
Exploring Thiazolopyridine AV25R: Unraveling of Biological Activities, Selective Anti-Cancer Properties and In Silico Target and Binding Prediction in Hematological Neoplasms

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Cell Biologic Characterisation of AVR25-Induced Effects
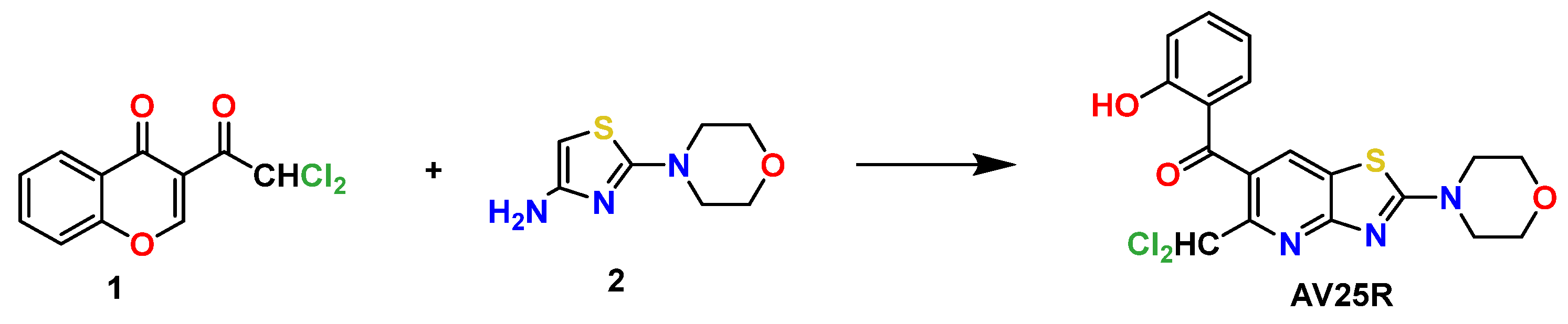
2.1.1. Hemolytic Activity
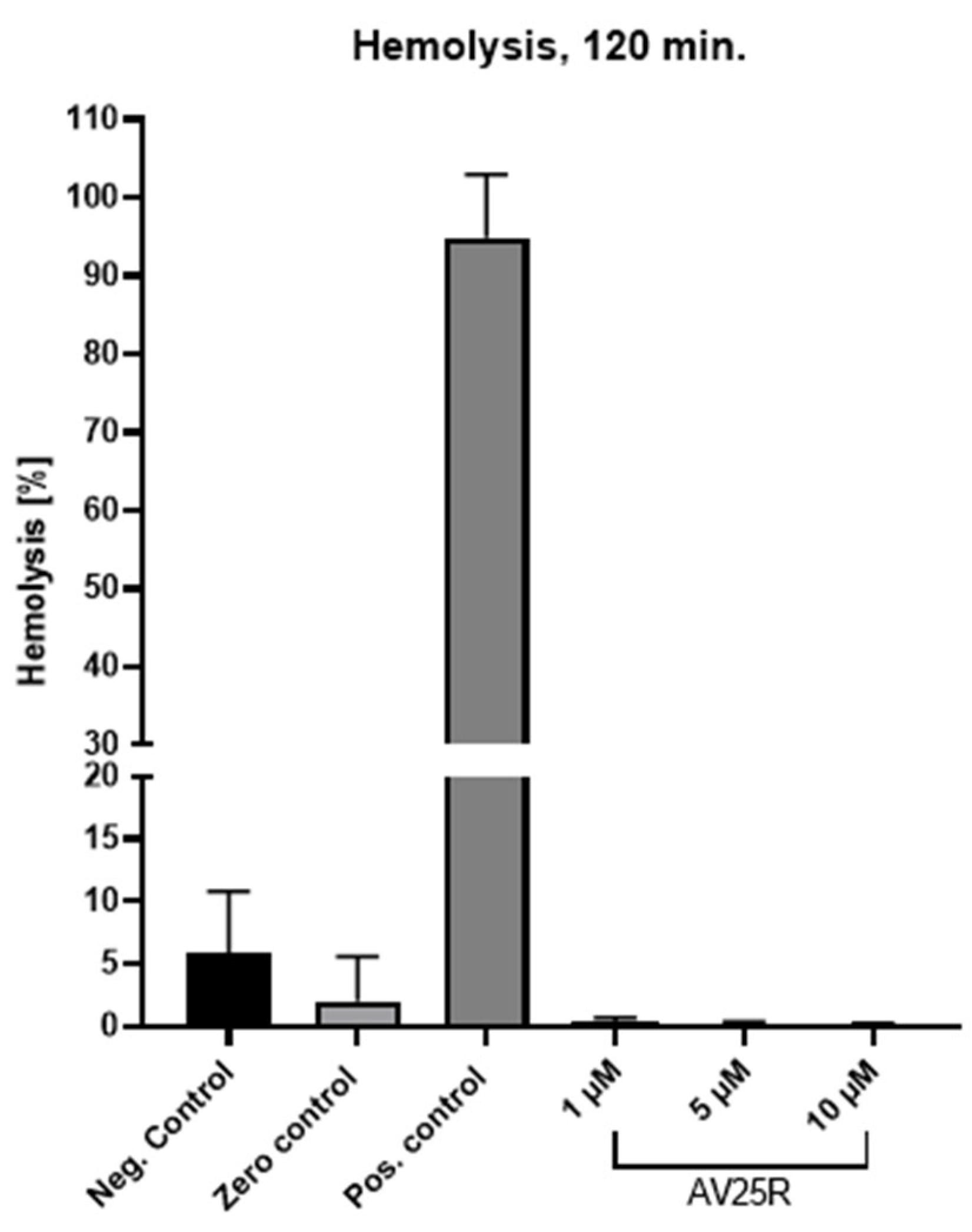
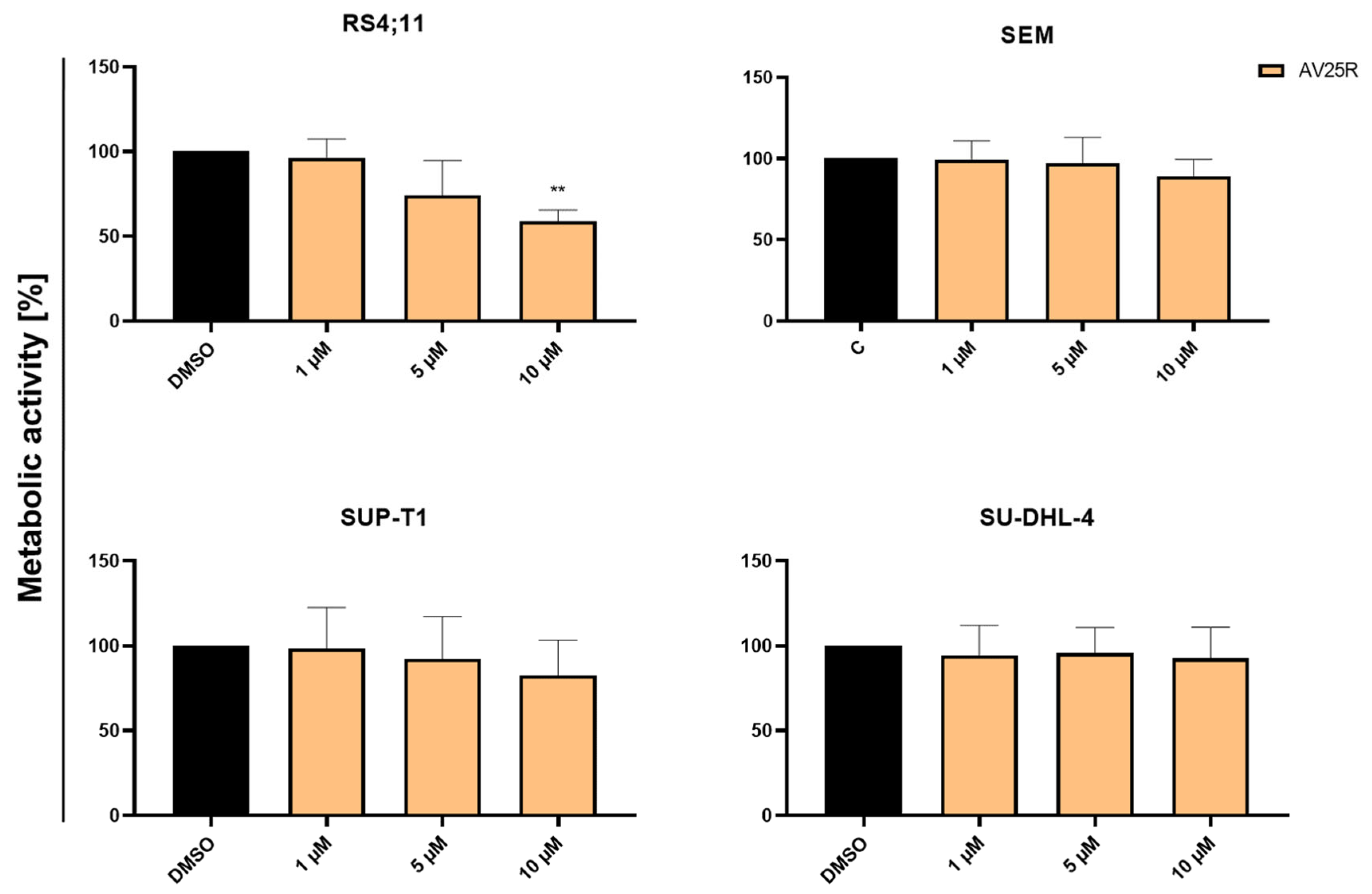
2.1.2. Evaluation of Proliferation and Metabolic Activity
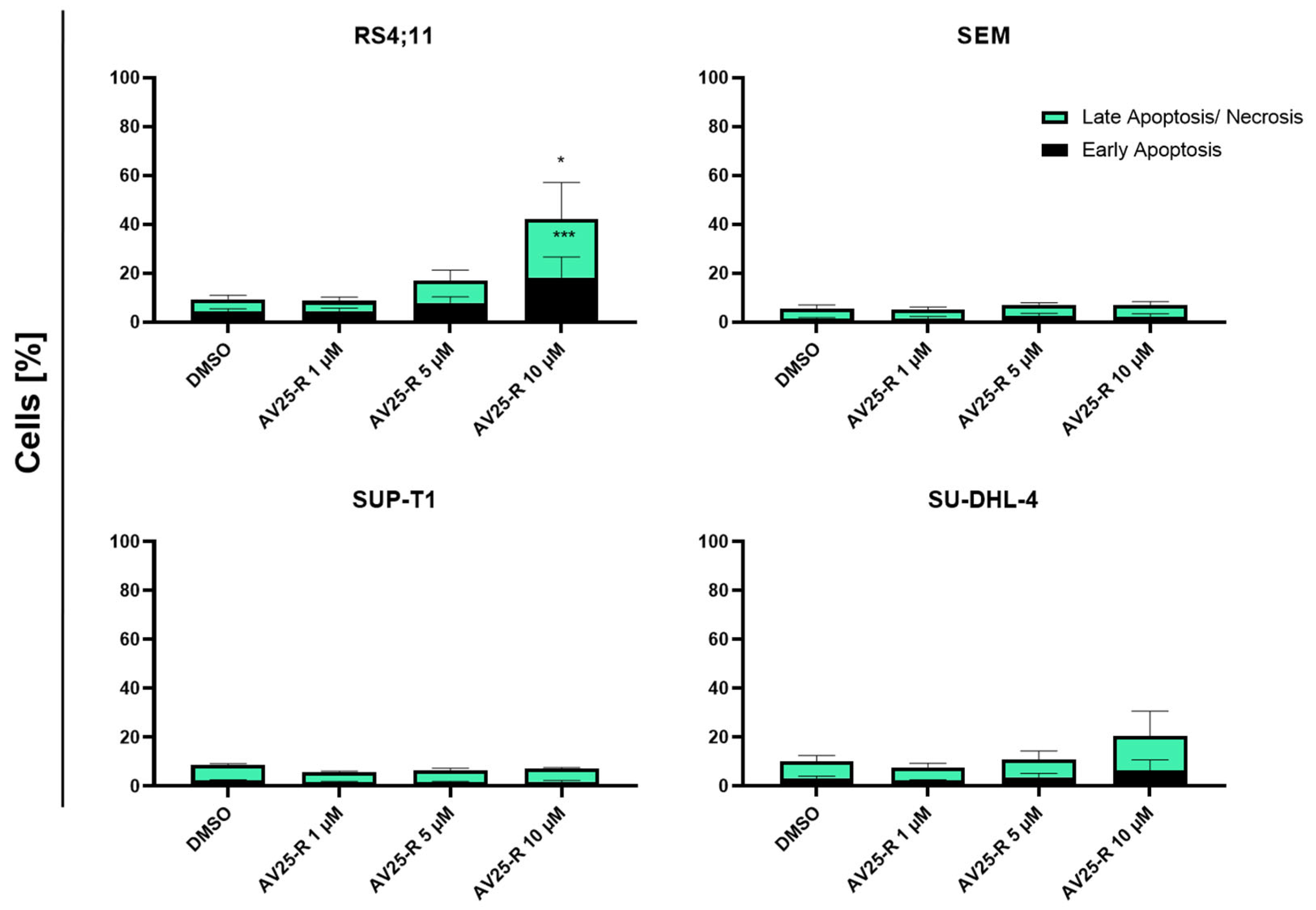
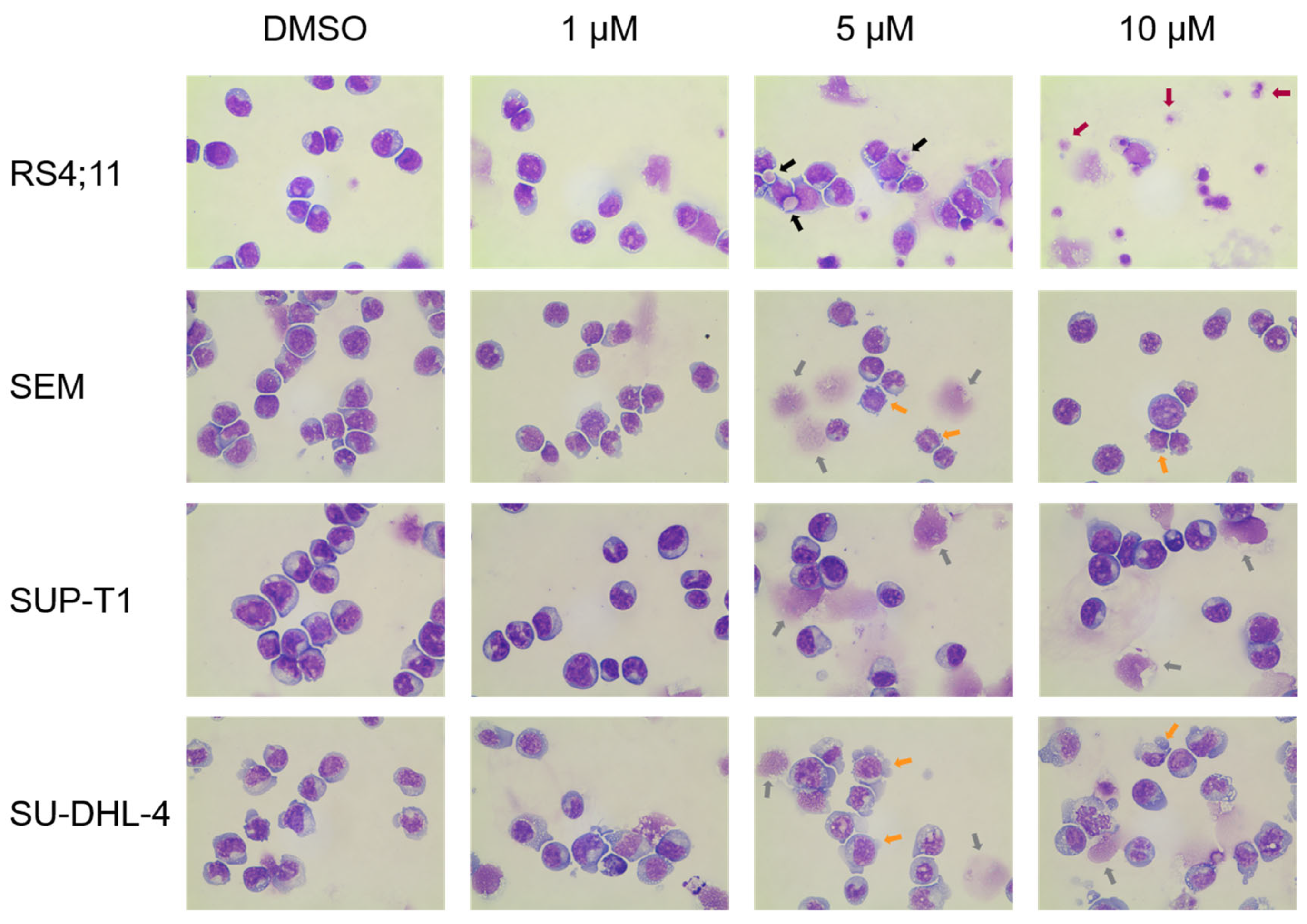
2.1.3. Induction of Apoptosis and Morphological Characterization
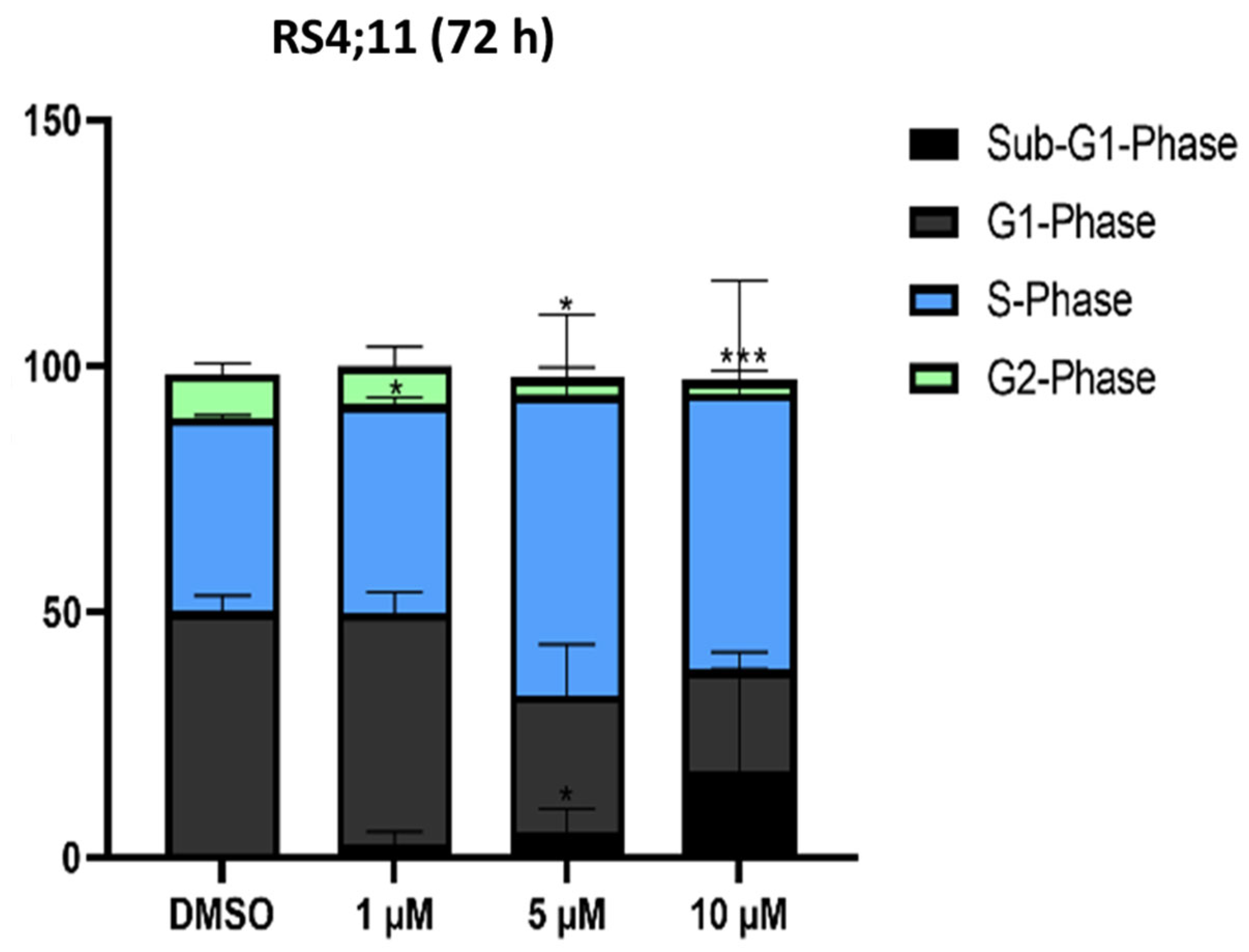
2.1.4. Cell Cycle Analysis
2.2. Evaluation of Gene Expression and Enrichtment Analysis
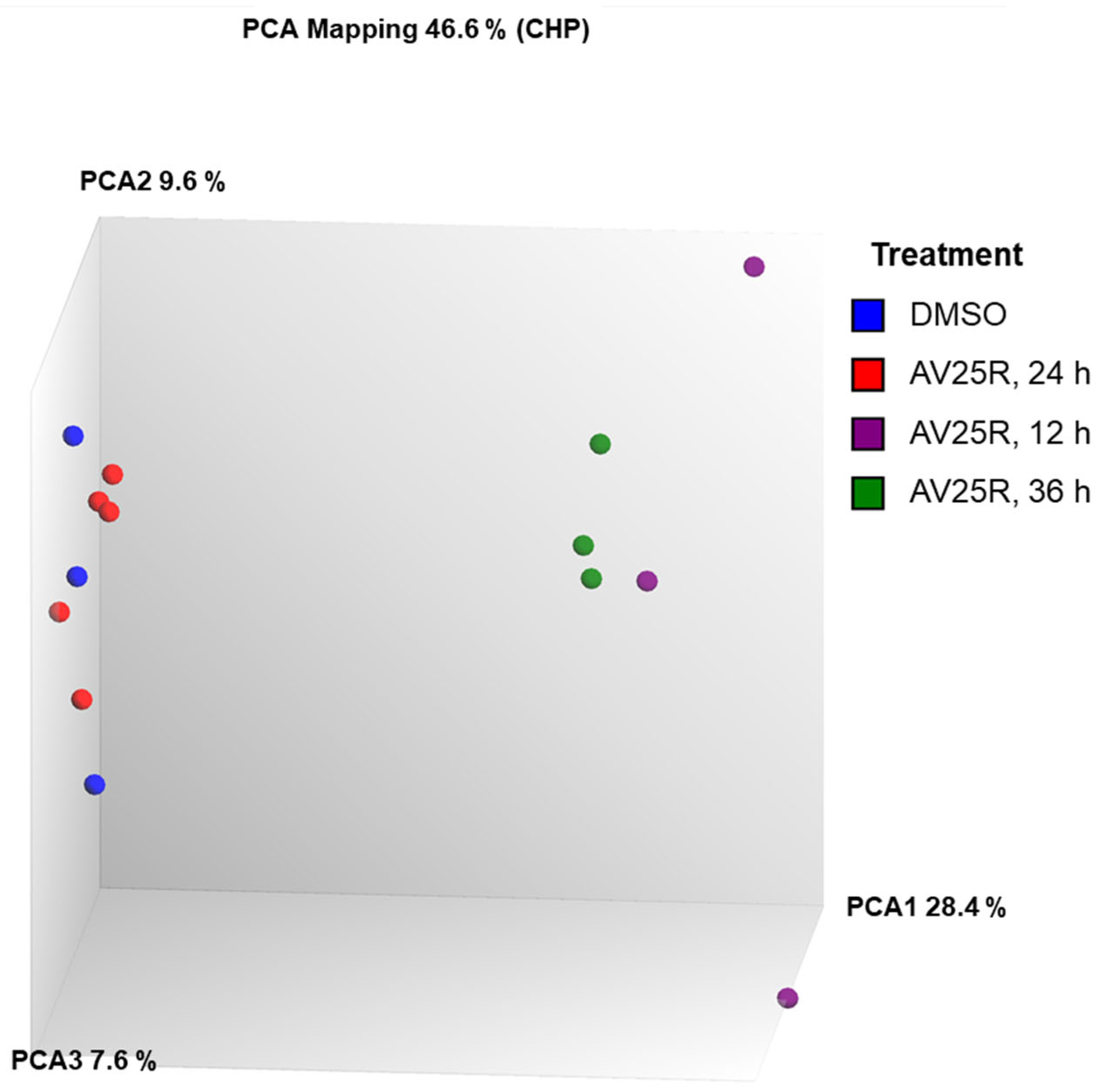
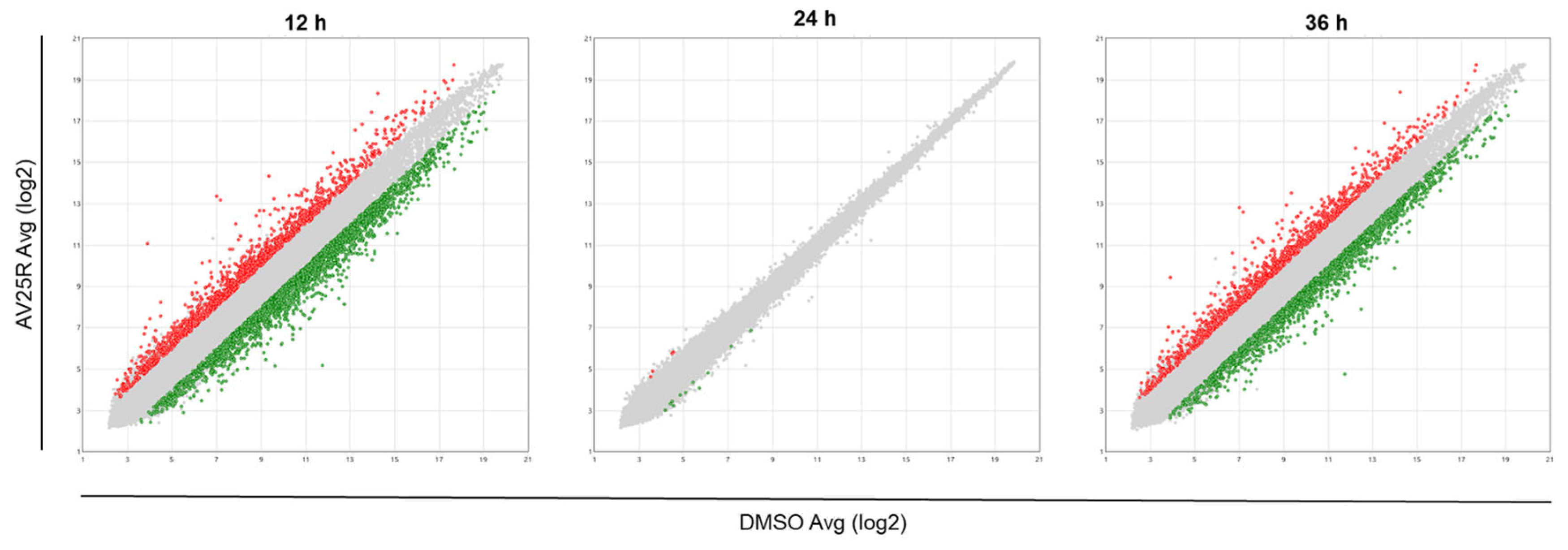
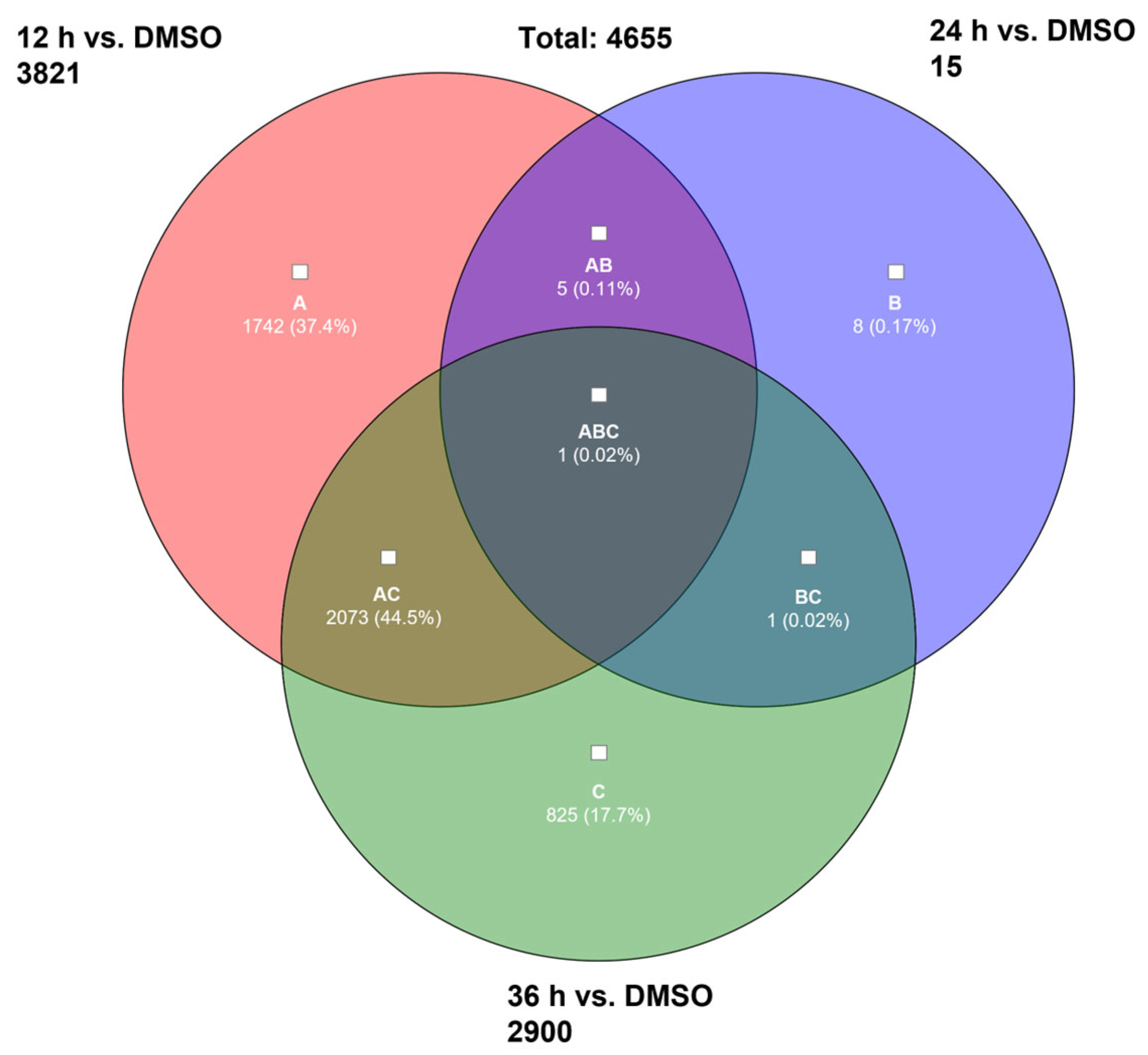
2.2.1. Microarray Analysis
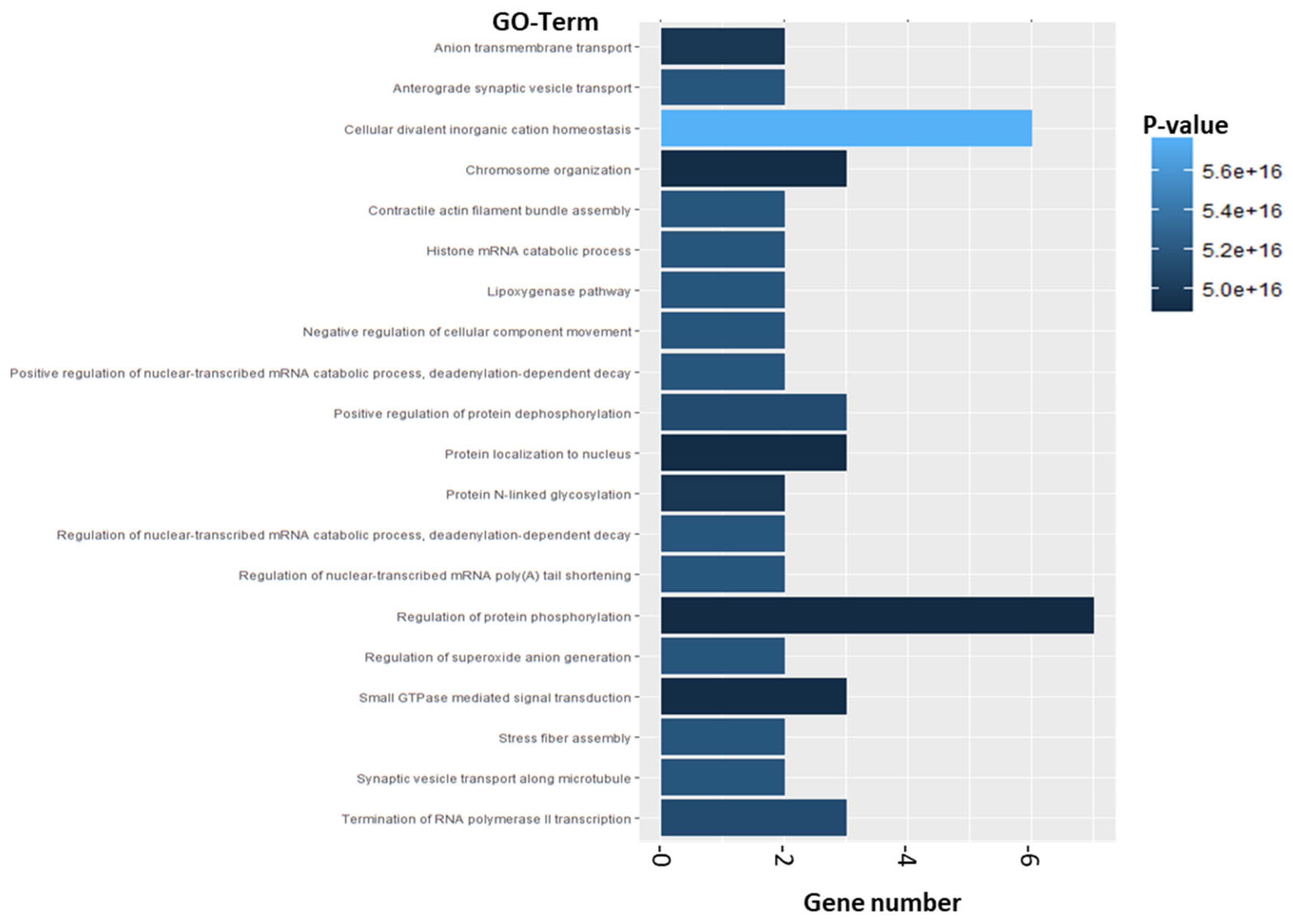
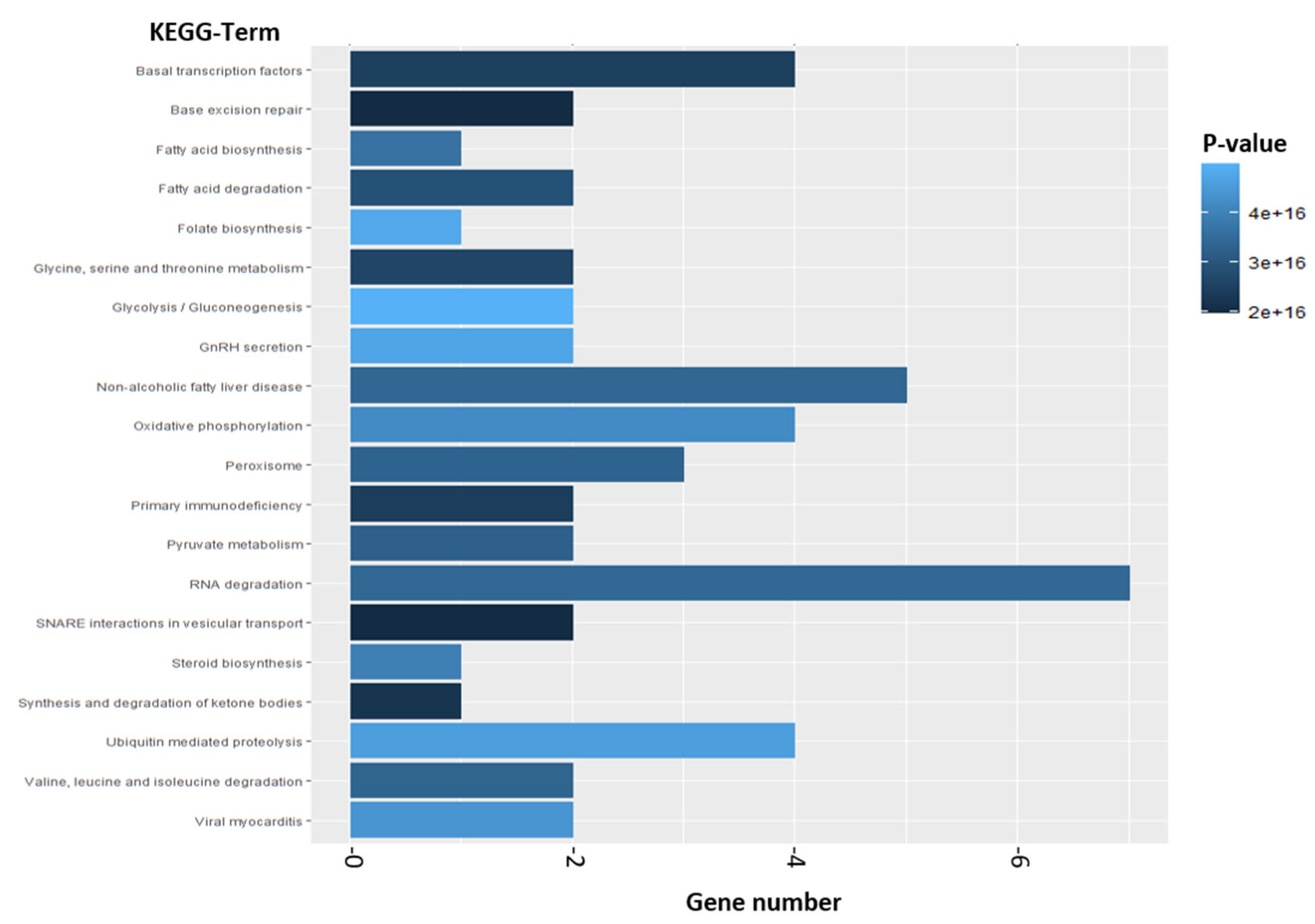
