Highlights
What are the main findings?
- Fluorinated telechelic bis(cyclocarbonate) was synthesized through carbonylation of a fluorinated diepoxide.
- Fluorinated polyhydroxyurethanes (FPHUs) with different molar masses were synthesized through polyaddition reaction.
- Analysis showed that the FPHUs have higher molar mass, glass transition, and decomposition temperatures compared to their hydrogenated homologues.
What is the implication of the main finding?
- New non-isocyanate polymers with improved properties.
- The synthesized FPHUs are potential candidates for applications requiring materials with these properties (E.g., adhesives).
Abstract
New fluorinated polyhydroxyurethanes (FPHUs) with various molar weights were synthesized via the polyaddition reaction of a fluorinated telechelic bis(cyclocarbonate) (bis-CC) with a diamine. The fluorinated bis-CC was initially synthesized by carbonylation of a fluorinated diepoxide, 1,4-bis(2′,3′-epoxypropyl)perfluorobutane, in the presence of LiBr catalyst, in high yield. Then, several reaction conditions were optimized through the model reactions of the fluorinated bis-CC with hexylamine. Subsequently, fluorinated polymers bearing hydroxyurethane moieties (FPHUs) were prepared by reacting the bis-CC with different hexamethylenediamine amounts in bulk at 80 °C and the presence of a catalyst. The chemoselective polymerization reaction yielded three isomers bearing primary and secondary hydroxyl groups in 61–82% yield. The synthesized fluorinated CCs and the corresponding FPHUs were characterized by 1H, 19F, and 13C NMR spectroscopy. They were compared to their hydrogenated homologues synthesized in similar conditions. The gel permeation chromatography (GPC), differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA) data of the FPHUs revealed a higher molar mass and a slight increase in glass transition and decomposition temperatures compared to those of the PHUs.
1. Introduction
Polyurethanes (PUs), which have been known for nearly 70 years, are used in various fields, such as formulations of adhesives, paints, foams, coatings, packaging components, fibers, and in some specialized applications, including biomedical surgeries where biocompatible, biodegradable, and non-toxic PUs are used. PUs have several interesting properties, such as excellent adhesion, high durability, good flexibility, resistance to corrosion and mechanical wear, and favorable optical properties [1].
The conventional method for the production of PUs is based on the reaction between isocyanates and the hydroxyl groups of oligomers [2]. It is a reaction consisting of the polyaddition of a polyisocyanate with polyols in the presence of a labile hydrogen atom. The rigidity of the final product changes depending on the nature of polyols and isocyanates.
The major problem of this chemistry is related to the toxicity and volatility of isocyanates [3,4]. The isocyanates are represented by two main families of aromatic structures: toluene diisocyanate (TDI), and methylene diphenyl diisocyanate (MDI) [5]. The NCO functional groups are introduced into the aromatic nuclei by phosgenation of toluene diamine to prepare TDI or aniline to obtain MDI. Nevertheless, in its turn, phosgene is a toxic reactive precursor involved in a dangerous reaction: phosgenation [6]. Therefore, the “isocyanate route” threatens the environment and operators’ health. Replacing or minimizing these hazardous substances has become both academic and industrial concerns [7].
Various approaches to the synthesis of isocyanate-free PUs were explored [8]. Among the most common and studied routes, ring-opening polyaddition of cyclocarbonates (CCs) and amines in bulk or solution is an excellent alternative to the conventional PUs based on isocyanate precursors [6,9,10,11,12,13]. The nucleophilic addition of the amines to the CCs leads to polyhydroxyurethanes (PHUs), also called non-isocyanate polyurethanes, which possess urethane linkages and primary and secondary hydroxyl groups that are reported to enhance the adhesive properties of the polymer (Figure 1) [14,15,16].
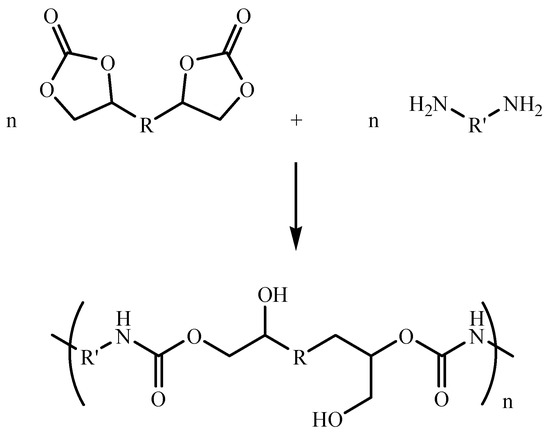
Figure 1.
Synthesis of PHUs by polyaddition of CCs and diamines.
In general, PHUs showed better chemical, thermal, and mechanical stability than PUs. In comparison to PUs, PHUs have a hydroxyl group adjacent to each carbamate group, which greatly enhances the concentration of hydrogen bonds through intra- or intermolecular interactions. The latter can strongly improve the mechanical and thermal properties and, therefore, the adhesive properties of these polymers [17]. Furthermore, the synthesis process of PHUs overcomes a critical drawback in synthesizing PUs: insensitivity to humidity [18]. On the other hand, the amines required for this aminolysis reaction are commercially available or can be yielded from bio-based resources. The CCs are considered superior to other reagents due to their ease of synthesis, high boiling point, biodegradability, and low toxicity [18,19,20,21]. To the best of our knowledge, only five-membered ring CCs are commercially available. In addition, five- and six-membered ring CCs were successfully and efficiently synthesized and reported in the literature [3,22,23]. The most common routes for synthesizing CCs with excellent selectivity and yields generally involve the insertion of carbon dioxide in epoxides [24,25,26,27,28,29]. Nevertheless, the significant downside in synthesizing PHUs is often the difficulty in reaching high molar masses [30]. Moreover, the polyaddition reaction is slow at room temperature and requires careful control of the stoichiometry of the reactants [31].
In terms of selectivity, the nucleophilic addition of the amine to five-membered CCs leads to the formation of two isomers [32]. However, a single product is obtained using six-membered CCs. In the case of five-membered CCs, both resulting isomers can be distinguished from one another by the presence of a primary or a secondary hydroxyl group [33,34]. Generally, the reaction of CCs with amines favors the formation of the isomer containing the secondary hydroxyl group [35]. As for the case of the 6-membered ring, only one hydroxyurethane product containing primary hydroxyl groups can be obtained [10].
