
Isolation and Anticancer Progression Evaluation of the Chemical Constituents from Bridelia balansae Tutcher

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
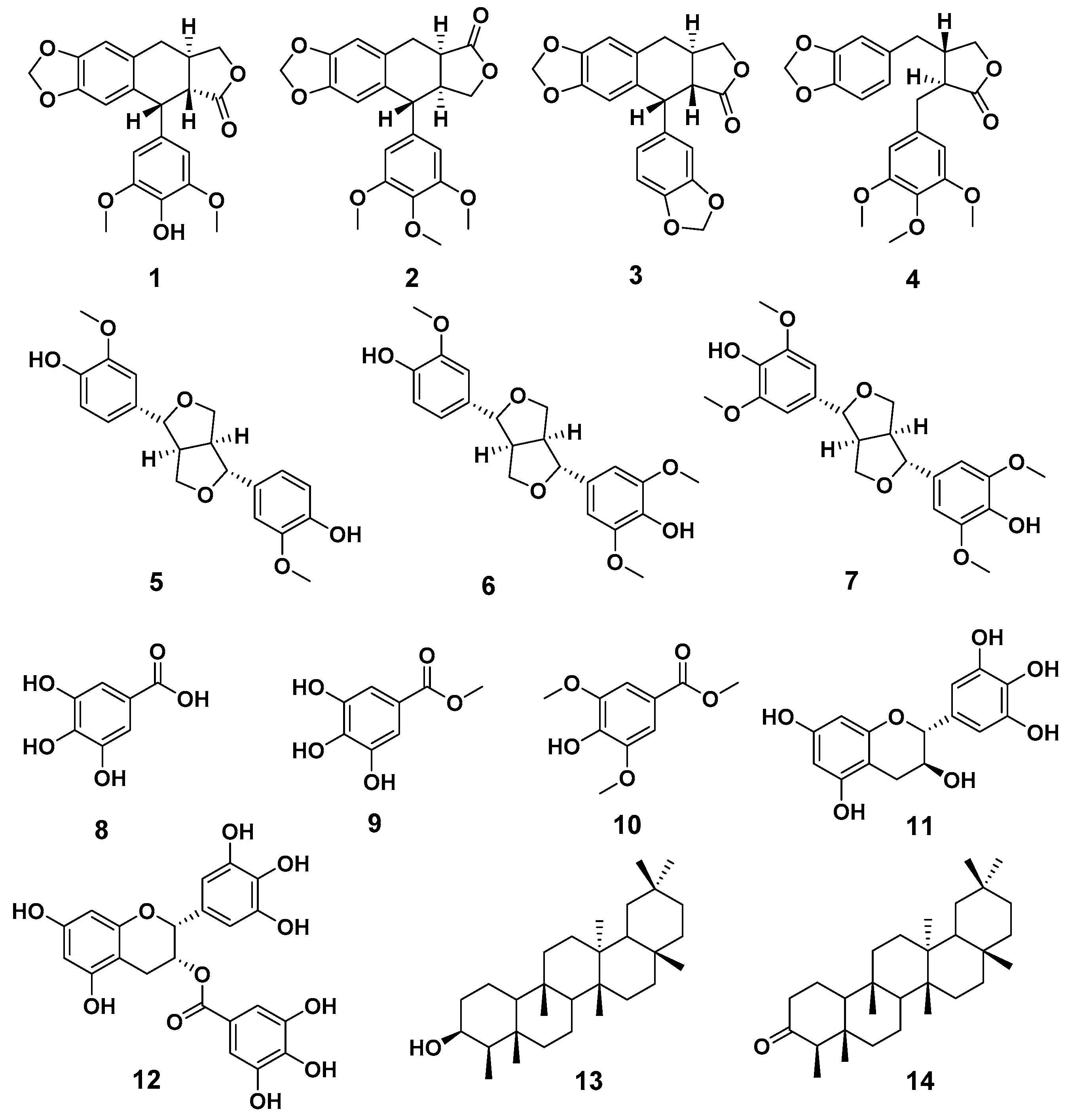
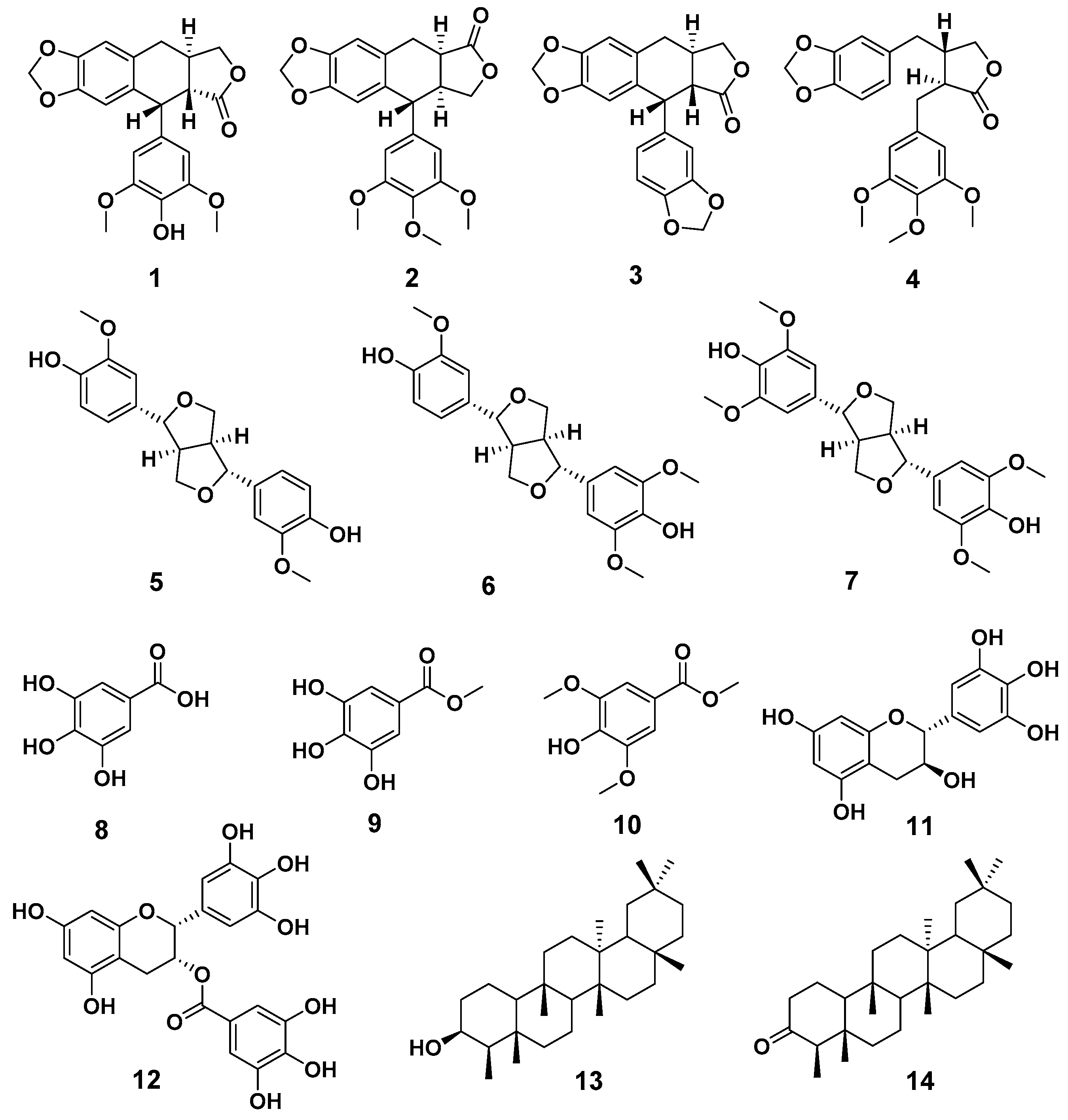
2.1. Compound Isolation and Structure Identification
2.2. Cytotoxic Effect of Isolated Compounds against HCT116 Cancer Cells
2.3. Compound 1 Induced Apoptosis in HCT116 Cancer Cells
2.4. Compound 1 Reduced Cellular Mobilization of HCT116 Cancer Cells
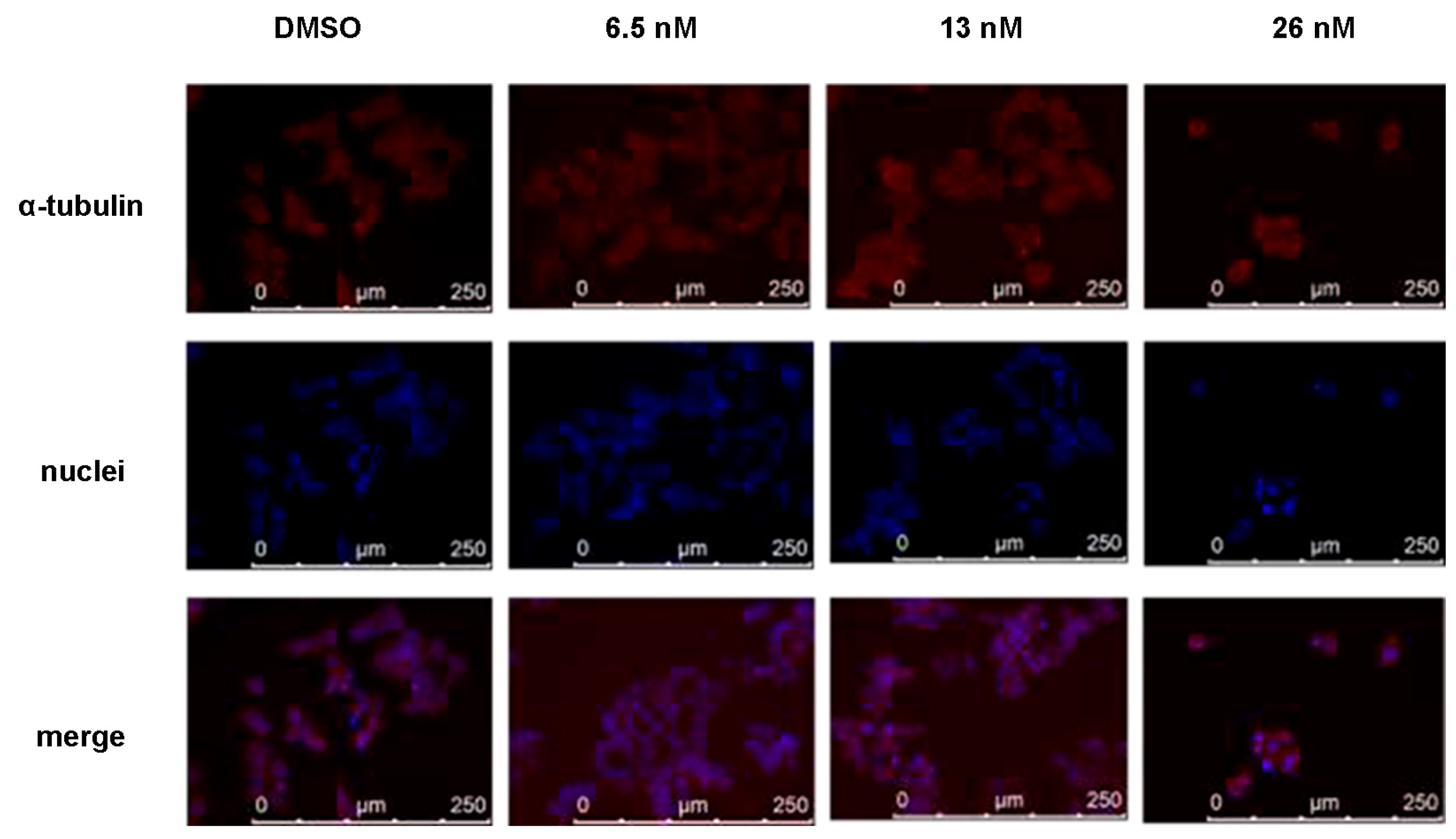
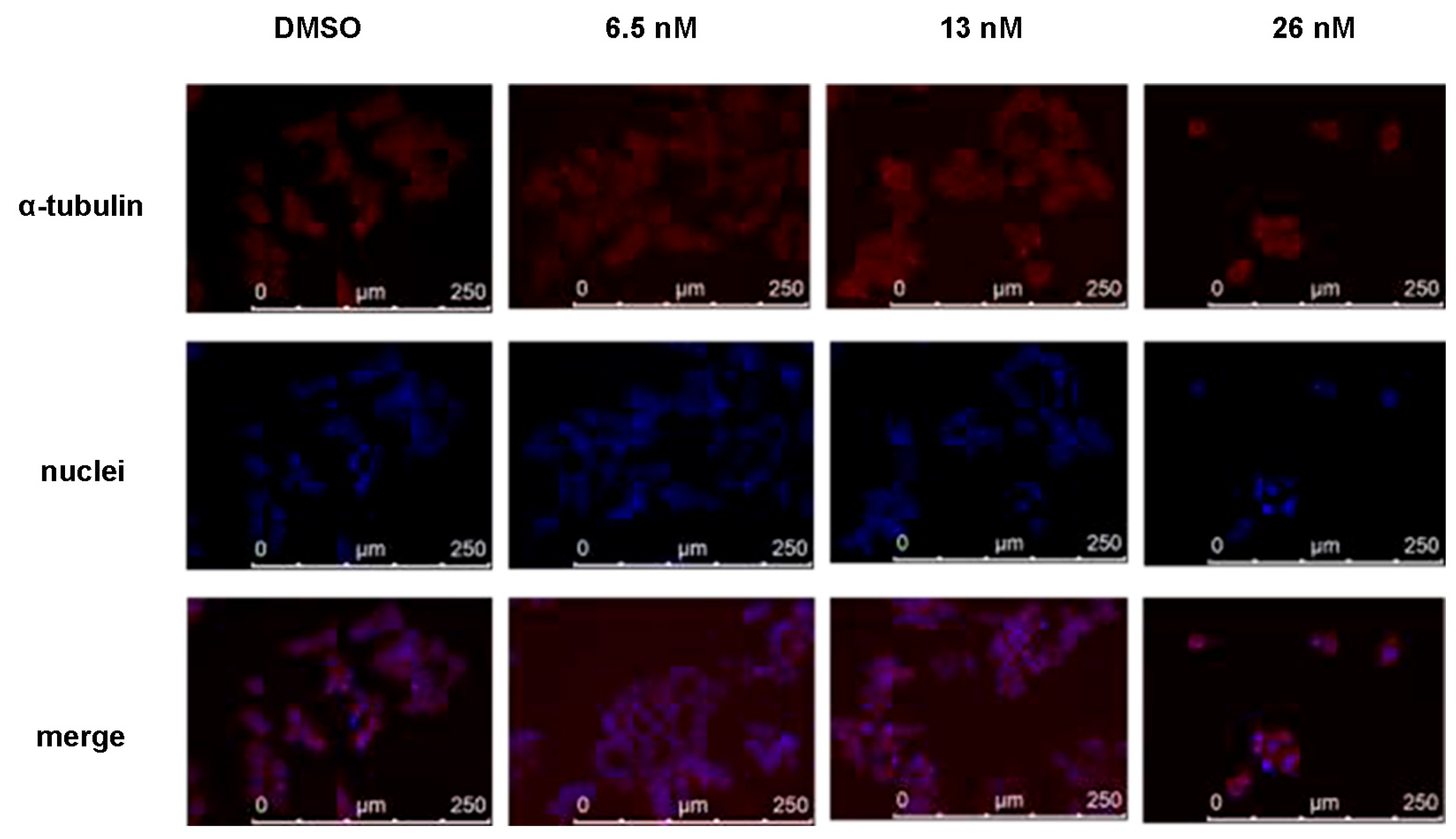
2.5. Compound 1 Disrupted Microtubules of HCT116 Cancer Cells
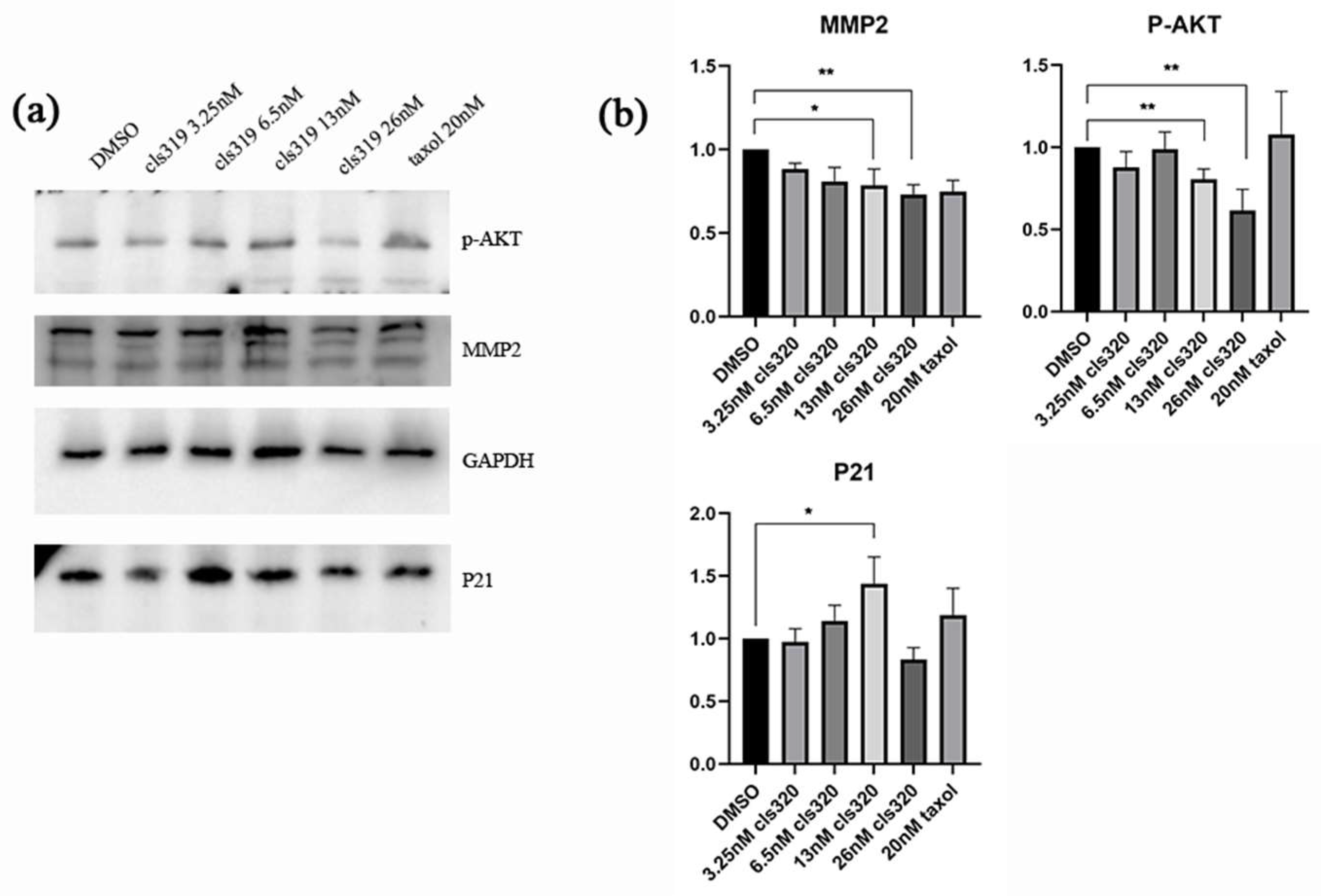
2.6. Effects of Compound 1 on Apoptosis and Migration Regulatory Proteins of HCT116 Cancer Cells
3. Discussion
4. Materials and Methods
4.1. General
4.2. Plant Material
4.3. Compound Isolation and Structure Identification
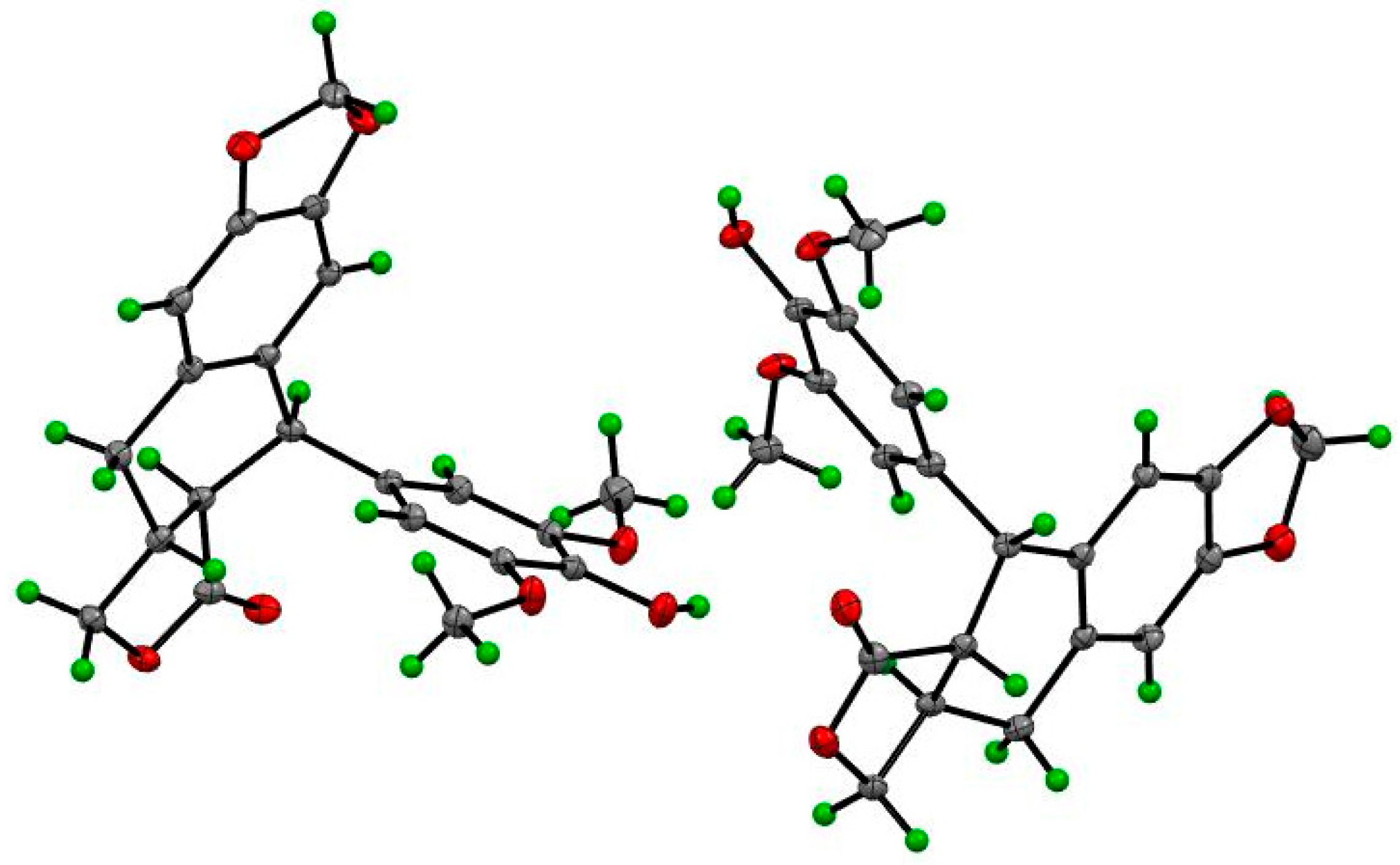
4.4. X-ray Crystallographic Data of 1
4.5. Cell Proliferation Assay
4.6. Flow Cytometry
4.7. Wound Healing Assay
4.8. Immunofluoresent Staining
