

Direct Homogeneous Synthesis of Compounds with Two O Atoms and Long-Chain Hydrocarbons from CO and H2: Co–Ru/N-methylpyrrolidone Catalyst



Abstract
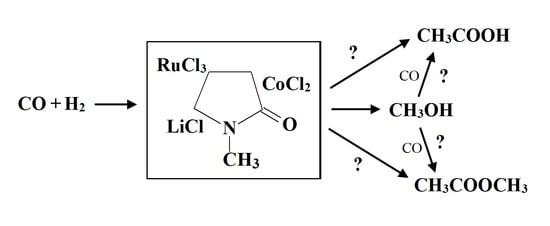
:
1. Introduction
- (a)
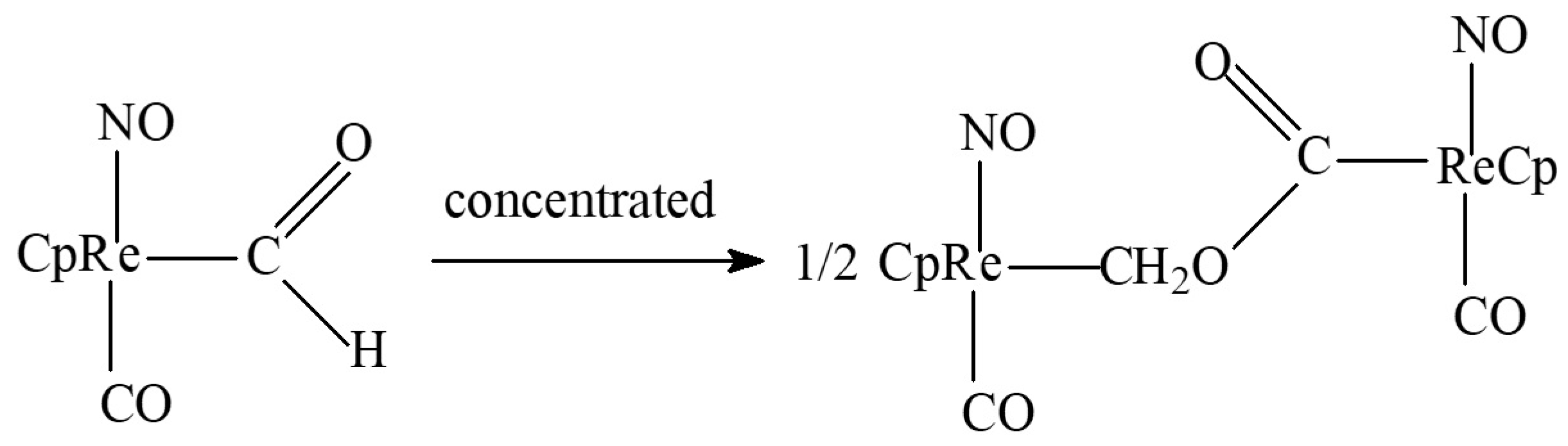
- For CO reduction, two-component carbonyl-late transition metal (Re, Fe, cluster, or polymetallic) hydride systems are required since single-centered hydride carbonylation is thermodynamically unfavorable.
- (b)
- For the effective formation of the C–C bond in homogeneous complexes, it is usually necessary to have Lewis-type promoters with optimal acidity. Polymetallic hydrides with a possible content of Ta, Ln, Y, and Ti promote chain growth without (ethenediolates) or with partial C–O breaking (allyl oxides, squarats, etc.) [12].
- (c)
- Long-chain product elimination requires the presence of sufficiently strong electrophiles and leads to the problem of the combination of protonic and hydride hydrogen forms at the same conditions. Therefore, it becomes necessary to use very high pressure and elevated temperatures.
2. Results
3. Discussion
3.1. Features of Oxygenate Formation
3.2. Features of Formation of Long-Chain Alkanes and Alcohols
4. Materials and Methods
5. Conclusions
- (a)
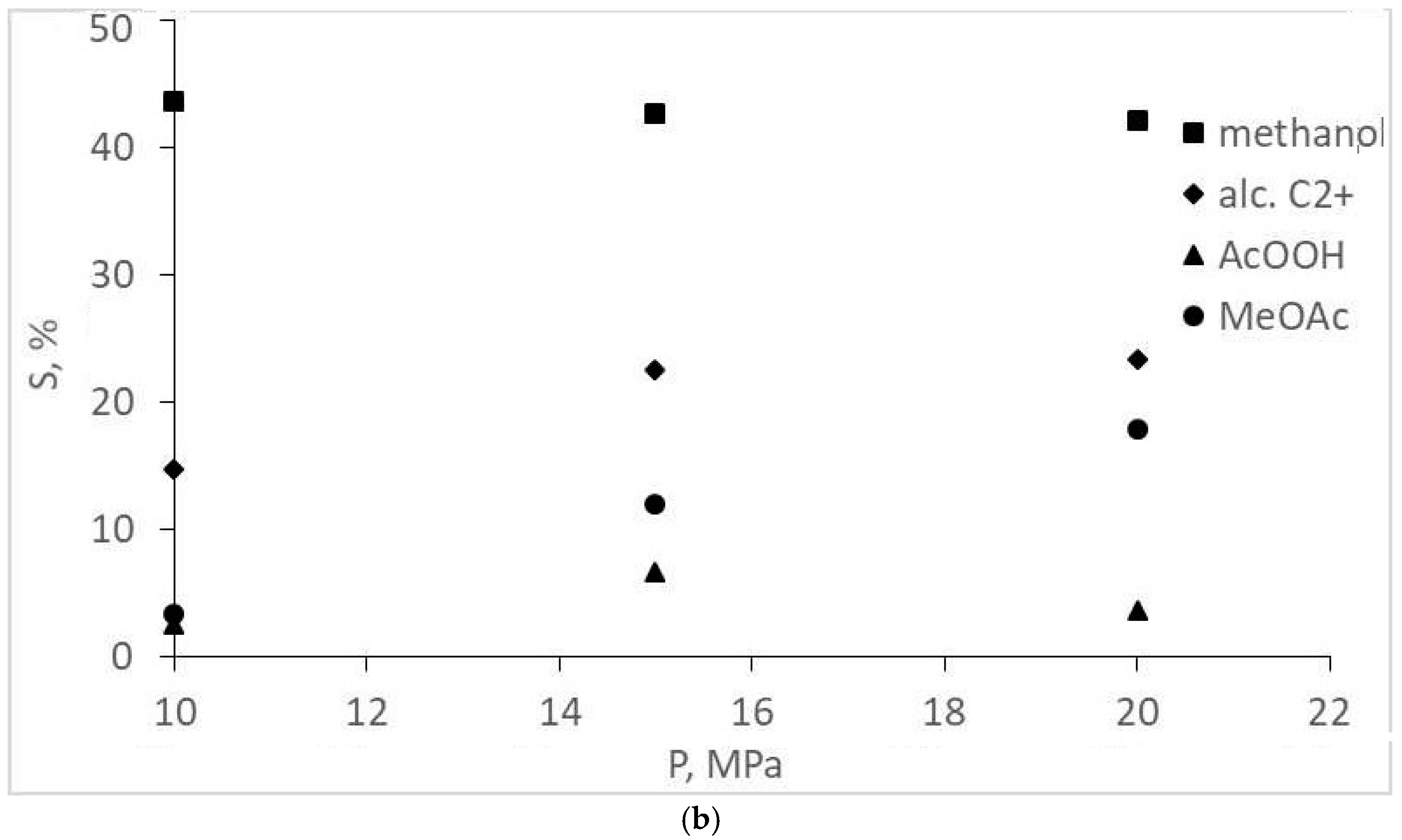
- Combinations of selectivity values of methanol, ethanol, methyl and ethyl acetate, and acetic acid, and the complete absence of acetaldehyde, can be interpreted as an indication of the formation of both acetates and acetic acid by direct independent CO and H2 conversion without any alcohol formation. This is very unusual for a homogeneous catalytic synthesis of two-oxygen-containing substances.