Ultimately, fluorinated polymers (FPs) are reported to demonstrate remarkable properties, including their chemical, thermal, and water resistance [36,37,38,39,40]. These properties are indispensable for multiple industries [41,42], especially for coatings. Most FPs are synthesized by radical (co)polymerization of fluoroalkenes. Two branches of FPs can be considered: (i) FPs, where the fluorinated groups are located within the polymer backbone, and (ii) FPs containing a fluorinated dangling group [39]. To the best of our knowledge, despite the synthesis and characterization of fluorinated PUs, fluorinated polyhydroxyurethanes (FPHUs) were rarely reported in literature. In 2014, FPs bearing five-membered CC pendant groups were synthesized by co- or ter-polymerization of chlorotrifluoroethylene with various vinyl ethers [36]. In 2019, fluorine-containing non-isocyanate polyurethane coatings were synthesized using bisphenol and perfluorooctyl CCs [37].
In this study, FPHUs were prepared via the polyaddition route, characterized by spectroscopy, and thermally analyzed. For this purpose, fluorinated telechelic bis-CC (Figure 2) derived from 1,4-bis(2′,3′ epoxypropyl)perfluorobutane was synthesized and reacted with mono- and diamine (Figure 2). The optimal reaction conditions were examined by synthesizing model fluorinated hydroxyurethanes from fluorinated bis-CC and hexylamine. Then, FPHUs were formed by reacting the fluorinated bis-CC with hexamethylenediamine using different molar ratios to provide a range of polymers of various molar masses. In light of this work, we synthesized PHUs using the same polymerization conditions and through a synthesized hydrogenated bis-CC (Figure 2). Finally, we compared the properties of both prepared polymers, FPHUs and PHUs.
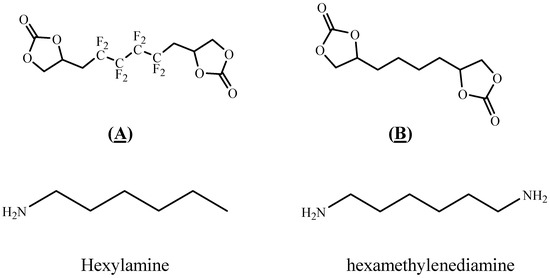
Figure 2.
The fluorinated and non-fluorinated biscyclocarbonates (A & B) and amines used in this work.
2. Results and Discussion
The syntheses of the fluorinated and non-fluorinated bis-CCs and the subsequently optimized syntheses of FPHUs and PHUs by addition with amines were achieved. The resulting products were characterized by spectroscopy and thermal analyses. In this work, the PHUs were prepared for properties comparison purposes.
2.1. Synthesis of Fluorinated Biscyclocarbonate (A) and Hydrogenated Biscyclocarbonate (B)
The syntheses of (A) and (B) were performed by direct carbonylation of the corresponding diepoxide with CO2 in the presence of a lithium bromide (LiBr) catalyst (Figure 3) [43].
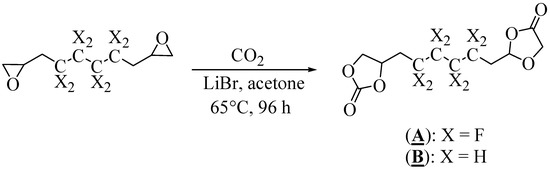
Figure 3.
Synthesis of (A) and (B) by carbonylation of the corresponding diepoxide.
After optimization of the reaction conditions, the carbonylation was conducted in a reactor in the presence of acetone at 65 °C for 96 h (Table 1). The fluorinated and non-fluorinated precursors (A) and (B) were then characterized and confirmed by 1H, 13C (and 19F for A) NMR spectroscopy (Figures S1–S5 in the Supplementary Materials).

Table 1.
Syntheses of (A) and (B) by carbonylation of the corresponding diepoxides a.
2.2. Model Reaction of (A) with Monoamine: Optimization of the Reaction Conditions
The hydroxyurethanes synthesis conditions were investigated and optimized by adding hexylamine to the synthesized bis-CC (A). The model reaction was studied by varying the temperature, solvent, time, (A)/amine molar ratio, and the presence of a catalyst (Table 2). Due to the asymmetry present in (A), the nucleophilic attack of hexylamine on both carbonyl groups led to three possible regioisomers, including the hydroxyurethane groups: (i) symmetric isomer with secondary hydroxyl groups (C), (ii) symmetric isomer with primary hydroxyl groups (C′), and (iii) asymmetric isomer having both primary and secondary hydroxyl groups (C″) (Figure 4).

Figure 4.
Synthesis of three possible regioisomer hydroxyurethanes by addition of hexylamine to (A).
Table 2.
Model reaction of (A) with hexylamine a: conditions optimization.
Table 2.
Model reaction of (A) with hexylamine a: conditions optimization.
| Entry | Solvent | (A): Amine | T (°C) | t (h) | Cat. (2 mol%) | Conv. (%) b | Yield (%) c | Secondary Alcohol (%) d | Primary Alcohol (%) d |
|---|---|---|---|---|---|---|---|---|---|
| 1 | DMSO | 1:2.0 | 25 | 48 | - | 55 | 48 e | 66 | 34 |
| 2 | DMSO | 1:2.0 | 60 | 18 | - | 72 | 57 e | 71 | 29 |
| 3 | DMSO | 1:2.0 | 80 | 18 | - | 81 | 55 e | 70 | 30 |
| 4 | DMF | 1:2.0 | 80 | 18 | - | 92 | 49 e | 73 | 27 |
| 5 | Acetonitrile | 1:2.0 | 80 | 18 | - | 79 | 52 | 65 | 35 |
| 6 | Dimethyl carbonate | 1:2.0 | 80 | 18 | - | 73 | 52 | 67 | 33 |
| 7 | 1,4-dioxane | 1:2.0 | 80 | 18 | - | 85 | 48 | 70 | 30 |
| 8 | Ethyl acetate | 1:2.0 | 80 | 18 | - | 88 | 42 | 65 | 35 |
| 9 | Trifluoro-toluene | 1:2.0 | 80 | 18 | - | 56 | 50 | 68 | 32 |
| 10 | - | 1:2.0 | 80 | 18 | - | 59 | 48 | 65 | 35 |
| 11 | - | 1:2.6 | 80 | 18 | - | 70 | 64 | 60 | 40 |
| 12 | - | 1:3.0 | 80 | 18 | - | 100 | 69 | 65 | 35 |
| 13 | - | 1:2.0 | 80 | 5 | NEt3 | 100 | 63 | 65 | 35 |
| 14 | - | 1:2.6 | 80 | 5 | NEt3 | 100 | 84 | 68 | 32 |
| 15 | - | 1:3.0 | 80 | 5 | NEt3 | 100 | 92 | 70 | 30 |
a Conditions: carried out in a 10 mL flask; (A)/solvent (if any) 0.5 g/2 mL. b Determined by 1H NMR: × 100; where stands for the integral of the signal centered at i ppm assigned to XY group (Figure 5 and Figure S1 in the Supplementary Materials). c The yield was calculated after purification. d The proportions of primary alcohols and secondary alcohols were calculated using 1H NMR spectra of the pure products: × 100; (Figure 5). e NMR reveals the presence of side products.