2.2.2. Enrichment Analysis
2.3. In Silico Profiling of AV25R
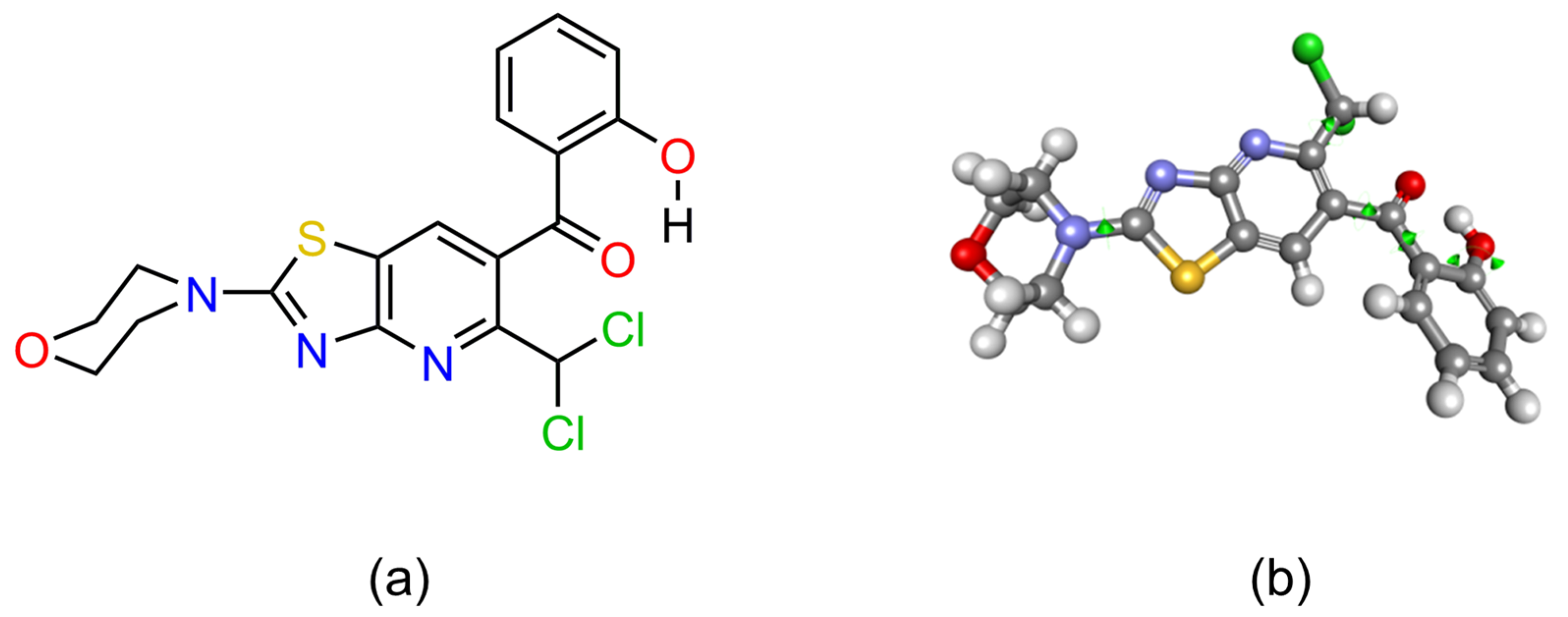
2.3.1. 3D Conformation of AV25R
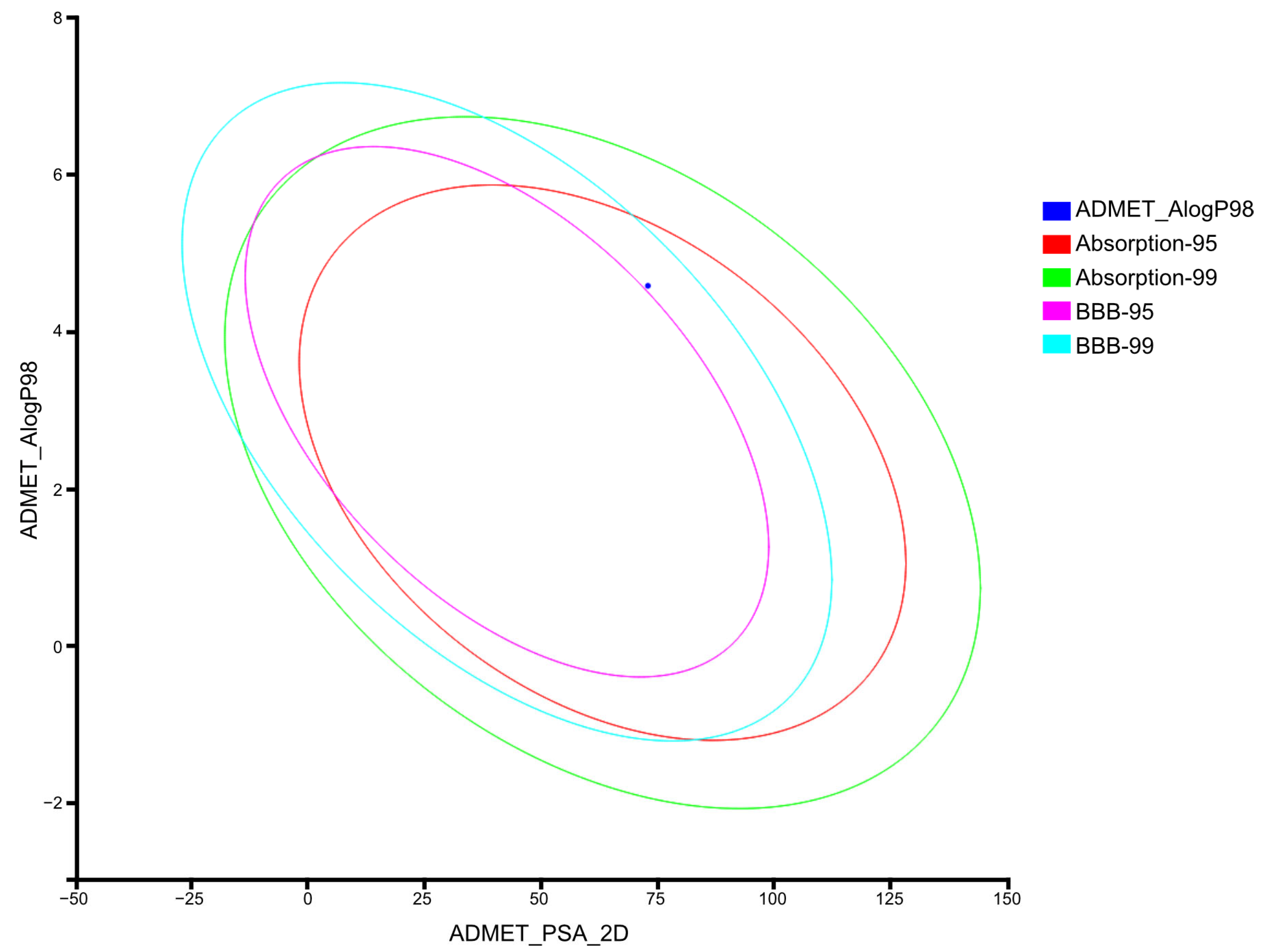
2.3.2. Computation of ADMET Descriptors for AV25R
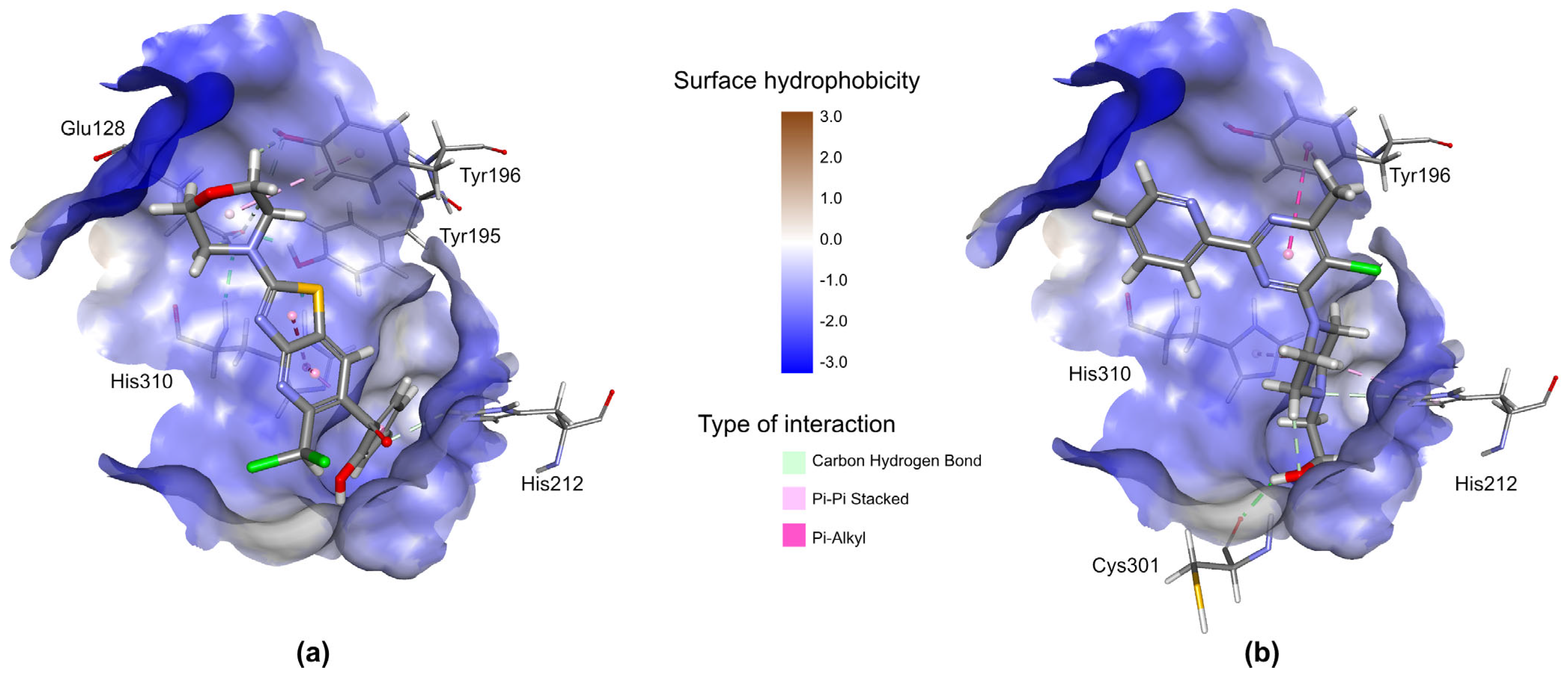
2.3.3. Prediction of Biological Targets of AV25R
3. Discussion
4. Materials and Methods
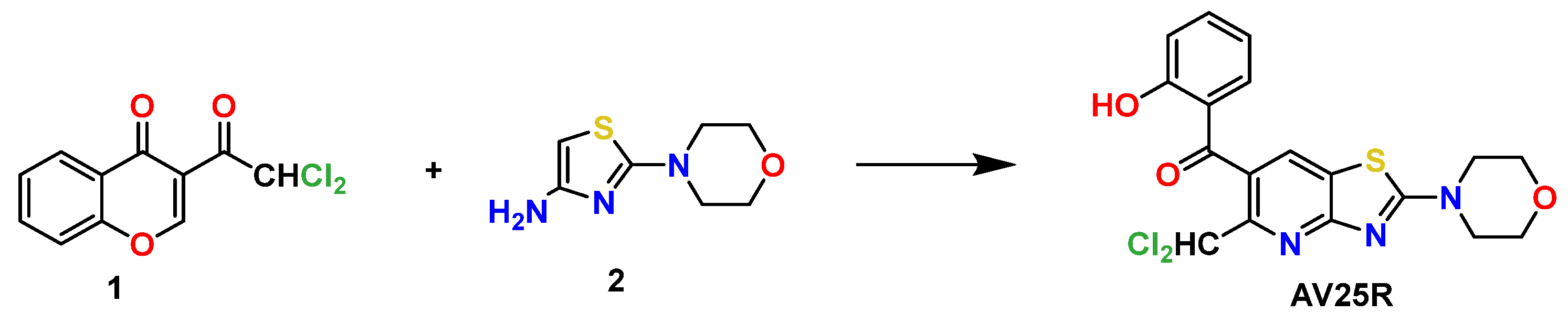
4.1. Chemical Substances
4.2. Cell lines and Cell Culture Methods
4.3. Drug Exposure Experiments
4.4. WST-1 Assay
4.5. Proliferation Assay
4.6. Calculation of IC50
4.7. Hemolysis Assay
4.8. Morphological Characterization
4.9. Analysis of Apoptosis
4.10. Cell Cycle Analysis
4.11. RNA Isolation for Microarray Analysis
4.12. Microarray Analysis
4.13. Enrichment Analysis
4.14. In Silico Profiling of AV25R
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, M.O.; Kavan, P.; Miller, W.H.; Panasci, L.; Assouline, S.; Johnson, N.; Cohen, V.; Patenaude, F.; Pollak, M.; Jagoe, R.T.; et al. Systemic cancer therapy: Achievements and challenges that lie ahead. Front. Pharmacol. 