | 8.8 | 4.0 | 2.19 |
| 43 | MHC class I antigen | 221148541 | HLA-A | <0.00010 | 25.5 | 11.8 | 2.17 |
| 44 | Proteasome subunit alpha type-1 (30 kDa prosomal protein) (PROS-30) (macropain subunit C2) (multicatalytic endopeptidase complex subunit C2) (proteasome component C2) (proteasome nu chain) | 13543551 | PSMA1 HC2 NU PROS30 PSC2 | 0.0036 | 9.0 | 4.3 | 2.12 |
| 45 | ADP-ribosylation factor 3 | 119626762 | ARF3 | 0.008 | 7.8 | 3.8 | 2.07 |
| 46 | ADP-ribosylation factor | 4502203 | ARF3 hCG_40390 | 0.0064 | 8.3 | 4.0 | 2.06 |
| 47 | Protein disulfide-isomerase A3 (EC 5.3.4.1) (58 kDa glucose-regulated protein) (58 kDa microsomal protein) (p58) (disulfide isomerase ER-60) (endoplasmic reticulum resident protein 57) (ER protein 57) (ERp57) (endoplasmic reticulum resident protein 60) (ER protein 60) (ERp60) | 114793397 | PDIA3 ERP57 ERP60 GRP58 | 0.00015 | 17.5 | 8.8 | 2.00 |
| 48 | MHC class I antigen | 255682810 | HLA-B | 0.0046 | 9.5 | 4.8 | 2.00 |
| 49 | 10 kDa heat shock protein, mitochondrial (Hsp10) (10 kDa chaperonin) (chaperonin 10) (CPN10) (early pregnancy factor) (EPF) | 4008131 | HSPE1 | <0.00010 | 33.8 | 18.3 | 1.85 |
| 50 | Gamma-enolase (EC 4.2.1.11) (2-phospho-D-glycerate hydro-lyase) (enolase 2) (neural enolase) (neuron-specific enolase) (NSE) | 182118 | ENO2 | <0.00010 | 24.0 | 13.0 | 1.85 |
| 51 | Thioredoxin (Trx) (ATL-derived factor) (ADF) (surface-associated sulfhydryl protein) (SASP) (allergen Hom s Trx) | 685425705 | TXN TRDX TRX TRX1 | 0.0036 | 12.0 | 6.5 | 1.85 |
| 52 | Adenylyl cyclase-associated protein | 5453595 | CAP1 hCG_2033246 | <0.00010 | 34.0 | 18.8 | 1.81 |
| 53 | Eukaryotic translation initiation factor 5A-1 (eIF-5A-1) (eIF-5A1) (eukaryotic initiation factor 5A isoform 1) (eIF-5A) (Rev-binding factor) (eIF-4D) | 4503545 | EIF5A | 0.007 | 8.3 | 15.0 | 0.55 |
| 54 | Tubulin beta-6 chain (tubulin beta class V) | 14210536 | TUBB6 | 0.00014 | 14.5 | 27.5 | 0.53 |
| 55 | 60S ribosomal protein L36 (large ribosomal subunit protein eL36) | 16117794 | RPL36 | 0.0042 | 7.0 | 13.8 | 0.51 |
| 56 | 14-3-3 protein gamma (protein kinase C inhibitor protein 1) (KCIP-1) [cleaved into: 14-3-3 protein gamma, N-terminally processed] | 380764684 | YWHAG | <0.00010 | 18.3 | 35.0 | 0.52 |
| 57 | 14-3-3 protein zeta/delta (protein kinase C inhibitor protein 1) (KCIP-1) | 347948616 | YWHAZ | <0.00010 | 25.3 | 51.8 | 0.49 |
| 58 | hCG2038942, partial | 119602344 | <0.00010 | 18.8 | 40.8 | 0.46 | |
| 59 | Elongation factor 1-gamma (EF-1-gamma) (eEF-1B gamma) | 15530265 | EEF1G EF1G PRO1608 | <0.00010 | 9.0 | 20.8 | 0.43 |
| 60 | HLA class II histocompatibility antigen gamma chain (HLA-DR antigens-associated invariant chain) (Ia antigen-associated invariant chain) (Ii) (CD antigen CD74) [cleaved into: class-II-associated invariant chain peptide (CLIP)] | 32132 | CD74 DHLAG | <0.00010 | 7.5 | 17.8 | 0.42 |