Figure 5.
1H NMR spectrum of the model hydroxyurethanes (C), (C′), and (C″) (DMSO-d6, 20 °C, 400 MHz).
First, the reaction was attempted in the absence of a catalyst. 55% conversion was obtained upon reacting (A) with 2.0 eq hexylamine at 25 °C for 48 h (Table 2, Entry 1). Subsequently, the effect of the temperature was evaluated using 2.0 eq amine in DMSO. At 60 °C, 18 h of reaction time was enough to increase the conversion of (A) to 72% (Table 2, Entry 2), which kept rising to 81% at 80 °C (Table 2, Entry 3). Therefore, the evolution of this reaction is highly dependent on temperature, proving the need for thermal activation of the aminolysis of (A). Nevertheless, in the presence of DMSO, it was observed that these reactions led to side products, evidenced by the 1H NMR spectrum and confirmed by LC-MS chromatography. Similarly, this was seen when DMF was implemented in the same conditions (Table 2, Entry 4). The addition reaction was then attempted in different solvents (Table 1, Entry 5–9), leading to the model oligomers in unsatisfactory yields and low to moderate conversions with no side products detected. Then, we focused on investigating the reaction in bulk, where there was no sign of any side products detected by 1H NMR. In the absence of any catalyst, the viscosity of the medium increased with the progress of the reaction, and a conversion reached 70% after 18 h at 80 °C with 2.6 eq of hexylamine (Table 2, Entry 11). This viscosity can be linked to the formation of a new hydroxyl group, driving the creation of hydrogen bonds and decreasing the mobility of the reagents. Moreover, it was necessary to use 3.0 eq hexylamine to completely convert the cyclocarbonate (A) in 18 h reaction time (Table 1, Entry 12). This may be due to the better stirring obtained upon increasing the liquid phase. Finally, the reaction time was optimized by adding 2% triethylamine as a catalyst. A 100% conversion and 92% yield was reached after a 5 h reaction at 80 °C with 3.0 eq of hexylamine (Table 1, Entry 15). In all the studied entries, it was noted that there was no significant difference in the proportions of the hydroxyl groups; 65–73% of secondary alcohols were quantified, consistent with the reported literature [10,35].
2.3. Characterization of the Model Hydroxyurethanes
The model hydroxyurethanes (C), (C′), and (C″) were characterized by 1H, 19F, and 13C NMR spectroscopy. By comparing Figure 5 with the 1H NMR spectrum of the fluorinated bis-CC (A) (Figure S1 in the Supplementary Materials), it is observed that the AB system corresponding to the two nonequivalent hydrogen atoms CHaHb of the carbonate groups has disappeared (at 4.6 and 4.2 ppm). Furthermore, the proton of the amide group (7, 7′) appeared at 7.2 ppm. The two peaks at 3.5 ppm (14, 14′) and 4.1 ppm (10, 10′) evidence the presence of the α-CH2 group of the primary and secondary alcohols, respectively. The multiplet appearing at 3.9 ppm (9, 9′) can be assigned to the carboxylate group’s α-CH2. The protons (12, 12′) in the CH2CF2 groups appeared as one multiplet (complex coupling with fluorine in positions α and β) at 2.2 ppm. In addition, the protons of the methyl (1, 1′) and methylene (2–5 and 2′–5′) groups, originally from the hexylamine, appear at 0.8 ppm and a multiplet at 1.3 ppm, respectively. The chemical shift at about 3 ppm can be attributed to the other two protons of the amine chain (6, 6′).
The 19F NMR spectrum of the hydroxyurethanes (Figure 6) is similar to that of (A) (Figure S2 in the Supplementary Materials). It shows the presence of an AB system located at δ − 112 ppm assigned to CH2CFAFBCF2 groups of the diastereoisomers due to the presence of the asymmetric carbon (10, 10′ and 13, 13′). Furthermore, the signal at δ − 123 ppm is assigned to the difluoromethylene groups (CH2CF2CF2).
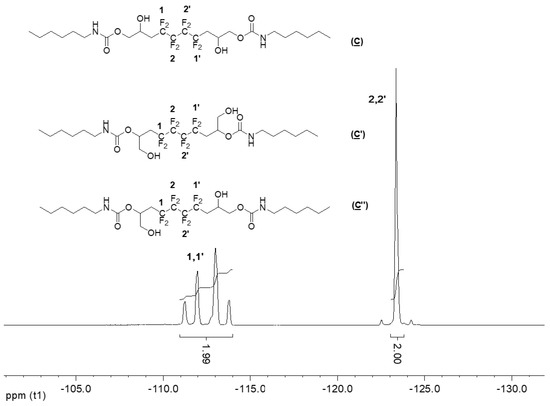
Figure 6.
19F NMR spectrum of the model hydroxyurethanes (C), (C′), and (C″) (DMSO-d6, 20 °C, 235.2 MHz).
Expectedly, the 13C NMR spectra (DEPT135) of (C), (C′), and (C″) (Figure 7) has many similar characteristics observed in (A) (Figure S3 in the Supplementary Materials). The signal at δ 155 ppm corresponds to the carbonyl groups (8, 8′). In addition, the signal of CF2 groups (7, 7′) appeared between δ 106 and δ 120 ppm. The carboxylate group’s α-CH2 (9, 9′) can be found at δ 67 ppm. The chemical shift at 31 ppm corresponds to the methylene group adjacent to CF2 groups (11, 11′). Most importantly, the signals of the carbon atoms bearing the hydroxyl group are characterized and distinguished: 10, 10′ hold the secondary OH, whereas 13, 13′ are the β-CH to the primary OH; they correspond to signals centered at 63 and 67 ppm, respectively. The low intensity of 13,13′ confirms the low amount of primary alcohol. As for the hexylamine used in the addition reaction, its presence as a part of the model products is highlighted by the peaks at 40 ppm, 22 ppm, and 30 ppm assigned to the CH2 adjacent to the amide (6, 6′) and CH2 groups of the amine (2–5/2′–5′), respectively. Finally, the signal of the CH3 (1, 1′) is noted at δ 14 ppm.