- (b)
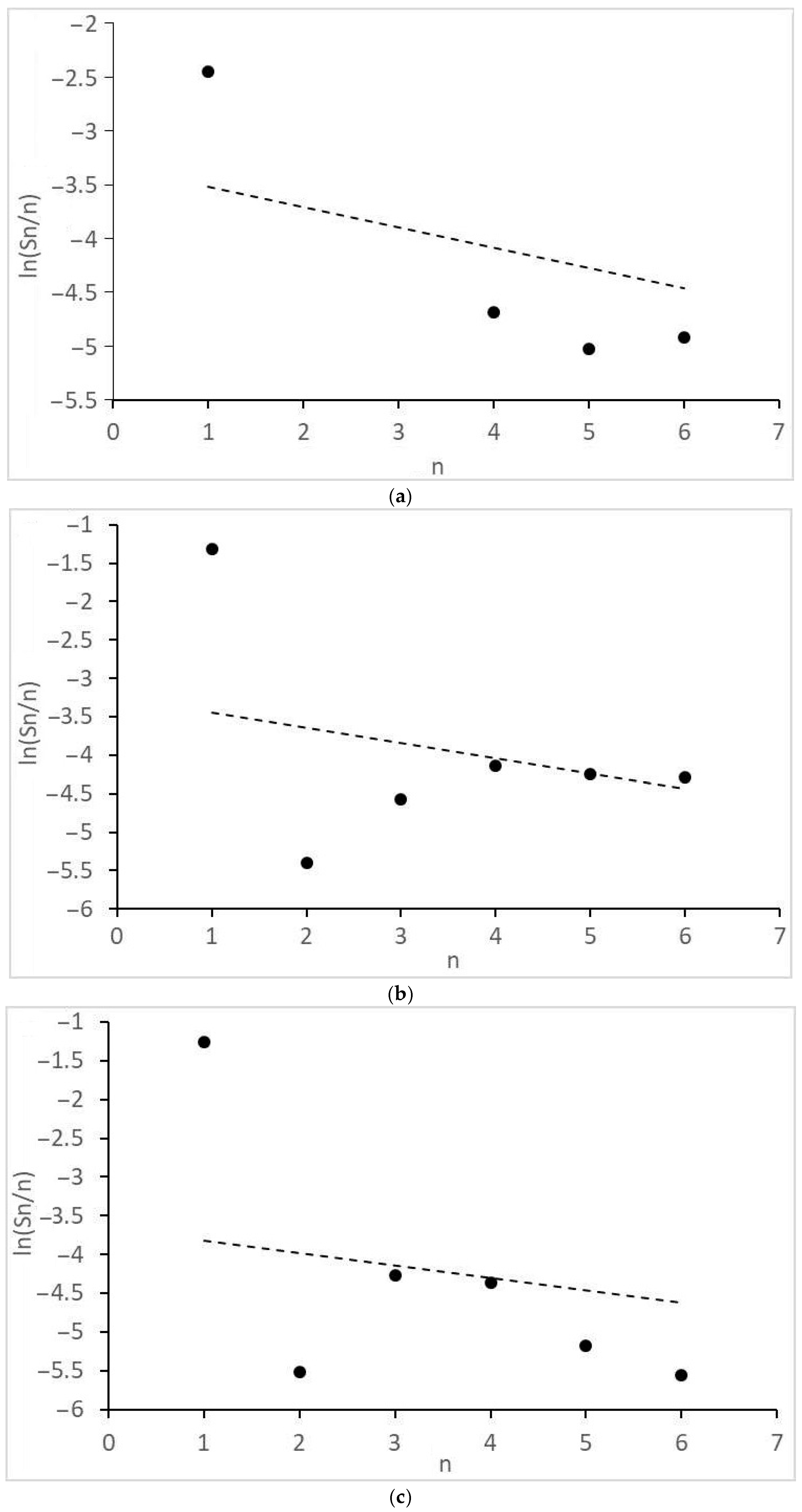
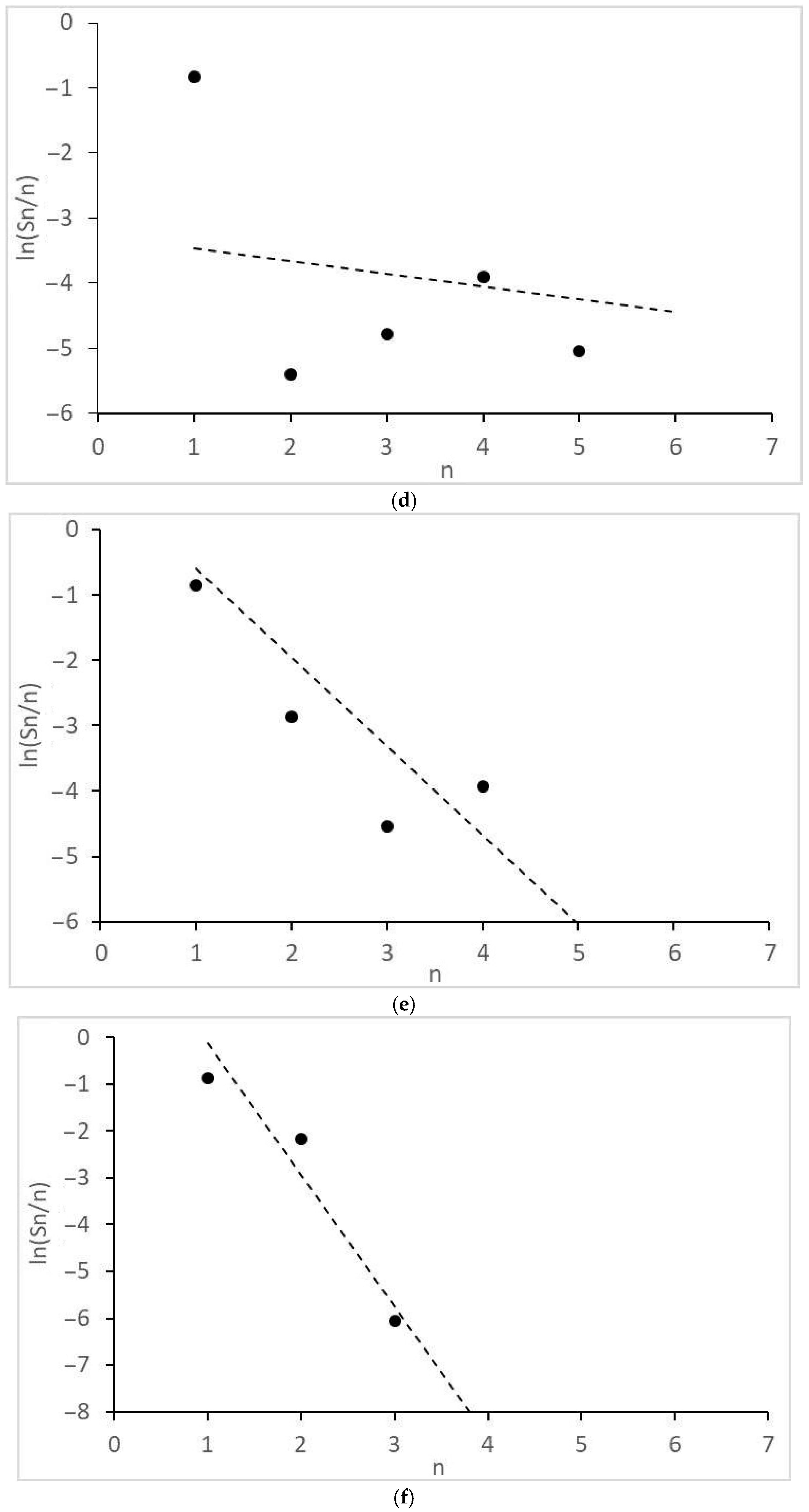
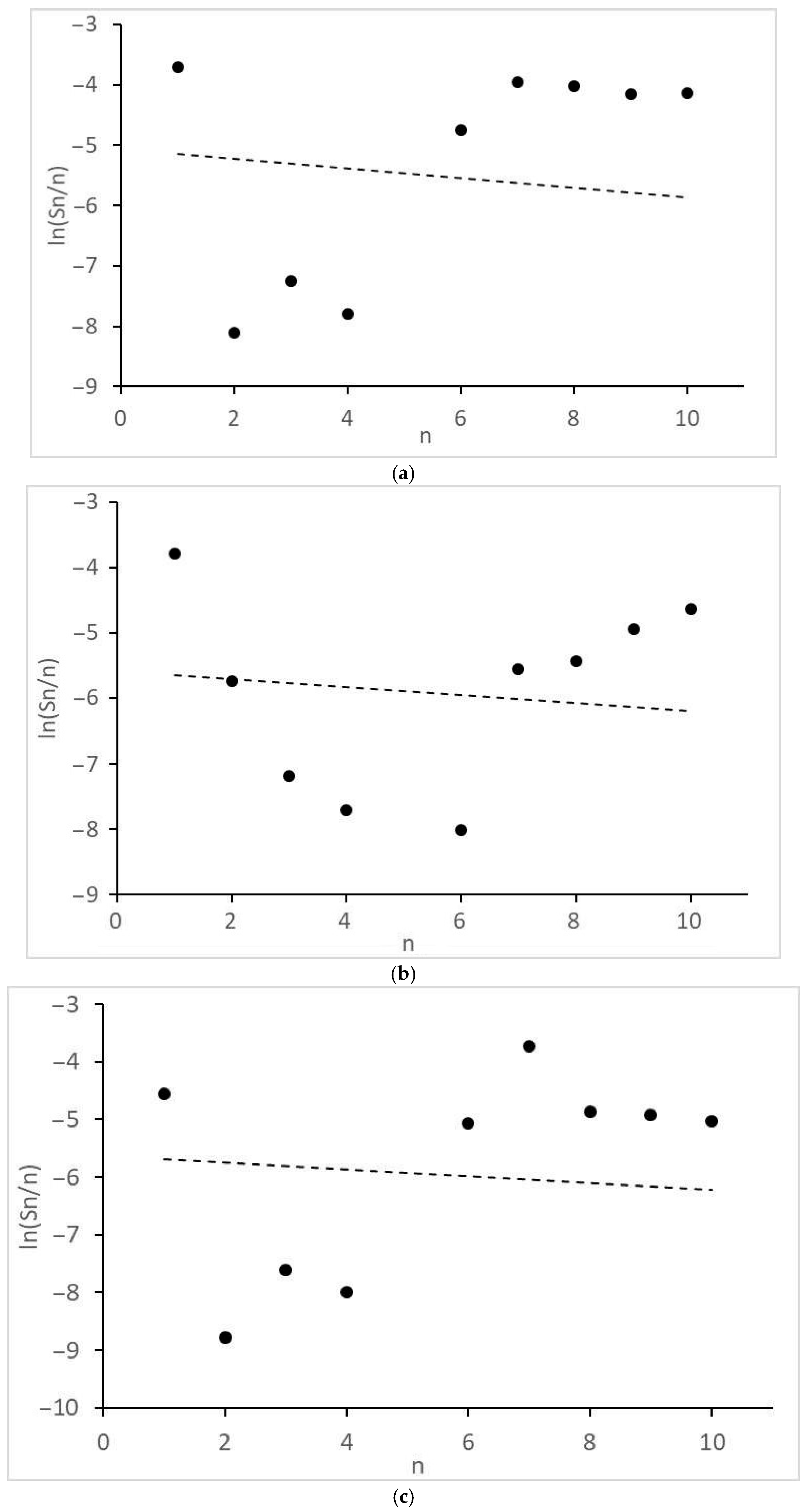
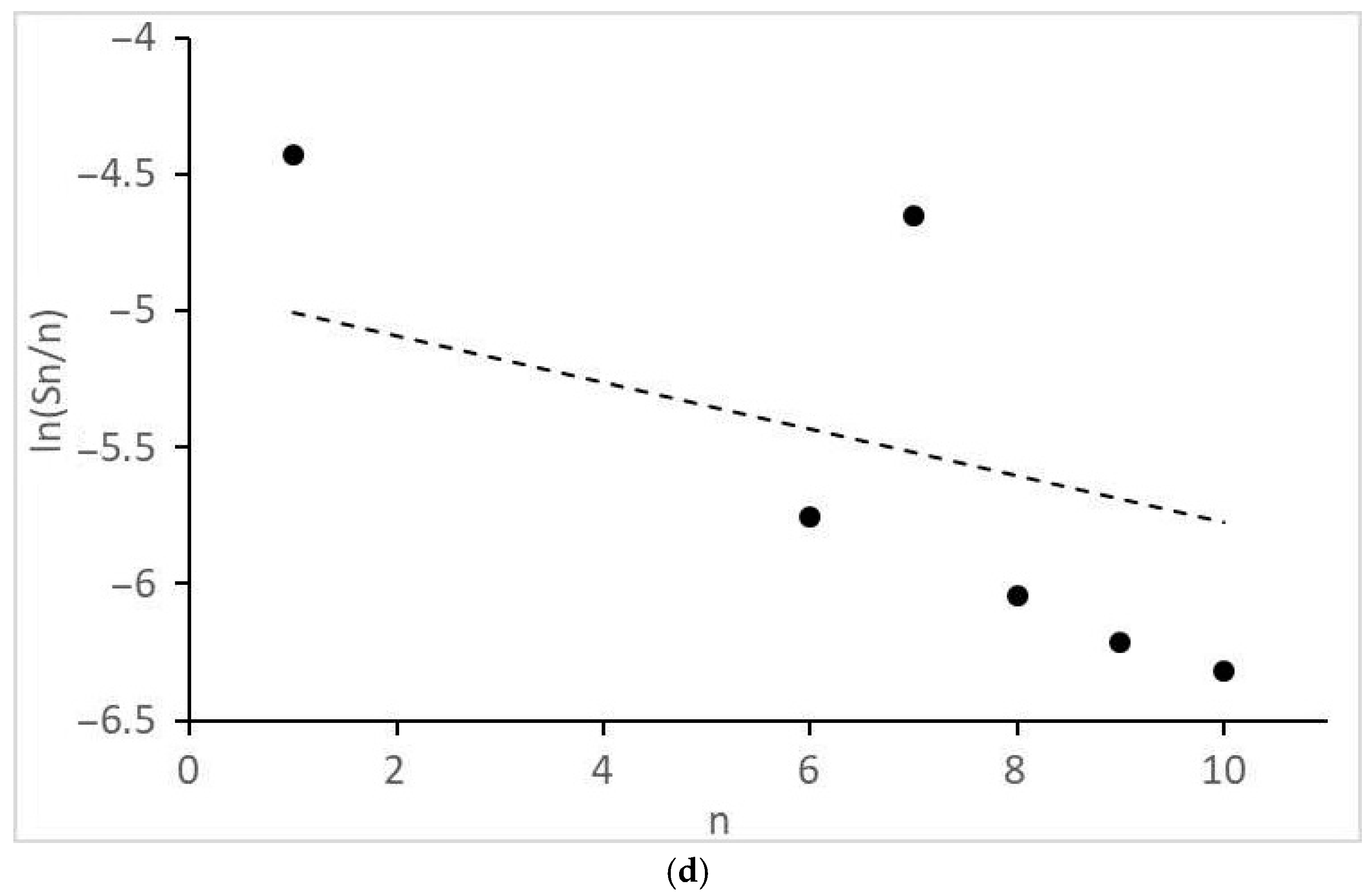
- Significant and different deviations of the molecular mass distributions of alkanes and alcohols from the Anderson-Schultz-Flory model, characterized by local maxima and minima, are observed. These tendencies may indicate significant restrictive control of the growth of carbon chains.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cornils, B.; Herrmann, W.A.; Rasch, M. Otto Roelen, Pioneer in Industrial Homogeneous Catalysis. Angew. Chem. Int. Ed. 1994, 33, 2144–2163. [Google Scholar] [CrossRef]
- Arpe, H.-J. Industrielle Organische Chemie: Bedeutende vor- und Zwischenprodukte; Wiley-VCH-Verlag: Weinheim, Germany, 2007. [Google Scholar]
- Franke, R.; Selent, D.; Börner, A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.H. The Cativa Process for the Manufacture Plant of Acetic Acid. Platinum Met. Rev. 2000, 44, 94–105. [Google Scholar]
- Ohkuma, T.; Ooka, H.; Ikariya, T.; Noyori, R. Preferential hydrogenation of aldehydes and ketones. J. Am. Chem. Soc. 1995, 117, 10417–10418. [Google Scholar] [CrossRef]
- Gorgas, N.; Stöger, B.; Veiros, L.F.; Kirchner, K. Highly Efficient and Selective Hydrogenation of Aldehydes: A Well-Defined Fe(II) Catalyst Exhibits Noble-Metal Activity. ACS Catal. 2016, 6, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Dombek, B.D. Homogeneous Catalytic Hydrogenation of Carbon Monoxide: Ethylene Glycol and Ethanol from Synthesis Gas. Adv. Catal. 1983, 32, 325–416. [Google Scholar] [CrossRef]