2013, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. Quo Vadis Cancer Therapy? Fascinating Discoveries of the Last 60 Years; LAP LAMBERT Academic Publishing: Saarbrucken, Germany, 2017. [Google Scholar]
- Malhotra, V.; Perry, M.C. Classical chemotherapy: Mechanisms, toxicities and the therapeutic window. Cancer Biol. Ther. 2003, 2 (Suppl. S1), S2–S4. [Google Scholar] [CrossRef] [PubMed]
- Morganti, S.; Tarantino, P.; Ferraro, E.; D’Amico, P.; Duso, B.A.; Curigliano, G. Next Generation Sequencing (NGS): A Revolutionary Technology in Pharmacogenomics and Personalized Medicine in Cancer. Adv. Exp. Med. Biol. 2019, 1168, 9–30. [Google Scholar] [PubMed]
- Kurzrock, R.; Giles, F.J. Precision oncology for patients with advanced cancer: The challenges of malignant snowflakes. Cell Cycle 2015, 14, 2219–2221. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Delord, J.P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Stacy, A.E.; Porter, G.M.; Merlot, A.M. Clinical development of targeted and immune based anti-cancer therapies. J. Experimental Clin. Cancer Res. 2019, 38, 156. [Google Scholar] [CrossRef] [PubMed]
- Gilead Statement on Zydelig®, U.S. Indication for Follicular Lymphoma and Small Lymphocytic Leukemia. Available online: https://www.gilead.com/news-and-press/company-statements/gilead-statement-on-zydelig-us-indication-for-follicular-lymphoma-and-small-lymphocytic-leukemia (accessed on 19 November 2023).
- FDA Alerts Healthcare Professionals about Clinical Trials with Zydelig (idelalisib) in Combination with Other Cancer Medicines [News Release]. Silver Springs, MD: U.S. Food and Drug Administration; 14 March 2016. Available online: http://www.fda.gov/Drugs/DrugSafety/ucm490618.htm?source=govdelivery&utm_medium=email&utm_source=govdelivery (accessed on 14 December 2023).
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef]