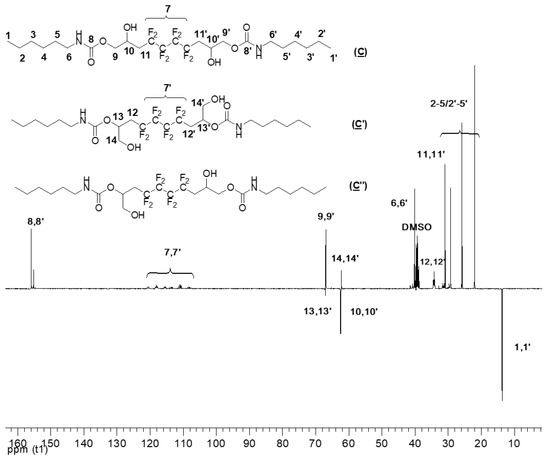
Figure 7.
13C NMR spectrum of the model hydroxyurethanes (C), (C′), and (C″) (DMSO, 20 °C, 100.6 MHz).
2.4. Synthesis and Characterization of the FPHUs and PHUs: Reaction with Diamine
The fluorinated polyhydroxyurethanes (FPHUs) were synthesized by ring opening of the fluorinated bis-CC (A) with different molar ratios of (A)/hexamethylenediamine, based on the optimized conditions obtained in the model reaction and taking into consideration the two active hydrogen atoms in the diamine groups (Figure 8).
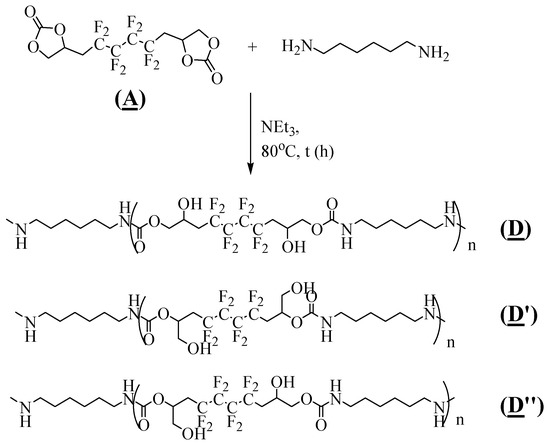
Figure 8.
Synthesis of FPHUs by polyaddition of (A) with hexamethylenediamine.
A series of bulk polymerization reactions were conducted at 80 °C in the presence of 2% triethylamine, implementing 1.5, 1.3, and 1.0 eq of the diamine (Table 3). Initially, (A) was fully converted after adding 1.5 eq of the hexamethylenediamine to give FPHUs P1 oligomers in 82% yield (Table 3, Entry 1). Then, a varying ratio of hexamethylenediamine was used in combination with (A) to produce FPHUs with different molar masses (Table 3, Entry 2–3). Adding 1.0 eq of hexamethylenediamine yielded FPHUs P3 polymers in 61% yield and 94% conversion after 18 h reaction (Table 3, Entry 3). Finally, the synthesis of PHUs P4 was performed to compare their properties to that of FPHUs. The hydrogenated polymers were synthesized using the same optimized conditions used in P3 synthesis (Table 3, Entry 4). (B) was 88% converted by reacting it with 1.0 eq of hexamethylenediamine for 18 h. P4 was produced in 57% yield, characterized, and confirmed via 1H NMR spectroscopy (Figure S6 in the Supplementary Materials). It is worth noting that the reactivity of the fluorinated bis-CC (A) was higher than the hydrogenated bis-CC (B) with the diamine under the same conditions.
Table 3.
FPHUs and PHUs synthesis from (A) or (B) with hexamethylenediamine a.
Next, the target FPHUs P3 were characterized by NMR spectroscopy. The 1H NMR spectrum of the pure P3 (Figure 9) is very similar to that of the products of the model reaction. The main difference is the absence of the CH3 signal of the terminal amine at 0.8 ppm.
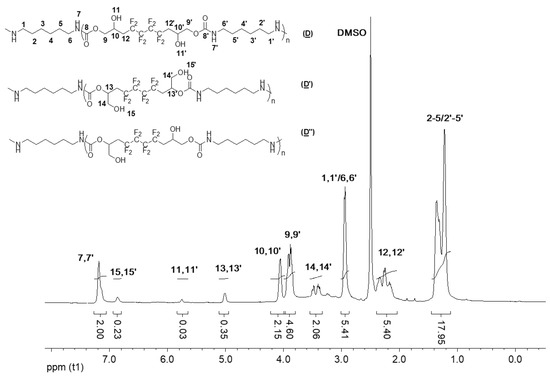
Figure 9.
1H NMR spectrum of the synthesized P3 (D), (D′), and (D″) (DMSO-d6, 20 °C, 400 MHz).
Comparably, the 19F NMR spectrum of P3 (Figure 10) is similar to that of the products of the model reaction and the synthesized bis-CC (A) (Figure S2 in the Supplementary Materials), showing the presence of the AB system at δ − 112 ppm, and the signal at δ − 123 ppm.
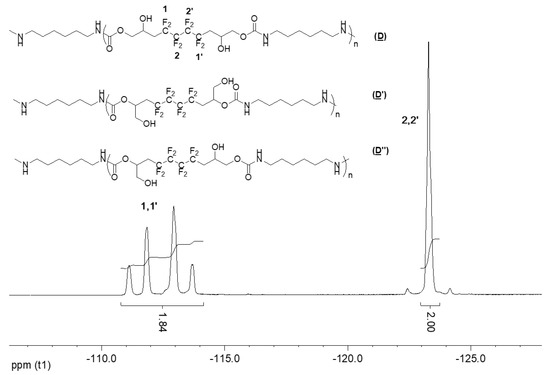
Figure 10.
19F NMR spectrum of the synthesized P3 (D), (D′), and (D″) (DMSO-d6, 20 °C, 235.2 MHz).
In turn, the 13C NMR spectrum (DEPT135) of P3 (Figure 11) revealed almost the same characteristics seen in the products of the model reaction, except for the absence of the CH3 peak of the amine.
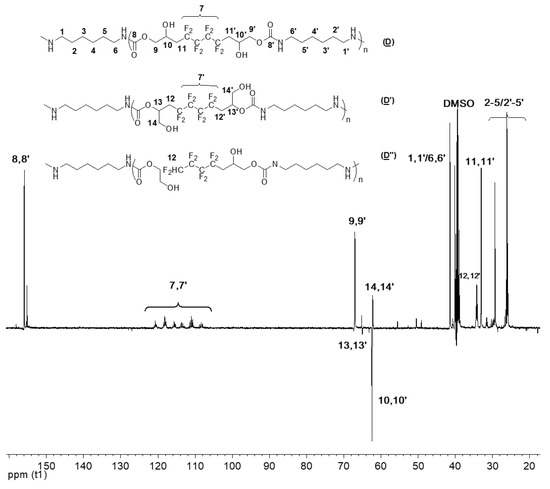
Figure 11.