- Marko, L. TMC Literature Highlights—30. Transit. Met. Chem. 1992, 17, 474–480. [Google Scholar] [CrossRef]
- Whyman, R.; Wright, A.P.; Iggo, J.A.; Heaton, B.T. Carbon monoxide activation in homogeneously catalysed reactions: The nature and roles of catalytic promoters. J. Chem. Soc. Dalton Trans. 2002, 771–777. [Google Scholar] [CrossRef]
- West, N.M.; Miller, A.J.M.; Labinger, J.A.; Bercaw, J.E. Homogeneous syngas conversion. Coord. Chem. Rev. 2011, 255, 881–898. [Google Scholar] [CrossRef]
- Labinger, J.A. Approaches to homogeneously catalyzed CO hydrogenation: A personal retrospective. J. Organometal. Chem. 2017, 847, 4–12. [Google Scholar] [CrossRef]
- Parr, J.M.; Crimmin, M.R. Carbon-Carbon Bond Formation from Carbon Monoxide and Hydride: The Role of Metal Formyl Intermediates. Angew. Chem. Int. Ed. 2023, 62, e202219203. [Google Scholar] [CrossRef]
- Sheldon, R.A. Chemicals from Synthesis Gas; Springer: Dordrecht, The Netherlands, 1983; pp. 185–196. [Google Scholar]
- Bhasin, M.M.; Bartley, W.J.; Ellgen, P.C.; Wilson, T.P. Synthesis gas conversion over supported rhodium and rhodium-iron catalysts. J. Catal. 1978, 54, 120–128. [Google Scholar] [CrossRef]
- Ellgen, P.C.; Bhasin, M.M. Process for Producing Ethanol, Acetic Acid and/or Acetaldehyde, from Synthesis Gas. U.S. Patent 4,096,164, 20 June 1978. [Google Scholar]
- Bhasin, M.M.; O’Conner, G.L. Process for Producing Acetic Acid, Ethanol, and Acetaldehyde from Synthesis Gas. US Patent 4,246,186, 20 January 1981. [Google Scholar]
- Xu, B.Q.; Sachtler, W.M.H. Rh/NaY: A Selective Catalyst for Direct Synthesis of Acetic Acid from Syngas. J. Catal. 1998, 180, 194–206. [Google Scholar] [CrossRef]