| 61 | 60S ribosomal protein L23a (Large ribosomal subunit protein uL23) | 17105394 | RPL23A | <0.00010 | 7.8 | 20.0 | 0.39 |
| 62 | MHC class I antigen | 388240742 | HLA-B | <0.00010 | 8.0 | 20.8 | 0.39 |
| 63 | MHC class I antigen | 563403983 | HLA-C | <0.00010 | 9.3 | 24.5 | 0.38 |
| 64 | 60S ribosomal protein L8 (large ribosomal subunit protein uL2) | 15082586 | RPL8 | 0.01 | 2.3 | 6.0 | 0.38 |
| 65 | Tyrosine-protein phosphatase non-receptor type 6 (EC 3.1.3.48) (hematopoietic cell protein-tyrosine phosphatase) (protein-tyrosine phosphatase 1C) (PTP-1C) (protein-tyrosine phosphatase SHP-1) (SH-PTP1) | 18104989 | PTPN6 HCP PTP1C | 0.0006 | 4.0 | 10.8 | 0.37 |
| 66 | Ribosomal protein L7a, isoform CRA_ | 119608467 | 0.0022 | 3.0 | 8.3 | 0.36 | |
| 67 | Fermitin family homolog 3 (Kindlin-3) (MIG2-like protein) (Unc-112-related protein 2) | 28626504 | FERMT3 KIND3 MIG2B URP2 | 0.014 | 1.8 | 5.0 | 0.35 |
| 68 | 40S ribosomal protein S6 (phosphoprotein NP33) (small ribosomal subunit protein eS6) | 337514 | RPS6 OK/SW-cl.2 | <0.00010 | 8.0 | 23.0 | 0.35 |
| 69 | Ig alpha-2 chain C region | 70058 | IGHA2 | 0.001 | 3.0 | 8.8 | 0.34 |
| 70 | cDNA, FLJ93036, highly similar to Homo sapiens tyrosine 3-monooxygenase/tryptophan 5-monooxygenaseactivation protein, eta polypeptide (YWHAH), mRNA | 189069195 | 0.00015 | 3.8 | 11.3 | 0.33 | |
| 71 | 5C5 | 3868714 | 0.0013 | 2.5 | 7.8 | 0.32 | |
| 72 | Ig M Fc | 222978 | 0.0036 | 2.0 | 6.3 | 0.32 | |
| 73 | MHC class I antigen | 572455377 | HLA-B | <0.00010 | 12.0 | 38.0 | 0.32 |
| 74 | Actin-like protein | 62421128 | ACT | <0.00010 | 16.8 | 53.5 | 0.31 |
| 75 | Beta-actin-like protein 2 (kappa-actin) | 62420949 | ACTBL2 | <0.00010 | 47.3 | 166.3 | 0.28 |
| 76 | 60S ribosomal protein L24 (60S ribosomal protein L30) (large ribosomal subunit protein eL24) | 4506619 | RPL24 | 0.0075 | 1.3 | 4.5 | 0.28 |
| 77 | Coatomer subunit beta (beta-coat protein) (beta-COP) | 7705369 | COPB1 COPB MSTP026 | 0.0029 | 1.5 | 5.5 | 0.27 |
| 78 | Glutamyl-prolyl-tRNA synthetase, isoform CRA_a | 119613715 | EPRS | 0.0007 | 1.8 | 6.8 | 0.26 |
| 79 | cDNA FLJ61136, highly similar to Ras-related protein Rab-11A | 194387154 | 0.00073 | 1.3 | 5.8 | 0.22 | |
| 80 | Unnamed protein product | 34526163 | 0.0019 | 1.0 | 4.8 | 0.21 | |
| 81 | Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit 1 | 14124942 | 0.0019 | 1.0 | 4.8 | 0.21 | |
| 82 | Beta-actin-like protein 2 (Kappa-actin) | 63055057 | ACTBL2 | <0.00010 | 31.5 | 156.3 | 0.20 |
| 83 | Guanine nucleotide-binding protein (G protein), beta polypeptide 2-like 1, isoform CRA_ | 119574081 | GNB2L1 | <0.00010 | 4.5 | 23.3 | 0.19 |
| 84 | Hypoxia up-regulated protein 1 (150 kDa oxygen-regulated protein) (ORP-150) (170 kDa glucose-regulated protein) (GRP-170) | 62897071 | HYOU1 GRP170 ORP150 | <0.00010 | 4.0 | 21.3 | 0.19 |