4.9. Western Blot Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Dias, D.A.; Urban, S.; Roessner, U. A historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.D.; Chaudhari, M.A.; Sapkale, P.V.; Chaudhari, R.B. A recent review on anticancer herbal drugs. J. Drug Discov. Ther. 2013, 1, 77–84. [Google Scholar]
- Waltenberger, B.; Mocan, A.; Šmejkal, K.; Heiss, E.H.; Atanasov, A.G. Natural Products to Counteract the Epidemic of Cardiovascular and Metabolic Disorders. Molecules 2016, 21, 807. [Google Scholar] [CrossRef]
- Tintore, M.; Vidal-Jordana, A.; Sastre-Garriga, J. Treatment of multiple sclerosis—Success from bench to bedside. Nat. Rev. Neurol. 2019, 15, 53–58. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Chabner, B.A. NCI-60 cell line screening: A radical departure in its time. J. Natl. Cancer Inst. 2016, 108, djv388. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Ngueyem, T.A.; Brusotti, G.; Caccialanza, G.; Finzi, P.V. The genus Bridelia: A phytochemical and ethnopharmacological review. J. Ethnopharmacol. 2009, 124, 339–349. [Google Scholar] [CrossRef]
- Karou, S.D.; Tchacondo, T.; Tchibozo, M.A.D.; Abdoul-Rahaman, S.; Anani, K.; Koudouvo, K.; Batawila, K.; Agbonon, A.; Simpore, J.; de Souza, C. Ethnobotanical study of medicinal plants used in the management of diabetes mellitus and hypertension in the Central Region of Togo. Pharm. Biol. 2011, 49, 1286–1297. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, G.N.; Owusu, F.W.A.; Archer, M.A.; Kyene, M.O.; Kumadoh, D.; Ayertey, F.; Mintah, S.O.; Atta-Adjei Junior, P.; Appiah, A.A. Bridelia ferruginea Benth; An ethnomedicinal, phytochemical, pharmacological and toxicological review. Heliyon 2022, 8, e10366. [Google Scholar] [CrossRef]
- Maroyi, A. Utilization of Bridelia mollis as herbal medicine, nutraceutical and functional food in southern Africa: A review. Trop. J. Pharm. Res. 2019, 18, 203–209. [Google Scholar] [CrossRef]
- Murthy, H.N.; Dalawai, D.; Mamatha, U.; Angadi, N.B.; Dewir, Y.H.; Al-Suhaibani, N.A.; El-Hendawy, S.; Al-Ali, A.M. Bioactive constituents and nutritional composition of Bridelia stipularis L. Blume fruits. Int. J. Food Prop. 2021, 24, 796–805. [Google Scholar] [CrossRef]
- Pettit, G.R.; Searcy, J.D.; Tan, R.; Cragg, G.M.; Melody, N.; Knight, J.C.; Chapuis, J.C. Antineoplastic Agents. 585. Isolation of Bridelia ferruginea Anticancer Podophyllotoxins and Synthesis of 4-Aza-podophyllotoxin Structural Modifications. J. Nat. Prod. 2016, 79, 507–518. [Google Scholar]