- Constantinescu, T.; Mihis, A.G. Two Important Anticancer Mechanisms of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2022, 23, 11595. [Google Scholar] [CrossRef]
- van der Zanden, S.Y.; Luimstra, J.J.; Neefjes, J.; Borst, J.; Ovaa, H. Opportunities for Small Molecules in Cancer Immunotherapy. Trends Immunol. 2020, 41, 493–511. [Google Scholar] [CrossRef]
- Zambre, V.P.; Petkar, N.N.; Dewoolkar, V.P.; Bhadke, S.V.; Sawant, S.D. Assessment of Structural Basis for Thiazolopyridine Derivatives as DNA Gyrase-B Inhibitors. Curr. Drug Discov. Technol. 2023, 20, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Kale, M.G.; Raichurkar, A.; P, S.H.; Waterson, D.; McKinney, D.; Manjunatha, M.R.; Kranthi, U.; Koushik, K.; Jena, L.K.; Shinde, V.; et al. Thiazolopyridine ureas as novel antitubercular agents acting through inhibition of DNA Gyrase B. J. Med. Chem. 2013, 56, 8834–8848. [Google Scholar] [CrossRef]
- Clark, R.B.; Lamppu, D.; Libertine, L.; McDonough, A.; Kumar, A.; LaRosa, G.; Rush, R.; Elbaum, D. Discovery of novel 2-((pyridin-3-yloxy)methyl)piperazines as α7 nicotinic acetylcholine receptor modulators for the treatment of inflammatory disorders. J. Med. Chem. 2014, 57, 3966–3983. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, S. Advances in the discovery of novel antimicrobials targeting the assembly of bacterial cell division protein FtsZ. Eur. J. Med. Chem. 2015, 95, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.E.A. Tryptoline-Based Benzothiazoles and Their Use as Antibiotics and Antibiotic Resistance-Modifying Agents. U.S. Patent 20220402914-A1, 22 December 2022. [Google Scholar]
- Patel, N.R.; Patel, D.V.; Murumkar, P.R.; Yadav, M.R. Contemporary developments in the discovery of selective factor Xa inhibitors: A review. Eur. J. Med. Chem. 2016, 121, 671–698. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.E.A. Capsid Inhibitors for the Treatment of HIV. U.S. Patent US-20230038823-A1. Unpublished. 2023. Available online: https://patents.google.com/patent/US20200038389A1/en (accessed on 13 December 2023).