| 85 | ABI gene family member 3 (new molecule including SH3) (nesh) | 12052938 | ABI3 NESH | 0.0031 | 0.8 | 4.0 | 0.19 |
| 86 | Activator of 90 kDa heat shock protein ATPase homolog 1 (AHA1) (p38) | 194374633 | AHSA1 C14orf3 HSPC322 | 0.014 | 0.5 | 2.8 | 0.18 |
| 87 | Phosphoribosylformylglycinamidine synthase (EC 6.3.5.3) (formylglycinamide ribonucleotide amidotransferase) (formylglycinamide ribotide amidotransferase) | 119610474 | PFAS hCG_31283 | 0.0049 | 0.5 | 3.3 | 0.15 |
| 88 | 60S ribosomal protein L13 (breast basic conserved protein 1) (large ribosomal subunit protein eL13) | 15431295 | RPL13 BBC1 OK/SW-cl.46 | <0.00010 | 1.0 | 7.8 | 0.13 |
| 89 | Glucose phosphate isomerase variant | 62088730 | 0.013 | 0.3 | 2.3 | 0.11 | |
| 90 | BH3-interacting domain death agonist (p22 BID) (BID) [cleaved into: BH3-interacting domain death agonist p15 (p15 BID); BH3-interacting domain death agonist p13 (p13 BID); BH3-interacting domain death agonist p11 (p11 BID)] | 159163783 | BID | 0.0074 | 0.3 | 2.5 | 0.10 |
| 91 | Cullin-associated NEDD8-dissociated protein 1 (Cullin-associated and neddylation-dissociated protein 1) (TBP-interacting protein of 120 kDa A) (TBP-interacting protein 120A) (p120 CAND1) | 34782987 | CAND1 KIAA0829 TIP120 TIP120A | <0.00010 | 1.0 | 13.0 | 0.08 |
| 92 | Tripeptidyl-peptidase 1 (TPP-1) (EC 3.4.14.9) (cell growth-inhibiting gene 1 protein) (Lysosomal pepstatin-insensitive protease) (LPIC) (tripeptidyl aminopeptidase) (tripeptidyl-peptidase I) (TPP-I) | 15928808 | TPP1 CLN2 GIG1 UNQ267/PRO304 | 0.00069 | 0.3 | 3.5 | 0.07 |
| 93 | cDNA FLJ54564, highly similar to 150 kDa oxygen-regulated protein (Orp150) | 221043650 | <0.00010 | 0.3 | 5.5 | 0.05 | |
| 94 | Amyloid beta (A4) precursor protein-binding, family B, member 1 interacting protein, isoform CRA_ | 119606501 | APBB1I | 0.0094 | 0.0 | 1.8 | −inf |
| 95 | NEDD8 (neddylin) (neural precursor cell expressed developmentally down-regulated protein 8) (NEDD-8) (ubiquitin-like protein Nedd8) | 208435631 | NEDD8 | 0.0094 | 0.0 | 1.8 | −inf |
| 96 | PREDICTED: rho GTPase-activating protein 17 isoform X5 [Homo sapiens] | 767988626 | 0.0094 | 0.0 | 1.8 | −inf | |
| 97 | Apoptosis inhibitor 5 (API-5) (anti apoptosis clone 11 protein) (AAC-11) (cell migration-inducing gene 8 protein) (fibroblast growth factor 2-interacting factor) (FIF) (protein XAGL) | 377656459 | API5 MIG8 | 0.0048 | 0.0 | 2.0 | −inf |
| 98 | Eukaryotic translation initiation factor 4 gamma 1 (eIF-4-gamma 1) (eIF-4G 1) (eIF-4G1) (p220) | 119598670 | EIF4G1 EIF4F EIF4G EIF4GI | 0.0048 | 0.0 | 2.0 | −inf |
| 99 | Xaa-Pro dipeptidase (X-Pro dipeptidase) (EC 3.4.13.9) (endopeptidase) (peptidase D) (proline dipeptidase) (prolidase) | 112491419 | PEPD PRD | 0.0025 | 0.0 | 2.3 | −inf |