- Ochwang’i, D.O.; Kimwele, C.N.; Oduma, J.A.; Gathumbi, P.K.; Kiama, S.G.; Efferth, T. Cytotoxic activity of medicinal plants of the Kakamega County (Kenya) against drug-sensitive and multidrug-resistant cancer cells. J. Ethnopharmacol. 2018, 215, 233–240. [Google Scholar] [CrossRef]
- Huong, D.T.; Kamperdick, C.; Van Sung, T. Homogentisic acid derivatives from Miliusa balansae. J. Nat. Prod. 2004, 67, 445–447. [Google Scholar] [CrossRef]
- Jackson, D.E.; Dewick, P.M. Aryltetralin lignans from Podophyllum hexandrum and Podophyllum peltatum. Phytochemistry 1984, 23, 1147–1152. [Google Scholar] [CrossRef]
- Xu, H.; Wang, J.; Sun, H.; Lv, M.; Tian, X.; Yao, X.; Zhang, X. Semisynthesis and quantitative structure- activity relationship (QSAR) study of novel aromatic esters of 4′-demethyl-4-deoxypodophyllotoxin as insecticidal agents. J. Agric. Food Chem. 2009, 57, 7919–7923. [Google Scholar] [CrossRef]
- Moujir, L.S.; Seca, A.M.L.; Silva, A.M.S.; Barreto, M.C. Cytotoxic Activity of Diterpenes and Extracts of Juniperus brevifolia. Planta Med. 2008, 74, 751–753. [Google Scholar] [CrossRef]