13C NMR (DEPT 135) spectrum of the synthesized P3 (D), (D′), and (D″) (DMSO, 20 °C, 100.6 MHz).
2.5. Gel Permeation Chromatography and Thermal Analysis of FPHUs and PHUs: Comparison
The synthesized FPHUs and PHUs were characterized and compared by GPC, TGA, and DSC (Table 4).
Table 4.
Comparison of the properties of FPHUs and PHUs.
This polymerization seemed highly dependent on the excess of the diamine initially implemented which is in accordance with the reported literature where the excess of diamine adjusted the degree of polymerization, and thus, their molar masses, based on Flory’s theory and Carothers’ equation [30,44]. For example, due to the stoichiometry used, P1, and P2 could only reach maximum degrees of polymerization of 5 and 8 respectively (at 100% extent of reaction). The different CC/diamine molar ratios used in the polyaddition reflect the variation of the molar masses of FHPUs P1–P3; indeed, the degree of polymerization upon the polyaddition decreased as soon as the diamine was in excess (Table 4, Entry 1–2). Higher molar mass was obtained by implementing a stoichiometric CC/diamine ratio (Table 4, Entry 3). As reported by Besse et al. in 2015, the relatively low molar masses of the FPHUs obtained can be attributed to numerous side reactions at the early stages of the polyaddition reaction, yielding undesired compounds. By comparing the fluorinated and non-fluorinated polymers, P3 and P4, FPHUs exhibited higher molar mass with 1.0 equivalent of the diamine, whereas the polydispersity indexes of both polymers were close (Table 4, Entry 3–4).
The thermogravimetric data gives an insight into the stability of the polymers and estimates the degradation’s initiation and completion required temperatures. The most relevant TGA results are listed in Table 4. Indeed, the degradation in P3 started (T5%) at a higher temperature than in P1 and P2. The complete degradation, estimated arbitrarily at 70 wt% loss, was observed at 450 °C in P3. By comparing the thermogravimetric results of both polymers FPHUs P3 and PHUs P4 (Figure S7), all the decomposition temperatures for P4 were lower than in the fluorinated counter, typically by 50–80 °C (Table 4). Relatively low values of the glass transition temperature, Tg, were observed for FHPUs P1–P3 and PHUs P4. However, the latter shows a lower Tg than FPHUs. This can be explained by the larger atomic size of fluorine compared to hydrogen.
3. Materials and Methods
3.1. Materials
1,4-bis(2′,3′-epoxypropyl)perfluorobutane (BEPFB) was provided by TOSOH Finechemical Corporation (Shunan, Japan). Lithium bromide (LiBr), acetone (analytical grade), hexylamine, hexamethylenediamine, 1,2,7,8-diepoxyoctane, Dimethylformamide (DMF), Dimethyl Sulfoxide (DMSO), acetonitrile, ethyl acetate, dimethyl carbonate, trifluorotoluene, and 1,4-dioxane were purchased from Sigma-Aldrich. The deuterated solvents were purchased from Euroiso-top (purity > 99.8%).
3.2. Characterization
3.2.1. Nuclear Magnetic Resonance (NMR)
The NMR spectra were recorded on a Bruker AC 400 instrument, using deuterated chloroform, d6-N,N-dimethylsulfoxide, and d6-acetone as solvents, and tetramethylsilane (TMS) (or CFCl3) as references for 1H (or 19F) nuclei. Coupling constants and chemical shifts are given in hertz (Hz) and parts per million (ppm), respectively. The experimental conditions for recording 1H, 13C, (or 19F) NMR spectra were as follows: flip angle 90° (or 30°), acquisition time 4.5 s (or 0.7 s), pulse delay 2 s (or 2 s), number of scans 128 (or 512), and a pulse width of 5 μs for 19F NMR.
3.2.2. Thermogravimetric Analysis (TGA)
TGA was performed with a TGA 51 apparatus from TA Instruments, under air, and at the heating rate of 10 °C/min from room temperature up to a maximum of 550 °C. The sample weight varied between 10 and 15 mg.
3.2.3. Differential Scanning Calorimetry (DSC)
DSC analysis was conducted on a Netzsch 200F3 DSC apparatus equipped with Proteus software under a nitrogen atmosphere at a heating rate of 20 °C/min. The temperature range was between −50 to +200 °C. The DSC system was first calibrated in temperature using indium and n-hexane. The second run led to the glass transition temperature, defined as the inflection point in the heat capacity jump. The sample weight was about 10 mg.
3.3. Synthetic Procedures
3.3.1. Synthesis of Fluorinated Biscyclocarbonate (A)
4-(2,2,3,3,4,4,5,5-octafluoro-6-(2-oxo-1,3-dioxolan-4-yl)hexyl)-1,3-dioxolan-2-one
The reactions were performed in an autoclave of 100 mL Parr Hastelloy. First, LiBr (276 mg, 3.18 mmol) dissolved in acetone (30 mL) was placed into the autoclave which was pressurized to 30 bars of nitrogen for 1 h to check for leaks. Once the nitrogen was evacuated, the reactor was placed under vacuum for 30 min, and then acetone (30 mL), 1,4-bis(2′,3′-epoxypropyl)perfluorobutane (10.02 g, 0.0318 mol), and carbon dioxide (15–20 bars) were added. The reactor was then heated progressively to 65 °C for 96 h, and the pressure and temperature evolution were recorded. Subsequently, the reactor was cooled and degassed (release of unreacted CO2), and the crude product was washed with acetone (100 mL), filtered, then dried under a vacuum.
1H NMR (400.1 MHz, DMSO, δ): 2.90 (m, CH2CF2, 4H); 4.63 and 4.22 (m, OCH2, 4H); 5.20 (m, CH, 1H).
19F NMR (235.2 MHz, DMSO, δ): −112.29 (AB system, CH2CFAFBCF2), −123.18 (s, CH2CF2CF2).
13C NMR (100.6 MHz, DMSO, δ): 154.38 (C=O); 106.56–120.72 (CF2); 70.24 (CH); 60.24 (OCH2); 39.26(CH2CF2).
3.3.2. Synthesis of Non-Fluorinated Biscyclocarbonate (B): 4-(6-(2-oxo-1,3-dioxolan-4-yl)hexyl)-1,3-dioxolan-2-one
The carbonylation of the hydrogenated epoxide 1,2,7,8-diepoxyoctane was performed using the same procedure as the fluorinated epoxide using the following quantities and conditions: LiBr (605.3 mg, 6.97 mmol), DMF (30 mL), 1,2,7,8-diepoxyoctane (9.911 g, 0.0697 mol), and carbon dioxide (15–20 bars). The reactor was heated progressively to 65 °C for 96 h. The crude product was distilled under a high vacuum pump at 40–60 °C and then placed under direct vacuum overnight to remove the DMF. However, the DMF was not totally removed until the product was extracted in H2O/ethyl acetate.