| 100 | RNA transcription, translation and transport factor protein (CLE7 homolog) (CLE) (hCLE) | 7706322 | RTRAF C14orf166 CGI-99 | 0.0025 | 0.0 | 2.3 | −inf |
| 101 | cDNA FLJ55804, highly similar to COP9 signalosome complex subunit 1 | 194388460 | 0.0025 | 0.0 | 2.3 | −inf | |
| 102 | 40S ribosomal protein S20 (small ribosomal subunit protein uS10) | 226246671 | RPS20 | 0.0013 | 0.0 | 2.5 | −inf |
| 103 | cDNA FLJ56765, highly similar to galactokinase (cDNA, FLJ79080, highly similar to galactokinase) | 194383498 | 0.0013 | 0.0 | 2.5 | −inf | |
| 104 | Trifunctional purine biosynthetic protein adenosine-3 [Includes: Phosphoribosylamine--glycine ligase (EC 6.3.4.13) (Glycinamide ribonucleotide synthetase) (GARS) (Phosphoribosylglycinamide synthetase); phosphoribosylformylglycinamidine cyclo-ligase (EC 6.3.3.1) (AIR synthase) (AIRS) (phosphoribosyl-aminoimidazole synthetase); phosphoribosylglycinamide formyltransferase (EC 2.1.2.2) (5’-phosphoribosylglycinamide transformylase) (GAR transformylase) (GART)] | 4503915 | GART PGFT PRGS | 0.0013 | 0.0 | 2.5 | −inf |
| 105 | ADP-ribosylation factor 4 | 4502205 | ARF4 ARF2 | 0.00065 | 0.0 | 2.8 | −inf |
| 106 | Aspartate--tRNA ligase, cytoplasmic (EC 6.1.1.12) (aspartyl-tRNA synthetase) (AspRS) (cell proliferation-inducing gene 40 protein) | 499142118 | DARS1 DARS PIG40 | 0.00033 | 0.0 | 3.0 | −inf |
| 107 | Mitochondrial-processing peptidase subunit alpha (alpha-MPP) (inactive zinc metalloprotease alpha) (P-55) | 545478969 | PMPCA INPP5E KIAA0123 MPPA | 0.00033 | 0.0 | 3.0 | −inf |
| 108 | Nucleosome assembly protein 1-like 1, isoform CRA_b | 119617721 | NAP1L1 hCG_2015037 | 0.00033 | 0.0 | 3.0 | −inf |
| 109 | 60S ribosomal protein L23 | 13097600 | 0.00033 | 0.0 | 3.0 | −inf | |
| 110 | COP9 constitutive photomorphogenic homolog subunit 2 (Arabidopsis), isoform CRA_a | 119597770 | COPS2 | 0.00017 | 0.0 | 3.3 | −inf |
| 111 | Glutamine--fructose-6-phosphate aminotransferase [isomerizing] 1 (EC 2.6.1.16) (D-fructose-6-phosphate amidotransferase 1) (Glutamine:fructose-6-phosphate amidotransferase 1) (GFAT 1) (GFAT1) (hexose phosphate aminotransferase 1) | 183082 | GFPT1 GFAT GFPT | 0.00017 | 0.0 | 3.3 | −inf |
| 112 | Protein diaphanous homolog 1 (cDNA FLJ61549, highly similar to protein diaphanous homolog 1) | 194385544 | DIAPH1 | <0.00010 | 0.0 | 3.5 | −inf |
| 113 | Phosphoribosyl pyrophosphate synthetase-associated protein 2, isoform CRA_ | 119570819 | PRPSAP2 | <0.00010 | 0.0 | 4.5 | −inf |
| 114 | Thymopoietin isoform X | 530400796 | TMPO | <0.00010 | 0.0 | 7.3 | −inf |
| 115 | Tubulin alpha chain-like 3 | 13376181 | TUBAL3 | <0.00010 | 0.0 | 7.8 | −inf |
| 116 | 60S ribosomal protein L13 (breast basic conserved protein 1) (large ribosomal subunit protein eL13) | 29383 | RPL13 BBC1 OK/SW-cl.46 | <0.00010 | 0.0 | 8.0 | −inf |