- Wada, K.; Munakata, K. (−) Parabenzlactone, a new piperolignanolide isolated from Parabenzoin trilobum nakai. Tetrahedron Lett. 1970, 11, 2017–2019. [Google Scholar] [CrossRef]
- Enders, D.; Lausberg, V.; Del Signore, G.; Berner, O.M. A general approach to the asymmetric synthesis of lignans:(−)-methyl piperitol,(−)-sesamin,(−)-aschantin,(+)-yatein,(+)-dihydroclusin,(+)-burseran, and (−)-isostegane. Synthesis 2002, 2002, 515–522. [Google Scholar] [CrossRef]
- Deyama, T.I.T.; Kitagawa, S.; Nishibe, S. The Constituents of Eucommia ulmoicles OLIV. V. Isolation of Dihydroxydehydrodiconiferyl Alcohol Isomers and Phenolic Compounds (Organic, Chemical). Chem. Pharm. Bull. 1987, 35, 1785–1789. [Google Scholar]
- Haajanen, K.; Botting, N.P. Synthesis of multiply 13C-labeled furofuran lignans using 13C-labeled cinnamyl alcohols as building blocks. Steroids 2006, 71, 231. [Google Scholar] [CrossRef] [PubMed]
- Das, B.V.B.; Kashinatham, A. Studies on phytochemicals.Part XXV. (+)-Syringaresinol from Parthenium hysterophorus. Fitoterapia 1999, 70, 2. [Google Scholar]
- Gottlieb, H.E.; Kumar, S.; Sahai, M.; Ray, A.B. Ethyl brevifolin carboxylate from Flueggea microcarpa. Phytochemistry 1991, 30, 2435–2438. [Google Scholar] [CrossRef]