1H NMR (400.1 MHz, DMSO, δ): 1.38 (m, CH2CH2, 4H); 1.69 (m, CH2CH, 4H); 4.56 and 4.12 (m, OCH2, 4H); 4.76 (m, CH, 1H).
13C NMR (100.6 MHz, DMSO, δ): 76.87 (CH); 69.15 (OCH2); 32.63(CHCH2CH2); 23.56(CH2CH2CH2).
3.3.3. Model Reaction of the Fluorinated Bis-CC (A) with Monoamine: Optimization of the Reaction Conditions
(A) (2.00 g, 4.97 mmol, 1.00 eq), hexylamine (2.00, 2.60, or 3.00 eq), and triethylamine (NEt3) (10 mg, 2–3 drops) were placed in a round bottom flask. The flask was heated to 80 °C to dissolve (A). The mixture was then stirred, and a sample was taken at t0. The flask was then placed in an oil bath at the desired temperature for a specific time (th). Finally, the model product (C, C′, and C″) was washed with distilled water and pentane, water was removed by decantation, and the material was dried at 60 °C under vacuum for 16 h.
1H NMR (400.1 MHz, DMSO, δ): 0.90 (m, CH3CH2, 6H); 1.18 and 1.35 (m, CH2, 16H); 2.22 (m, CH2, 4H); 2.9 (m, CH2NH, 4H); 3.45 (m, CH2OH, 4H); 3.9 (m, CHCH2O, 4H); 4.05 (s, CHOH, 2H); 5 (s, OCHCH2, 2H); 5.3 (s, CHOH, 2H); 6.8 (s, CH2OH, 2H); 7.15 (m, NHCOO, 2H).
19F NMR (235.2 MHz, DMSO, δ): −112.39 (AB system, CH2CFAFB CF2); −123.36 (s, CH2CF2CF2).
13C NMR (100.6 MHz, DMSO, δ): 155.59 (C=O); 107.75–120.95 (CF2); 66.96 (OCH2); 62.26 (CH2OH); 63.04/63.27 (CHOH/ CH2O); 40.16 (CH2NH); 30.91 (CH2CF2); 21.99–29.28 (CH2CH2); 14.46 (CH3CH2).
3.3.4. The Reaction of the Fluorinated Bis-CC (A) with a Diamine: Formation of the FPHUs (D, D′, and D″)
Similarly to the model reaction, using the following quantities: (A) (1.00 g, 2.48 mmol, 1.00 eq), hexamethylenediamine (1.00, 1.30, or 1.50 eq), and triethylamine (NEt3) (5 mg).
1H (400.1 MHz, DMSO, δ): 1.21–1.37 (m, CH2CH2); 2.24 (m, CH2CF2); 2.94 (m, CH2NH); 3.44 (m, CH2OH); 3.88 (m, CHCH2O); 4.05 (s, CHOH); 5 (s, OCHCH2); 5.74 (s, CHOH); 6.86 (s, CH2OH); 7.19 (m, NHCOO).
19F NMR (235.2 MHz, DMSO, δ): −112.5 (AB system, CH2CFAFB CF2); −123.29 (s, CH2CF2CF2).
13C NMR (100.6 MHz, DMSO, δ): 155.94 (C=O); 106.74–121.03 (CF2), 67.00 (OCH2CH); 62.93/ 63.17 (CH2CHOH/OCHCH2); 62.21 (CH2OH); 41.43 (CH2NH); 33.04 (CH2CF2); 25.84–29.36 (CH2).
3.3.5. The Reaction of the Non-Fluorinated Bis-CC (B) with a Diamine: Formation of the PHUs (E, E′, and E″)
Similarly to the synthesis of the above FPHUs, the following quantities were used: (B) (0.53 g, 2.33 mmol, 1.00 eq), hexamethylenediamine (0.270 g, 2.33 mmol, 1.0 eq), and triethylamine (NEt3) (10–12 mg).
1H NMR (400.1 MHz, DMSO, δ): 0.85 (m, CH3CH2, 6H); 1.22 to 1.53 (m, CH2, 24H); 2.93 (m, CH2NH, 4H); 3.37 (m, CH2OH, 4H); 3.54 (m, CHO, 2H); 3.78 (m, CHOH, 2H); 4.55 (m, CHCH2OH, 2H); 6.64 (s, CH2OH, 2H); 6.68 (s, CH2OH, 2H); 7.03 (m, NHCOO, 2H).
4. Conclusions
FPHUs were synthesized via the polyaddition of a synthesized fluorinated bis(cyclocarbonate) (bis-CC) with a telechelic diamine. First, the bis-CC was prepared by carbonylation of a fluorinated telechelic bisepoxide. Then, model additions of n-hexylamine onto the fluorinated CC were optimized; complete conversion and good yield were obtained when a 3-fold excess of hexylamine was used in bulk, catalyzed by NEt3. All attempts showed that secondary and primary alcohols were produced with about two-thirds of the former isomer. Polyaddition of the fluorinated bis-CC with hexamethylenediamine was conducted using the optimized conditions. FPHUs with various molar weights were formed using different CC/diamine molar ratios. The synthesized FPHUs were deeply characterized by NMR spectroscopy, and the PHUs were synthesized using the same optimized conditions and characterized for comparison purposes. The data collected by GPC, TGA, and DSC led to the same conclusion; the presence of the fluorinated chains provided a slightly higher thermal stability to the resulting polymers.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28041795/s1, Figure S1: 1H NMR spectra of (A) (top) and BEPFB (bottom) (DMSO-d6, 20 °C, 400 MHz); Figure S2: 19F NMR spectra of (A) (top) and BEPFB (bottom) (DMSO-d6, 20 °C, 235.2 MHz); Figure S3: 13C NMR (DEPT 135) spectra of (A) (top) and BEPFB (bottom) (DMSO, 20 °C, 100.6 MHz); Figure S4: 1H NMR spectrum of (B) (top) and 1,2,7,8-diepoxyoctane (bottom) (DMSO-d6, 20 °C, 400 MHz); Figure S5: 13C NMR (DEPT 135) spectrum of (B) (top) and 1,2,7,8-diepoxyoctane (bottom) (DMSO, 20 °C, 100.6 MHz); Figure S6: 1H NMR spectrum of the synthesized PHUs (DMSO-d6, 20 °C, 400 MHz); Figure S7: TGA thermograms of P3 and P4.