| 117 | Myosin light polypeptide 6 (myosin, light polypeptide 6, alkali, smooth muscle and non-muscle, isoform CRA_c) | 119617307 | MYL6 hCG_2039617 | <0.00010 | 0.0 | 9.8 | −inf |
| 118 | Myl6 protein (myosin, light polypeptide 6, alkali, smooth muscle and non-muscle) | 33620739 | Myl6 | <0.00010 | 0.0 | 10.0 | −inf |
| 119 | Interferon-induced GTP-binding protein Mx1 (interferon-induced protein p78) (IFI-78K) (interferon-regulated resistance GTP-binding protein MxA) (myxoma resistance protein 1) (myxovirus resistance protein 1) [cleaved into: interferon-induced GTP-binding protein Mx1, N-terminally processed] | 544711185 | MX1 | <0.00010 | 0.0 | 21.3 | −inf |
| 120 | MHC class I antigen | 350281590 | HLA-A | <0.00010 | 0.0 | 26.3 | −inf |
| *Accession number gi-NCBI genInfo Identifer (https://www.ncbi.nlm.nih.gov/protein, accessed on 15 September 2022) |
| Ingenuity Canonical Pathways * | −log(p-Value) | Ratio | |
|---|---|---|---|
| 1 | EIF2 signaling * | 13.5 | 0.065 |
| 2 | Antigen presentation pathway * | 9.7 | 0.179 |
| 3 | Crosstalk between dendritic cells and natural killer cells | 7.0 | 0.076 |
| 4 | Virus entry via endocytic pathways | 6.5 | 0.064 |
| 5 | Caveolar-mediated endocytosis signaling | 6.1 | 0.078 |
| 6 | RAN signaling * | 6.0 | 0.211 |
| 7 | B cell development | 5.9 | 0.109 |
| 8 | IL-4 signaling | 5.6 | 0.063 |
| 9 | PD-1, PD-L1 cancer immunotherapy pathway | 5.3 | 0.056 |
| 10 | Th1 pathway | 4.9 | 0.049 |
| 11 | Protein ubiquitination pathway * | 4.7 | 0.029 |
| 12 | Multiple sclerosis signaling pathway | 4.3 | 0.030 |
| 13 | Phagosome maturation | 4.2 | 0.036 |
| 14 | FAT10 signaling pathway | 4.0 | 0.068 |
| 15 | ILK signaling | 3.7 | 0.030 |
| 16 | Coronavirus pathogenesis pathway | 3.7 | 0.029 |
| 17 | Th2 pathway | 3.6 | 0.037 |
| 18 | Interferon signaling * | 3.4 | 0.083 |
| 19 | Neuroinflammation signaling pathway | 3.4 | 0.021 |
| 20 | Actin cytoskeleton signaling * | 3.3 | 0.024 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitham, D.; Belenkiy, E.; Darie, C.C.; Radu, A. Proteomics Analysis of Lymphoblastoid Cell Lines from Patients with Amyotrophic Lateral Sclerosis. Molecules 2023, 28, 2014. https://doi.org/10.3390/molecules28052014
Whitham D, Belenkiy E, Darie CC, Radu A. Proteomics Analysis of Lymphoblastoid Cell Lines from Patients with Amyotrophic Lateral Sclerosis. Molecules. 2023; 28(5):2014. https://doi.org/10.3390/molecules28052014
Chicago/Turabian StyleWhitham, Danielle, Eugene Belenkiy, Costel C. Darie, and Aurelian Radu. 2023. "Proteomics Analysis of Lymphoblastoid Cell Lines from Patients with Amyotrophic Lateral Sclerosis" Molecules 28, no. 5: 2014. https://doi.org/10.3390/molecules28052014
APA StyleWhitham, D., Belenkiy, E., Darie, C. C., & Radu, A. (2023). Proteomics Analysis of Lymphoblastoid Cell Lines from Patients with Amyotrophic Lateral Sclerosis. Molecules, 28(5), 2014. https://doi.org/10.3390/molecules28052014