- Mostafa, M.; Nahar, N.; Mosihuzzaman, M.; Sokeng, S.D.; Fatima, N.; Rahman, A.U.; Choudhary, M.I. Phosphodiesterase-I inhibitor quinovic acid glycosides from Bridelia ndellensis. Nat. Prod. Res. 2006, 20, 686–692. [Google Scholar] [CrossRef]
- Russell, K.M.; Molan, P.C.; Wilkins, A.L.; Holland, P.T. Identification of some antibacterial constituents of New Zealand manuka honey. J. Agric. Food Chem. 1990, 38, 4. [Google Scholar] [CrossRef]
- Foo, L.; Newman, R.; Waghorn, G.; McNabb, W.; Ulyatt, M. Proanthocyanidins from Lotus corniculatus. Phytochemistry 1996, 41, 617–624. [Google Scholar] [CrossRef]
- Yang, L.-L.; Chang, C.C.; Chen, L.G.; Wang, C.C. Antitumor principle constituents of Myrica rubra var. acuminata. J. Agric. Food Chem. 2003, 51, 2974–2979. [Google Scholar] [CrossRef]
- Li, L.; Huang, X.; Sattler, I.; Fu, H.; Grabley, S.; Lin, W. Structure elucidation of a new friedelane triterpene from the mangrove plant Hibiscus tiliaceus. Magn. Reson. Chem. 2006, 44, 5. [Google Scholar] [CrossRef]