Author Contributions
Conceptualization, B.A.; methodology, validation, formal analysis, writing—review and editing, W.E.M., V.L., A.A. and B.A.; investigation, data curation, L.H., W.E.M. and A.A.; resources, V.L. and B.A.; writing—original draft preparation, visualization, L.H. and W.E.M.; supervision, project administration, A.A. and B.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Material.
Acknowledgments
The authors are grateful to the Institut de Chimie of the CNRS and TOSOH Finechemicals Corporation (Shunan, Japan) for supplying free materials. The authors thank Ecole Nat. Sup. de Chimie de Montpellier (ENSCM) and Université Libanaise (UL). They also thank Caillol (ENSCM) and Naoufal (UL).
Conflicts of Interest
The authors declare that there is no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Avar, G.; Meier-Westhues, U.; Casselmann, H.; Achten, D. 10.24—Polyurethanes. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 411–441. ISBN 978-0-08-087862-1. [Google Scholar]
- Asplund, J.O.B.; Bowden, T.; Mathisen, T.; Hilborn, J. Synthesis of Highly Elastic Biodegradable Poly(urethane urea). Biomacromolecules 2007, 8, 905–911. [Google Scholar] [CrossRef]
- Pyo, S.-H.; Persson, P.; Mollaahmad, M.A.; Sörensen, K.; Lundmark, S.; Hatti-Kaul, R. Cyclic carbonates as monomers for phosgene- and isocyanate-free polyurethanes and polycarbonates. Pure Appl. Chem. 2011, 84, 637–661. [Google Scholar] [CrossRef]
- Kapp, R.W. Isocyanates. In Encyclopedia of Toxicology (Third Edition); Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 1112–1131. ISBN 978-0-12-386455-0. [Google Scholar]
- Parodi, F. 23—Isocyanate-derived Polymers. In Comprehensive Polymer Science and Supplements; Allen, G., Bevington, J.C., Eds.; Pergamon: Amsterdam, The Netherlands, 1989; pp. 387–412. ISBN 978-0-08-096701-1. [Google Scholar]
- Blattmann, H.; Fleischer, M.; Bähr, M.; Mülhaupt, R. Isocyanate- and Phosgene-Free Routes to Polyfunctional Cyclic Carbonates and Green Polyurethanes by Fixation of Carbon Dioxide. Macromol. Rapid Commun. 2014, 35, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Carré, C.; Ecochard, Y.; Caillol, S.; Avérous, L. From the Synthesis of Biobased Cyclic Carbonate to Polyhydroxyurethanes: A Promising Route towards Renewable Non-Isocyanate Polyurethanes. ChemSusChem 2019, 12, 3410–3430. [Google Scholar] [CrossRef] [PubMed]
- Brocas, A.-L.; Cendejas, G.; Caillol, S.; Deffieux, A.; Carlotti, S. Controlled synthesis of polyepichlorohydrin with pendant cyclic carbonate functions for isocyanate-free polyurethane networks. J. Polym. Sci. Part Polym. Chem. 2011, 49, 2677–2684. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y.; Fan, Y.; Rager, M.-N.; Guérineau, V.; Bouteiller, L.; Li, M.-H.; Thomas, C.M. Polymerization of Cyclic Carbamates: A Practical Route to Aliphatic Polyurethanes. Macromolecules 2019, 52, 2719–2724. [Google Scholar] [CrossRef]
- Maisonneuve, L.; Lamarzelle, O.; Rix, E.; Grau, E.; Cramail, H. Isocyanate-Free Routes to Polyurethanes and Poly(hydroxy Urethane)s. Chem. Rev. 2015, 115, 12407–12439. [Google Scholar] [CrossRef]
- Tomita, H.; Sanda, F.; Endo, T. Reactivity comparison of five- and six-membered cyclic carbonates with amines: Basic evaluation for synthesis of poly(hydroxyurethane). J. Polym. Sci. Part Polym. Chem. 2001, 39, 162–168. [Google Scholar] [CrossRef]
- Kotanen, S.; Laaksonen, T.; Sarlin, E. Feasibility of polyamines and cyclic carbonate terminated prepolymers in polyurethane/polyhydroxyurethane synthesis. Mater. Today Commun. 2020, 23, 100863. [Google Scholar] [CrossRef]
- Sessini, V.; Thai, C.N.; Amorín, H.; Jiménez, R.; Samuel, C.; Caillol, S.; Cornil, J.; Hoyas, S.; Barrau, S.; Dubois, P.; et al. Solvent-Free Design of Biobased Non-isocyanate Polyurethanes with Ferroelectric Properties. ACS Sustain. Chem. Eng. 2021, 9, 14946–14958. [Google Scholar] [CrossRef]
- Cornille, A.; Michaud, G.; Simon, F.; Fouquay, S.; Auvergne, R.; Boutevin, B.; Caillol, S. Promising mechanical and adhesive properties of isocyanate-free poly(hydroxyurethane). Eur. Polym. J. 2016, 84, 404–420. [Google Scholar] [CrossRef]
- Cornille, A.; Blain, M.; Auvergne, R.; Andrioletti, B.; Boutevin, B.; Caillol, S. A study of cyclic carbonate aminolysis at room temperature: Effect of cyclic carbonate structures and solvents on polyhydroxyurethane synthesis. Polym. Chem. 2017, 8, 592–604. [Google Scholar] [CrossRef]
- Quienne, B.; Poli, R.; Pinaud, J.; Caillol, S. Enhanced aminolysis of cyclic carbonates by β-hydroxylamines for the production of fully biobased polyhydroxyurethanes. Green Chem. 2021, 23, 1678–1690. [Google Scholar] [CrossRef]
- Leitsch, E.K.; Heath, W.H.; Torkelson, J.M. Polyurethane/polyhydroxyurethane hybrid polymers and their applications as adhesive bonding agents. Int. J. Adhes. Adhes. 2016, 64, 1–8. [Google Scholar] [CrossRef]
- Bilal, M.; Ihsanullah, I.; Younas, M.; Ul Hassan Shah, M. Recent advances in applications of low-cost adsorbents for the removal of heavy metals from water: A critical review. Sep. Purif. Technol. 2021, 278, 119510. [Google Scholar] [CrossRef]
- Clements, J.H. Reactive Applications of Cyclic Alkylene Carbonates. Ind. Eng. Chem. Res. 2003, 42, 663–674. [Google Scholar] [CrossRef]
- Lombardo, V.M.; Dhulst, E.A.; Leitsch, E.K.; Wilmot, N.; Heath, W.H.; Gies, A.P.; Miller, M.D.; Torkelson, J.M.; Scheidt, K.A. Cooperative Catalysis of Cyclic Carbonate Ring Opening: Application Towards Non-Isocyanate Polyurethane Materials. Eur. J. Org. Chem. 2015, 2015, 2791–2795. [Google Scholar] [CrossRef]