- Knifton, J.F. Syngas reactions: IX. Acetic acid from synthesis gas. J. Catal. 1985, 96, 439–453. [Google Scholar] [CrossRef]
- Knifton, J.F.; Lin, J.J. Syngas reactions XII. The selective preparation of acetaldehyde, alkanols, esters and acetic acid from synthesis gas. Appl. Organometal. Chem. 1989, 3, 557–562. [Google Scholar] [CrossRef]
- Peng, J.-B.; Geng, H.-Q.; Wu, X.-F. The Chemistry of CO: Carbonylation. Chem 2019, 5, 526–552. [Google Scholar] [CrossRef]
- Yin, Z.; Xu, J.-X.; Wu, X.-F. No Making Without Breaking: Nitrogen-Centered Carbonylation Reactions. ACS Catal. 2020, 10, 6510–6531. [Google Scholar] [CrossRef]
- Qingli, Q.; Meng, C.; Jingjing, Z.; Junfeng, X.; Jinliang, S.; Guanying, Y.; Buxing, H. Synthesis of ethanol via a reaction of dimethyl ether with CO2 and H2. Green Chem. 2018, 20, 206–213. [Google Scholar] [CrossRef]
- Bodnar, T.; Coman, E.; Menard, K.; Cutler, A. Homogeneous reduction of ligated carbon dioxide and carbon monoxide to alkoxymethyl ligands. Inorg. Chem. 1982, 21, 1275–1277. [Google Scholar] [CrossRef]
- Cutler, A.R.; Hanna, P.K.; Vites, J.C. Carbon monoxide and carbon dioxide fixation: Relevant C1 and C2 ligand reactions emphasizing (η5-C5H5)Fe-containing complexes. Chem. Rev. 1988, 88, 1363–1403. [Google Scholar] [CrossRef]
- Santacesaria, E.; Di Serio, M.; Gelosa, D.; Carrà, S. Kinetics of methanol homologation: Part I. Behaviour of cobalt-phosphine-iodine catalysts. J. Mol. Catal. 1990, 58, 27–42. [Google Scholar] [CrossRef]