- Charlton, J.L.; Ploude, G.L.; Koh, K.; Secco, A.S. Asemmetric synthesis of podophyllotoxin analogs. Can. J. Chem. 1990, 68, 2022–2027. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Su, M.; Zhou, Y.; Chen, Y.; Li, J.; Lu, W. Design, synthesis and biological evaluation of 4′-demethyl-4-deoxypodophyllotoxin derivatives as novel tubulin and histone deacetylase dual inhibitors. RSC Adv. 2014, 4, 40444–40448. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Shah, M.A. Targeting the cell cycle: A new approach to cancer therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef]
- Schilstra, M.J.; Martin, S.R.; Bayley, P.M. The effect of Podophyllotoxin on Microtubule Dynamics. J. Biol. Chem. 1989, 264, 8827–8834. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.N.; Ju, J.A.; Ory, E.C.; Pratt, S.J.P.; Lee, R.M.; Mathias, T.J.; Chang, K.T.; Lee, C.J.; Goloubeva, O.G.; Bailey, P.C.; et al. Microtubule disruption reduces metastasis more effectively than primary tumor growth. Breast Cancer Res. 2022, 24, 13. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-H.; Robertson, K.L.; Don, S.S.L.; Taylor, S.R.; Farkas, M.E. Chemical modulation of circadian rhythms and assessment of cellular behavior via indirubin and derivatives. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2020; Volume 639, pp. 115–140. [Google Scholar]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibition Rate | Compound | Inhibition Rate |