- Kadow, J.F.; Naidu, B.N.; Wang, T.; Yin, Z.; Zhang, Z. 5-(N-[6,5]-Fused Bicyclic Aryl Tetrahydroisoquinolin-6-yl) Pyridin-3-yl-Acetic Acid Derivatives as Inhibitors of Human Immunodeficiency Virus. U.S. Patent 20180222890-A1, 20 June 2018. [Google Scholar]
- Morigi, R.; Locatelli, A.; Leoni, A.; Rambaldi, M. Recent Patents on Thiazole Derivatives Endowed with Antitumor Activity. Recent Pat. Anti-Cancer Drug Discov. 2015, 10, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Al-Said, M.S. Antitumor activity of novel pyridine, thiophene and thiazole derivatives. Arch. Pharmacal Res. 2012, 35, 965–973. [Google Scholar] [CrossRef] [PubMed]
- De, S.K.; Chen, L.H.; Stebbins, J.L.; Machleidt, T.; Riel-Mehan, M.; Dahl, R.; Chen, V.; Yuan, H.; Barile, E.; Emdadi, A.; et al. Discovery of 2-(5-nitrothiazol-2-ylthio)benzodthiazoles as novel c-Jun N-terminal kinase inhibitors. Bioorganic Med. Chem. 2009, 17, 2712–2717. [Google Scholar] [CrossRef]
- Asai, A.E.A. 1,3,4-Oxadiazole-2-Carboxamide Compound. EU Patent 2520575A1, 7 November 2012. [Google Scholar]
- Bärfacker, L.; Prechtl, S.; Siemeister, G.; Wengner, A.M.; Ackerstaff, J.; Nowak-Reppel, K.; Bader, B.; Lienau, P.; Stöckigt, D.; Heinrich, T. Amino-Substituted Isothiazoles. U.S. Patent 20150368260-A1, 15 June 2015. [Google Scholar]
- Gummadi, V.R.; Samajdar, S. Method of Preparing Oxazolo[4,5-B]Pyridine and Thiazolo[4,5-B]Pyridine Derivatives as IRAK4 Inhibitors for Treating Cancer. EU Patent EP3466955, 10 April 2019. [Google Scholar]
- Gummadi; Rao, V.; Samajdar, S. Bicyclic Heterocyclyl Derivatives as IRAK4 Inhibitors. U.S. Patent 10995100-B2, 19 March 2021. [Google Scholar]
- Crew, A.P.; Araujo, E.; Arvinas Operations Inc. Compounds and Methods for the Targeted Degradation of Interleukin-1-Receptor-Associated Kinase 4 Polypeptides. U.S. Patent 20220331297-A1, 20 July 2022. [Google Scholar]
- Ackler, S.L.; Bennett, N.B.; Boghaert, E.R.; Cullen, S.C.; Doherty, G.; Frey, R.R.; Haight, A.R.; Judd, A.S.; Kunzer, A.R.; Shen, X.; et al. BCL-XL Inhibitory Compounds and Antibody Drug Conjugates Including the Same. U.S. Patent 20200239553-A1, 20 August 2020. [Google Scholar]
- Jones, A.; Kemp, M.I.; Stockley, M.L.; Gibson, K.R.; Whitlock, G.A.; Madin, A. Novel Compounds. U.S. Patent 20230052191-A1, 19 September 2023. [Google Scholar]
- Iaroshenko, V.O.; Langer, P.; Mkrtchyan, S.; Ghazaryan, G.; Hakobyan, A.; Maalik, A.; Supe, L.; Villinger, A.; Tolmachev, A.; Ostrovskyi, D.; et al. 3-(Dichloroacetyl)chromone; A New Building Block for the Synthesis of Formylated Purine Isosteres: Design and Synthesis of Fused α-(Formyl)pyridines. Synthesis 2011, 2011, 469–479. [Google Scholar] [CrossRef]
- Azam, M.A.; Thathan, J.; Jubie, S. Dual targeting DNA gyrase B (GyrB) and topoisomerse IV (ParE) inhibitors: A review. Bioorganic Chem. 2015, 62, 41–63. [Google Scholar] [CrossRef]
- Park, H.R.; Kim, J.; Kim, T.; Jo, S.; Yeom, M.; Moon, B.; Choo, I.H.; Lee, J.; Lim, E.J.; Park, K.D.; et al. Oxazolopyridines and thiazolopyridines as monoamine oxidase B inhibitors for the treatment of Parkinson’s disease. Bioorganic Med. Chem. 2013, 21, 5480–5487. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Chen, J.; Wang, J.; Liu, J.; Jiang, Y. Targeting Akt in cancer for precision therapy. J. Hematol. Oncol. 2021, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Cimino, G.; Alder, H.; Nakamura, T.; Prasad, R.; Canaani, O.; Moir, D.T.; Jones, C.; Nowell, P.C.; Croce, C.M. The (4;11)(q21;q23) chromosome translocations in acute leukemias involve the VDJ recombinase. Proc. Natl. Acad. Sci. USA 1992, 89, 10464–10468. [Google Scholar] [CrossRef]
- Ragusa, D.; Makarov, E.M.; Britten, O.; Moralli, D.; Green, C.M.; Tosi, S. The RS4;11 cell line as a model for leukaemia with t(4;11)(q21;q23): Revised characterisation of cytogenetic features. Cancer Rep. 2019, 2, e1207. [Google Scholar] [CrossRef] [PubMed]
- Greil, J.; Gramatzki, M.; Burger, R.; Marschalek, R.; Peltner, M.; Trautmann, U.; Hansen-Hagge, T.E.; Bartram, C.R.; Fey, G.H.; Stehr, K.; et al. The acute lymphoblastic leukaemia cell line SEM with t(4;11) chromosomal rearrangement is biphenotypic and responsive to interleukin-7. Br. J. Haematol. 1994, 86, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Lockshin, R.A.; Zakeri, Z. Cell death in health and disease. J. Cell. Mol. Med. 2007, 11, 1214–1224. [Google Scholar] [CrossRef]
- Ashkenazi, A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 2008, 19, 325–331. [Google Scholar] [CrossRef]
- Voss, A.K.; Strasser, A. The essentials of developmental apoptosis. F1000Research 2020, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Estrada, C.C.; Maldonado, A.; Mallipattu, S.K. Therapeutic Inhibition of VEGF Signaling and Associated Nephrotoxicities. J. Am. Soc. Nephrol. JASN 2019, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L.; Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Santos, E.; Nogueira, K.A.B.; Fernandes, L.C.C.; Martins, J.R.P.; Reis, A.V.F.; Neto, J.D.B.V.; da Silva Júnior, I.J.; Pessoa, C.; Petrilli, R.; Eloy, J.O. EGFR targeting for cancer therapy: Pharmacology and immunoconjugates with drugs and nanoparticles. Int. J. Pharm. 2021, 592, 120082. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, H.D.; Sulaiman, R.S.; Qi, X.; Shetty, T.; Babu, S.S.P.; Sishtla, K.L.; Lee, B.; Quigley, J.; Alkhairy, S.; Briggs, C.M.; et al. Ferrochelatase is a therapeutic target for ocular neovascularization. EMBO Mol. Med. 2017, 9, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Babu, S.P.S.P.; White, D.; Corson, T.W. Ferrochelatase regulates retinal neovascularization. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 12419–12435. [Google Scholar]
- Cooper, A.C.; Karp, R.M.; Clark, E.J.; Taghizadeh, N.R.; Hoyt, J.G.; Labenski, M.T.; Murray, M.J.; Hannig, G.; Westlin, W.F.; Thompson, C.D. A novel methionine aminopeptidase-2 inhibitor, PPI-2458, inhibits non-Hodgkin’s lymphoma cell proliferation in vitro and in vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 2583–2590. [Google Scholar] [CrossRef]
- Steinberg, E.; Esa, R.; Schwob, O.; Stern, T.; Orehov, N.; Zamir, G.; Hubert, A.; Panigrahy, D.; Benny, O. Methionine aminopeptidase 2 as a potential target in pancreatic ductal adenocarcinoma. Am. J. Transl. Res. 2022, 14, 6243–6255. [Google Scholar]
- Gao, X.; Leone, G.W.; Wang, H. Cyclin D-CDK4/6 functions in cancer. Adv. Cancer Res. 2020, 148, 147–169. [Google Scholar]
- Federici, L.; Lo Sterzo, C.; Pezzola, S.; Di Matteo, A.; Scaloni, F.; Federici, G.; Caccuri, A.M. Structural basis for the binding of the anticancer compound 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol to human glutathione s-transferases. Cancer Res. 2009, 69, 8025–8034. [Google Scholar] [CrossRef]