- Liu, Y.; Lotero, E.; Goodwin, J.G., Jr. A comparison of the esterification of acetic acid with methanol using heterogeneous versus homogeneous acid catalysis. J. Catal. 2006, 242, 278–286. [Google Scholar] [CrossRef]
- Mekala, M.; Goli, V.R. Kinetics of esterification of methanol and acetic acid with mineral homogeneous acid catalyst. Chin. J. Chem. Eng. 2015, 23, 100–105. [Google Scholar] [CrossRef]
- Cui, M.; Qian, Q.; Zhang, J.; Wang, Y.; Asare Bediako, B.B.; Liu, H.; Han, B. Liquid fuel synthesis via carbon dioxde hydrogenation by coupling homogeneous and heterogeneous catalysis. Chem 2021, 7, 726–737. [Google Scholar] [CrossRef]
- Nijs, H.H.; Jacobs, P.A. New evidence for the mechanism of the Fischer-Tropsch synthesis of hydrocarbons. J. Catal. 1980, 66, 401–411. [Google Scholar] [CrossRef]
- Madon, R.J. On the growth of hydrocarbon chains in the Fischer-Tropsch synthesis. J. Catal. 1979, 57, 183–186. [Google Scholar] [CrossRef]
- Vanhove, D.; Makambo, L.; Blanchard, M. Selective catalytic synthesis of linear paraffins from CO and H2 over cobalt supported catalysts. J. Chem. Soc. Chem. Commun. 1979, 605–606. [Google Scholar] [CrossRef]
- Nijs, H.H.; Jacobs, P.A.; Uytterhoeven, J.B. Selective Fischer-Tropsch synthesis of hydrocarbons: Particle size effect of ruthenium metal in Faujasite-type zeolites. J. Chem. Soc. Chem. Commun. 1979, 1095–1096. [Google Scholar] [CrossRef]
- Fraenkel, D.; Gates, B. Shape-selective Fischer-Tropsch synthesis catalyzed by zeolite-entrapped cobalt clusters. J. Am. Chem. Soc. 1980, 102, 2478–2480. [Google Scholar] [CrossRef]