- Cornille, A.; Auvergne, R.; Figovsky, O.; Boutevin, B.; Caillol, S. A perspective approach to sustainable routes for non-isocyanate polyurethanes. Eur. Polym. J. 2017, 87, 535–552. [Google Scholar] [CrossRef]
- Matsukizono, H.; Endo, T. Reworkable Polyhydroxyurethane Films with Reversible Acetal Networks Obtained from Multifunctional Six-Membered Cyclic Carbonates. J. Am. Chem. Soc. 2018, 140, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Matsukizono, H.; Endo, T. Phosgene-Free Syntheses and Hydrolytic Properties of Water-Soluble Polyhydroxyurethanes with Ester–Carbonate–Ether Structures in Their Main Chains. Macromol. Chem. Phys. 2017, 218, 1700043. [Google Scholar] [CrossRef]
- Pescarmona, P.P. Cyclic carbonates synthesised from CO2: Applications, challenges and recent research trends. Curr. Opin. Green Sustain. Chem. 2021, 29, 100457. [Google Scholar] [CrossRef]
- Hosseinian, A.; Farshbaf, S.; Mohammadi, R.; Monfared, A.; Vessally, E. Advancements in six-membered cyclic carbonate (1,3-dioxan-2-one) synthesis utilizing carbon dioxide as a C1 source. RSC Adv. 2018, 8, 17976–17988. [Google Scholar] [CrossRef] [PubMed]
- Grignard, B.; Gennen, S.; Jérôme, C.; Kleij, A.W.; Detrembleur, C. Advances in the use of CO2 as a renewable feedstock for the synthesis of polymers. Chem. Soc. Rev. 2019, 48, 4466–4514. [Google Scholar] [CrossRef] [PubMed]
- Ballamine, A.; Kotni, A.; Llored, J.-P.; Caillol, S. Valuing CO2 in the development of polymer materials. Sci. Technol. Energy Transit. 2022, 77, 1. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Nocito, F.; Pastore, C. A study on the carboxylation of glycerol to glycerol carbonate with carbon dioxide: The role of the catalyst, solvent and reaction conditions. J. Mol. Catal. Chem. 2006, 257, 149–153. [Google Scholar] [CrossRef]
- Ochiai, B.; Hatano, Y.; Endo, T. Facile synthesis of polymers bearing cyclic carbonate structure through radical solution and precipitation polymerizations accompanied by concurrent carbon dioxide fixation. J. Polym. Sci. Part Polym. Chem. 2009, 47, 3170–3176. [Google Scholar] [CrossRef]
- Besse, V.; Camara, F.; Méchin, F.; Fleury, E.; Caillol, S.; Pascault, J.-P.; Boutevin, B. How to explain low molar masses in PolyHydroxyUrethanes (PHUs). Eur. Polym. J. 2015, 71, 1–11. [Google Scholar] [CrossRef]
- Cornille, A.; Serres, J.; Michaud, G.; Simon, F.; Fouquay, S.; Boutevin, B.; Caillol, S. Syntheses of epoxyurethane polymers from isocyanate free oligo-polyhydroxyurethane. Eur. Polym. J. 2016, 75, 175–189. [Google Scholar] [CrossRef]
- Ubaghs, L.; Fricke, N.; Keul, H.; Höcker, H. Polyurethanes with Pendant Hydroxyl Groups: Synthesis and Characterization. Macromol. Rapid Commun. 2004, 25, 517–521. [Google Scholar] [CrossRef]
- Bassam, N.; Laure, C.; Jean-François, B.; Yann, R.; Zephirin, M. Aza-Michael versus aminolysis reactions of glycerol carbonate acrylate. Green Chem. 2013, 15, 1900–1909. [Google Scholar] [CrossRef]
- Hahn, C.; Keul, H.; Möller, M. Hydroxyl-functional polyurethanes and polyesters: Synthesis, properties and potential biomedical application. Polym. Int. 2012, 61, 1048–1060. [Google Scholar] [CrossRef]
- Pasquier, N.; Keul, H.; Moeller, M. Polymers with specific adhesion properties for surface modification: Synthesis, characterization and applications. Des. Monomers Polym. 2005, 8, 679–703. [Google Scholar] [CrossRef]
- Hamiye, R.; Alaaeddine, A.; Awada, M.; Campagne, B.; Caillol, S.; Guillaume, S.M.; Ameduri, B.; Carpentier, J.-F. From glycidyl carbonate to hydroxyurethane side-groups in alternating fluorinated copolymers. Polym. Chem. 2014, 5, 5089–5099. [Google Scholar] [CrossRef]
- Wu, Z.; Tang, L.; Dai, J.; Qu, J. Synthesis and properties of fluorinated non-isocyanate polyurethanes coatings with good hydrophobic and oleophobic properties. J. Coat. Technol. Res. 2019, 16, 1233–1241. [Google Scholar] [CrossRef]
- Dolui, S.; Kumar, D.; Banerjee, S.; Ameduri, B. Well-Defined Fluorinated Copolymers: Current Status and Future Perspectives. Acc. Mater. Res. 2021, 2, 242–251. [Google Scholar] [CrossRef]
- Améduri, B. The Promising Future of Fluoropolymers. Macromol. Chem. Phys. 2020, 221, 1900573. [Google Scholar] [CrossRef]
- Ameduri, B. Fluoropolymers: The Right Material for the Right Applications. Chem. Eur. J. 2018, 24, 18830–18841. [Google Scholar] [CrossRef]
- Smart, M.C.; Ratnakumar, B.V.; Ryan-Mowrey, V.S.; Surampudi, S.; Prakash, G.K.S.; Hu, J.; Cheung, I. Improved performance of lithium-ion cells with the use of fluorinated carbonate-based electrolytes. J. Power Sources 2003, 119–121, 359–367. [Google Scholar] [CrossRef]
- McMillan, R.; Slegr, H.; Shu, Z.X.; Wang, W. Fluoroethylene carbonate electrolyte and its use in lithium ion batteries with graphite anodes. J. Power Sources 1999, 81–82, 20–26. [Google Scholar] [CrossRef]
- Kihara, N.; Hara, N.; Endo, T. Catalytic activity of various salts in the reaction of 2,3-epoxypropyl phenyl ether and carbon dioxide under atmospheric pressure. J. Org. Chem. 1993, 58, 6198–6202. [Google Scholar] [CrossRef]
- Cornille, A.; Ecochard, Y.; Blain, M.; Boutevin, B.; Caillol, S. Synthesis of hybrid polyhydroxyurethanes by Michael addition. Eur. Polym. J. 2017, 96, 370–382. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).