- Darnell, J.E. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhang, Y.; Xu, W.; Liu, D.; Jin, W.; Chen, X.; Hong, N. The chromatin accessibility dynamics during cell fate specifications in zebrafish early embryogenesis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Park, S.; Chapuis, N.; Tamburini, J.; Bardet, V.; Cornillet-Lefebvre, P.; Willems, L.; Green, A.; Mayeux, P.; Lacombe, C.; Bouscary, D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica 2010, 95, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting multiple signaling pathways: The new approach to acute myeloid leukemia therapy. Signal Transduct. Target. Ther. 2020, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Wang, S.; Chen, F.-E. Small-molecule MDM2 inhibitors in clinical trials for cancer therapy. Eur. J. Med. Chem. 2022, 236, 114334. [Google Scholar] [CrossRef]
- Koczan, D.; Fitzner, B.; Zettl, U.K.; Hecker, M. Microarray data of transcriptome shifts in blood cell subsets during S1P receptor modulator therapy. Sci. Data 2018, 5, 180145. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 Value (µM) |
|---|---|
| SEM | 20.98 |
| RS4;11 | 1.95 |
| SUP-T1 | 12.17 |
| SU-DHL-4 | 12.72 |
| Pathway | Total Number of DEGs | Up-Regulated DEGs | Down-Regulated DEGs | Significance | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12 h | 24 h | 36 h | 12 h | 24 h | 36 h | 12 h | 24 h | 36 h | 12 h | 24 h | 36 h | |
| VEGFA-VEGFR2 signaling | 87 | n.i. | 58 | 23 | n.i. | 23 | 64 | n.i. | 35 | 32.71 | n.i. | 18.73 |
| Ciliary landscape | 56 | n.i. | 39 | 5 | n.i. | 6 | 51 | n.i. | 33 | 27.85 | n.i. | 17.72 |
| Electron transport chain: OXPHOS system in mitochondria | 39 | n.i. | 22 | 5 | n.i. | 5 | 34 | n.i. | 17 | 26.12 | n.i. | 11.68 |
| Nonalcoholic fatty liver disease | 44 | n.i. | 27 | 7 | n.i. | 8 | 37 | n.i. | 19 | 23.63 | n.i. | 12.01 |
| Retinoblastoma gene in cancer | 32 | n.i. | n.i. | 0 | n.i. | n.i. | 32 | n.i. | n.i. | 20.74 | n.i. | n.i. |
| miR-targeted genes in lymphocytes | 75 | n.i. | 62 | 18 | n.i. | 22 | 57 | n.i. | 40 | 20.32 | n.i. | 18.36 |
| Proteasome degradation | 27 | n.i. | n.i. | 1 | n.i. | n.i. | 26 | n.i. | n.i. | 19.70 | n.i. | n.i. |
| Alzheimer’s disease | 51 | n.i. | 41 | 10 | n.i. | 10 | 41 | n.i. | 31 | 18.50 | n.i. | 15.54 |
| miR-targeted genes in epithelium | 57 | n.i. | 45 | 13 | n.i. | 15 | 44 | n.i. | 30 | 17.46 | n.i. | 14.23 |
| EGF/EGFR signaling pathway | 37 | n.i. | n.i. | 19 | n.i. | n.i. | 18 | n.i. | n.i. | 16.55 | n.i. | n.i. |
| Elabela signaling pathway | n.i. | 1 | n.i. | n.i. | 0 | n.i. | n.i. | 1 | n.i. | n.i. | 3.15 | n.i. |
| Perturbations to host-cell autophagy, induced by SARS-CoV-2 proteins | n.i. | 1 | n.i. | n.i. | 0 | n.i. | n.i. | 1 | n.i. | n.i. | 2.71 | n.i. |
| Steatosis adverse outcome pathway | n.i. | 1 | n.i. | n.i. | 0 | n.i. | n.i. | 1 | n.i. | n.i. | 2.58 | n.i. |
| Autophagy | n.i. | 1 | n.i. | n.i. | 0 | n.i. | n.i. | 1 | n.i. | n.i. | 2.51 | n.i. |
| JAK-STAT signaling in the regulation of Beta-cells | n.i. | 1 | n.i. | n.i. | 0 | n.i. | n.i. | 1 | n.i. | n.i. | 2.45 | n.i. |
| Target of rapamycin signaling | n.i. | 1 | n.i. | n.i. | 0 | n.i. | n.i. | 1 | n.i. | n.i. | 2.44 | n.i. |
| Factors and pathways affecting insulin-like growth factor (IGF1)-Akt signaling | n.i. | 1 | n.i. | n.i. | 0 | n.i. | n.i. | 1 | n.i. | n.i. | 2.41 | n.i. |
| Neurodegeneration with brain iron accumulation (NBIA) subtypes pathway | n.i. | 1 | n.i. | n.i. | 0 | n.i. | n.i. | 1 | n.i. | n.i. | 2.35 | n.i. |
| MTOR signaling | n.i. | 1 | n.i. | n.i. | 0 | n.i. | n.i. | 1 | n.i. | n.i. | 2.30 | n.i. |
| DYRK1A involvement regarding cell proliferation in brain development | n.i. | 1 | n.i. | n.i. | 0 | n.i. | n.i. | 1 | n.i. | n.i. | 2.19 | n.i. |
| Alzheimer’s disease and miRNA effects | n.i. | n.i. | 42 | n.i. | n.i. | 10 | n.i. | n.i. | 32 | n.i. | n.i. | 12.88 |
| RNA pol II transcription—Initiation and elongation | n.i. | n.i. | 21 | n.i. | n.i. | 6 | n.i. | n.i. | 15 | n.i. | n.i. | 12.27 |
| miR-targeted genes in muscle cell | n.i. | n.i. | 46 | n.i. | n.i. | 16 | n.i. | n.i. | 30 | n.i. | n.i. | 12.08 |
| Rank | PDB ID | Target Name | Target Full Name | Target Class | Fit Value |
|---|---|---|---|---|---|
| 1 | 2HRC | FECH | Ferrochelatase | Lyase | 0.843442 |
| 2 | 4IKR | MAP11 | Methionine aminopeptidase 1 | Aminopeptidase | 0.769355 |
| 3 | 3IE3 | GSTP1 | Glutathione S-transferase P | Transferase | 0.736885 |
| 4 | 1FKG | FKB1A | Peptidyl-prolyl cis-trans isomerase FKBP1A | Isomerase | 0.736476 |
| 5 | 3BYZ | DHI1 | 11-beta-hydroxysteroid dehydrogenase 1 | Oxidoreductase | 0.702364 |
| 6 | 3ET7 | FAK2 | Protein-tyrosine kinase 2-beta | Tyrosine-protein kinase | 0.649484 |
| 7 | 4NRA | BAZ2B | Bromodomain adjacent to zinc finger domain protein 2B | DNA-binding | 0.587567 |
| 8 | 4OJ4 | PPARG | Peroxisome proliferator-activated receptor gamma | DNA-binding | 0.577672 |
| 9 | 3FZM | HSPA8 | Heat shock cognate 71 kDa protein | Chaperone | 0.563491 |
| 10 | 3TJS | CYP3A4 | Cytochrome P450 3A4 | Oxidoreductase | 0.482263 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ladwig, A.; Gupta, S.; Ehlers, P.; Sekora, A.; Alammar, M.; Koczan, D.; Wolkenhauer, O.; Junghanss, C.; Langer, P.; Murua Escobar, H. Exploring Thiazolopyridine AV25R: Unraveling of Biological Activities, Selective Anti-Cancer Properties and In Silico Target and Binding Prediction in Hematological Neoplasms. Molecules 2023, 28, 8120. https://doi.org/10.3390/molecules28248120
Ladwig A, Gupta S, Ehlers P, Sekora A, Alammar M, Koczan D, Wolkenhauer O, Junghanss C, Langer P, Murua Escobar H. Exploring Thiazolopyridine AV25R: Unraveling of Biological Activities, Selective Anti-Cancer Properties and In Silico Target and Binding Prediction in Hematological Neoplasms. Molecules. 2023; 28(24):8120. https://doi.org/10.3390/molecules28248120
Chicago/Turabian StyleLadwig, Annika, Shailendra Gupta, Peter Ehlers, Anett Sekora, Moosheer Alammar, Dirk Koczan, Olaf Wolkenhauer, Christian Junghanss, Peter Langer, and Hugo Murua Escobar. 2023. "Exploring Thiazolopyridine AV25R: Unraveling of Biological Activities, Selective Anti-Cancer Properties and In Silico Target and Binding Prediction in Hematological Neoplasms" Molecules 28, no. 24: 8120. https://doi.org/10.3390/molecules28248120
APA StyleLadwig, A., Gupta, S., Ehlers, P., Sekora, A., Alammar, M., Koczan, D., Wolkenhauer, O., Junghanss, C., Langer, P., & Murua Escobar, H. (2023). Exploring Thiazolopyridine AV25R: Unraveling of Biological Activities, Selective Anti-Cancer Properties and In Silico Target and Binding Prediction in Hematological Neoplasms. Molecules, 28(24), 8120. https://doi.org/10.3390/molecules28248120