- Ballivet-Tkatchenko, D.; Tkatchenko, I. Small particles in zeolites as selective catalysts for the hydrocondensation of carbon monoxide. J. Mol. Catal. 1981, 13, 1–10. [Google Scholar] [CrossRef]
- Ungar, R.K.; Baird, M.C. Co(CO)3NO encapsulated in zeolite NaY; a novel shape-selective Fischer-Tropsch catalyst. J. Chem. Soc. Chem. Commun. 1986, 643–645. [Google Scholar] [CrossRef]
- Kaya, C.; Karakas, D.; Ustun, E. A new approach to predicting the carbonyl stretching frequencies of Co2(CO)8 with D3d symmetry. Indian J. Chem. A 2007, 46, 1388–1392. [Google Scholar]
- Sayan, Ş.; Kantcheva, M.; Suzer, S.; Uner, D.O. FTIR characterization of Ru/SiO2 catalyst for ammonia synthesis. J. Mol. Struct. 1999, 480–481, 241–245. [Google Scholar] [CrossRef]
- Tannenbaum, R.; Bor, G. Infrared spectroscopic studies of the reactions between Co2(CO)8, H2 and D2. Inorg. Chim. Acta 1992, 201, 87–93. [Google Scholar] [CrossRef]
- Henrici-Olivé, G.; Olivé, S. The Chemistry of the Catalyzed Hydrogenation of Carbon Monoxide; Springer: Berlin/Heidelberg, Germany, 1984; pp. 143–196. [Google Scholar]
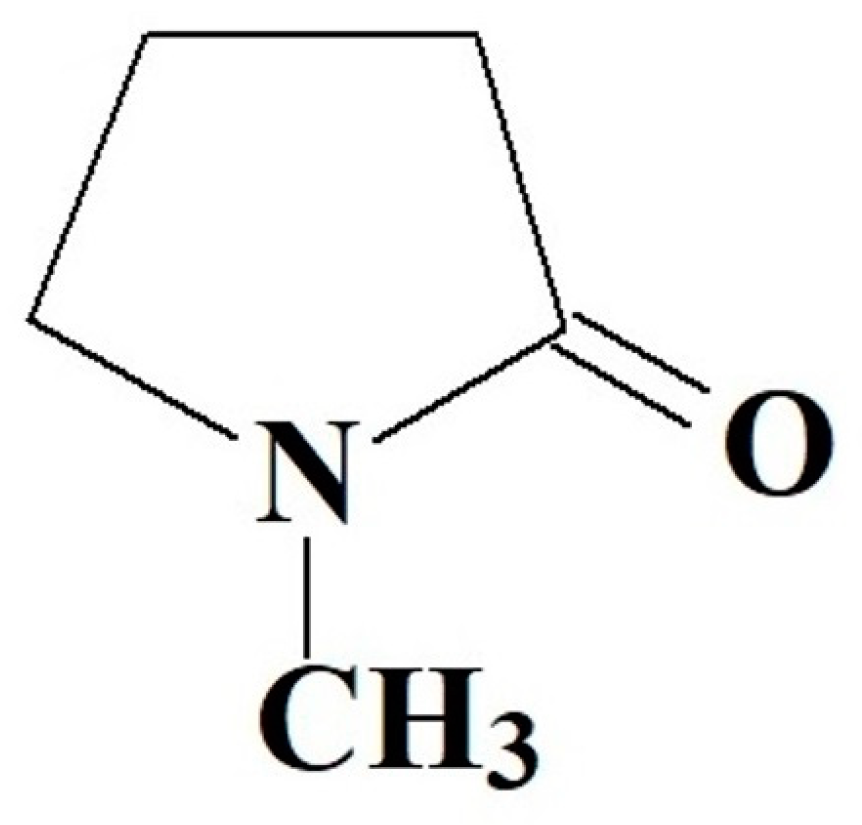


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
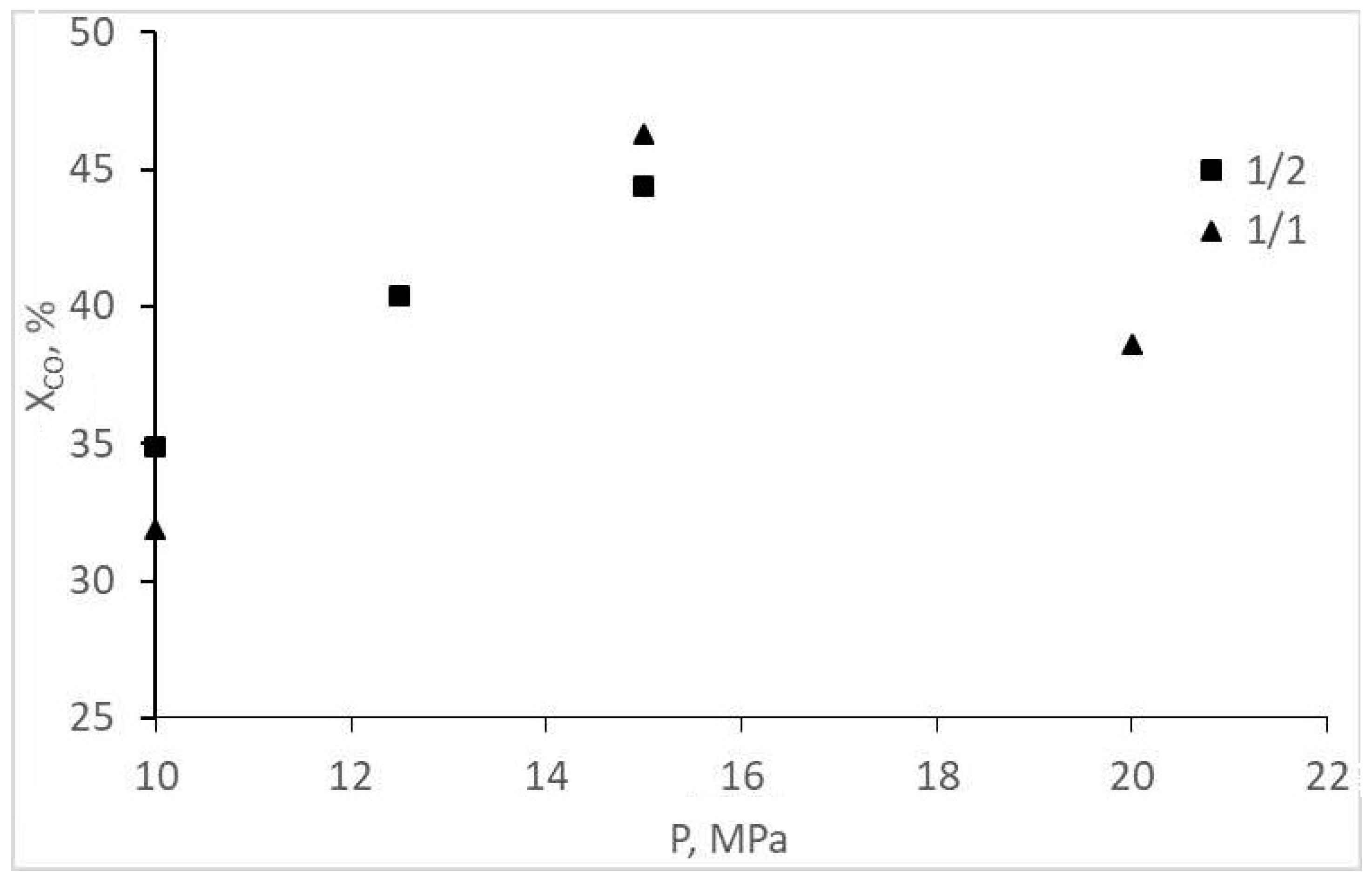
| Initial CO:H2 | 1/2 | 1/2 | 1/2 | 1/1 | 1/1 | 1/1 |
|---|---|---|---|---|---|---|
| Initial pressure, MPa | 10.0 | 12.5 | 15.0 | 10.0 | 15.0 | 20.0 |
| CO conversion, % | 34.9 | 40.4 | 44.4 | 31.9 | 46.3 | 38.6 |
| TOF, s−1 (Equation (10)) | 0.005 | 0.008 | 0.010 | 0.007 | 0.002 | 0.002 |
| Selectivity, % | ||||||
| CO2 | 12.5 | 17.2 | 7.3 | 20.5 | 12.5 | 10.1 |
| CH4 | 2.5 | 2.3 | 1.1 | 1.2 | 0.74 | 0.31 |
| Ethane | 0.031 | 0.32 | 0.016 | 0.0 | 0.026 | 0.0 |
| Propane | 0.072 | 0.075 | 0.055 | 0.0 | 0.0 | 0.0 |
| Butanes | 0.042 | 0.045 | 0.035 | 0.0 | 0.0 | 0.0 |
| Hexanes | 5.2 | 0.18 | 3.8 | 1.9 | 0.0 | 0.0 |
| Heptanes | 13.5 | 2.7 | 16.8 | 6.7 | 0.0 | 0.4 |
| Octanes | 14.3 | 3.5 | 6.2 | 1.9 | 0.0 | 0.0 |
| Nonanes | 14.2 | 6.5 | 6.6 | 1.8 | 0.0 | 0.0 |
| Decanes | 15.9 | 9.7 | 6.5 | 1.8 | 0.0 | 0.0 |
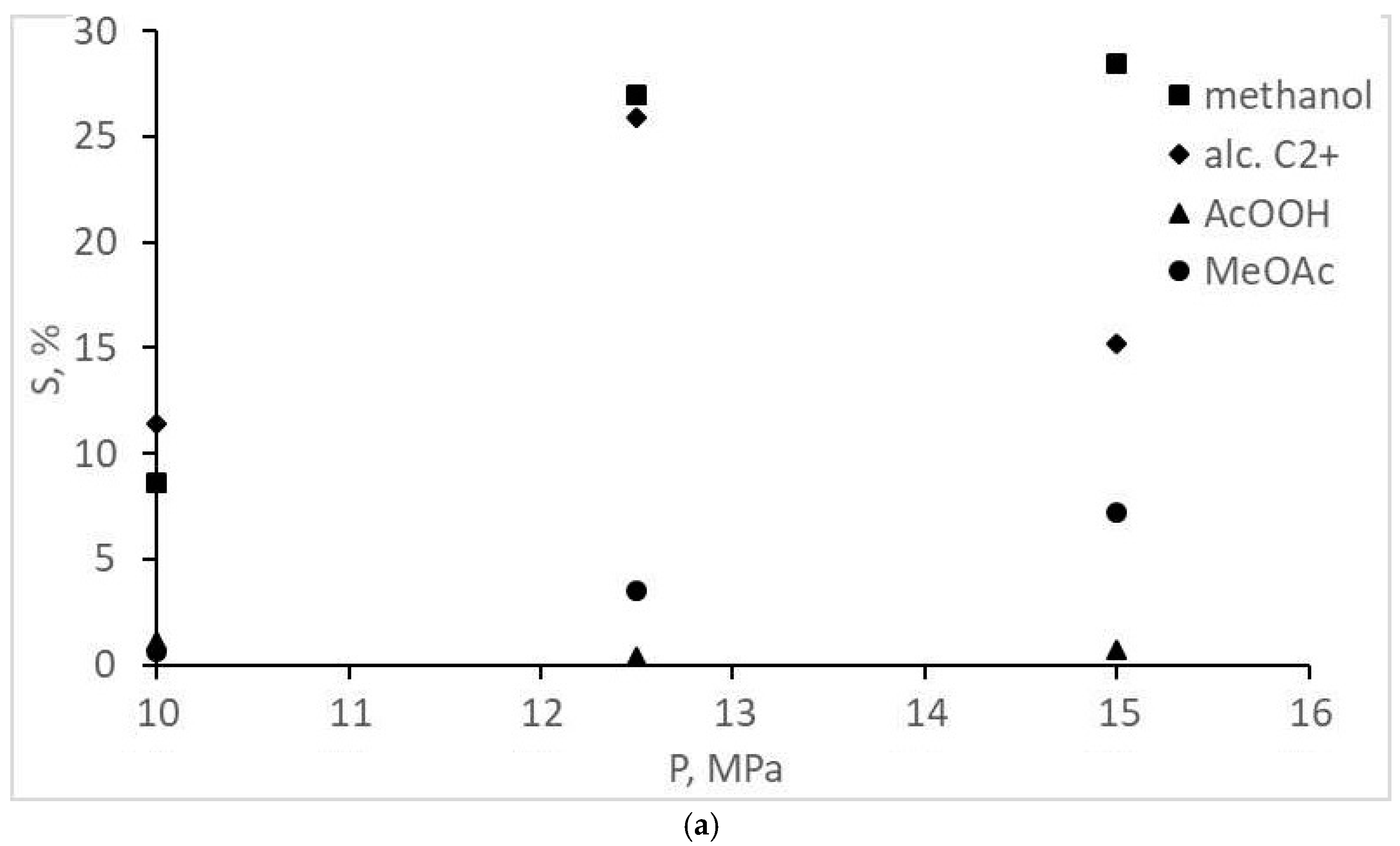
| Methanol | 8.6 | 27.0 | 28.5 | 43.7 | 42.7 | 42.2 |
| Ethanol | 0.0 | 0.88 | 0.76 | 0.93 | 11.4 | 22.7 |
| Propanol | 0.0 | 3.1 | 4.2 | 2.5 | 3.2 | 0.69 |
| Butanol+iso-propylmethylketone | 3.7 | 6.4 | 5.1 | 8.1 | 7.9 | 0.0 |
| Pentanol | 3.3 | 7.2 | 2.8 | 3.2 | 0.0 | 0.0 |
| Hexanol | 4.4 | 8.3 | 2.3 | 0.0 | 0.0 | 0.0 |
| Acetic acid | 1.1 | 0.43 | 0.71 | 2.5 | 6.6 | 3.6 |
| Methyl acetate | 0.64 | 3.5 | 7.2 | 3.3 | 12.0 | 17.9 |
| Ethyl acetate | 0.0 | 0.0 | 0.0 | 0.0 | 2.9 | 2.1 |
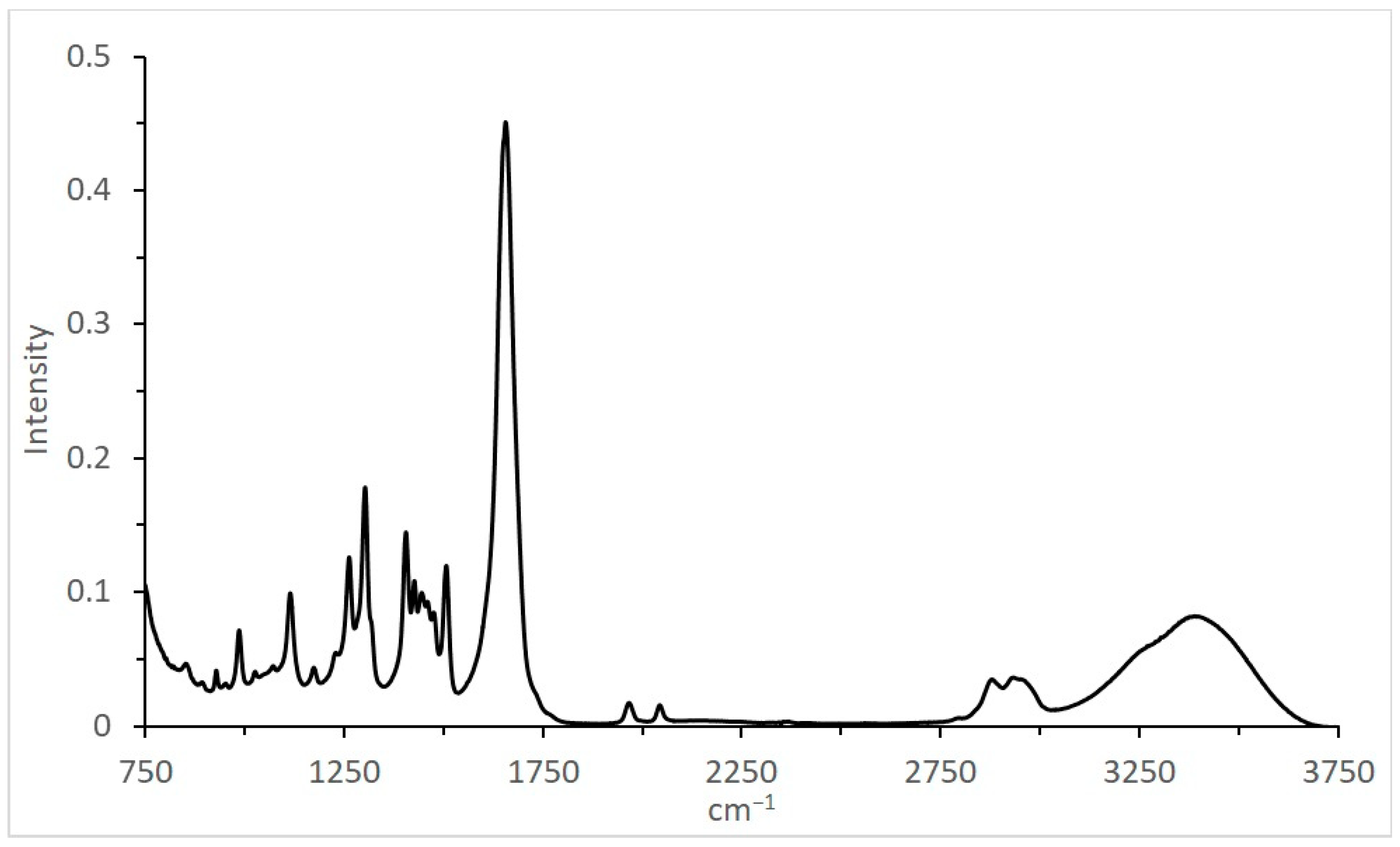
| cm−1 | Intensity |
|---|---|
| 3399 | 0.0818 |
| 2951 | 0.0351 |
| 2879 | 0.0348 |
| 2044 | 0.0156 |
| 1967 | 0.0173 |
| 1656 | 0.451 |
| 1506 | 0.120 |
| 1475 | 0.0848 |
| 1460 | 0.0929 |
| 1445 | 0.0995 |
| 1427 | 0.108 |
| 1405 | 0.144 |
| 1302 | 0.178 |
| 1262 | 0.126 |
| 1229 | 0.0548 |
| 1173 | 0.0436 |
| 1114 | 0.0992 |
| 1071 | 0.0449 |
| 1026 | 0.0406 |
| 985 | 0.0715 |
| 928 | 0.0415 |
| 851 | 0.0467 |
| 802 | 0.0507 |
| Catalyst | This Study | Ru3(CO)12/CoI2/ Bu4PBr [18] * | Ru3(CO)12/ Co2(CO), Bu4PBr [19] * | |||
|---|---|---|---|---|---|---|
| T, °C | 200 | 200 | 200 | 200 | 220 | 220 |
| CO:H2 | 1/2 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 |
| Pressure, MПa | 15.0 | 15.0 | 48.0 | 48.0 | 55.0 | 55.0 |
| Co/Ru | 1.25 | 1.25 | 1 | 1.5 | 1 | 2 |
| Selectivity, % * | ||||||
| Methanol | 28.5 | 42.7 | 2 | ~0 | 25 | 8 |
| Ethanol | 0.76 | 11.4 | ~0 | ~0 | 21 | 8 |
| Acetic acid | 0.71 | 6.6 | 64 | 85 | 26 | 56 |
| Methyl acetate | 7.2 | 12.0 | 3 | 15 | 10 | 10 |
| Ethyl acetate | 0.0 | 2.9 | 10 | ~0 | 13 | 16 |
| CH4 | 1.1 | 0.74 | 20 | ~0 | ~0 | ~0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maximov, A.L.; Kulikova, M.V.; Kuz’min, A.E.; Ivantsov, M.I. Direct Homogeneous Synthesis of Compounds with Two O Atoms and Long-Chain Hydrocarbons from CO and H2: Co–Ru/N-methylpyrrolidone Catalyst. Molecules 2023, 28, 6341. https://doi.org/10.3390/molecules28176341
Maximov AL, Kulikova MV, Kuz’min AE, Ivantsov MI. Direct Homogeneous Synthesis of Compounds with Two O Atoms and Long-Chain Hydrocarbons from CO and H2: Co–Ru/N-methylpyrrolidone Catalyst. Molecules. 2023; 28(17):6341. https://doi.org/10.3390/molecules28176341
Chicago/Turabian StyleMaximov, Anton Lvovich, Mayya V. Kulikova, Alexey E. Kuz’min, and Mikhail I. Ivantsov. 2023. "Direct Homogeneous Synthesis of Compounds with Two O Atoms and Long-Chain Hydrocarbons from CO and H2: Co–Ru/N-methylpyrrolidone Catalyst" Molecules 28, no. 17: 6341. https://doi.org/10.3390/molecules28176341
APA StyleMaximov, A. L., Kulikova, M. V., Kuz’min, A. E., & Ivantsov, M. I. (2023). Direct Homogeneous Synthesis of Compounds with Two O Atoms and Long-Chain Hydrocarbons from CO and H2: Co–Ru/N-methylpyrrolidone Catalyst. Molecules, 28(17), 6341. https://doi.org/10.3390/molecules28176341