|---|---|---|---|
| 1 | 96.1% | 9 | 76.1% |
| 3 | 11.7% | 10 | 3.4% |
| 8 | 100% | 11 | 24.5% |
| Compound | IC50 |
|---|---|
| 1 | 0.02 ± 0.003 |
| 8 | 20 ± 0.006 |
| 9 | 16.3 ± 0.003 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Xie, W.-J.; Du, Y.-X.; Xia, Y.-X.; Liu, K.-L.; Ku, C.F.; Ou, Z.; Wang, M.-Z.; Zhang, H.-J. Isolation and Anticancer Progression Evaluation of the Chemical Constituents from Bridelia balansae Tutcher. Molecules 2023, 28, 6165. https://doi.org/10.3390/molecules28166165
Zhao L, Xie W-J, Du Y-X, Xia Y-X, Liu K-L, Ku CF, Ou Z, Wang M-Z, Zhang H-J. Isolation and Anticancer Progression Evaluation of the Chemical Constituents from Bridelia balansae Tutcher. Molecules. 2023; 28(16):6165. https://doi.org/10.3390/molecules28166165
Chicago/Turabian StyleZhao, Lihan, Wen-Jian Xie, Yin-Xiao Du, Yi-Xuan Xia, Kang-Lun Liu, Chuen Fai Ku, Zihao Ou, Ming-Zhong Wang, and Hong-Jie Zhang. 2023. "Isolation and Anticancer Progression Evaluation of the Chemical Constituents from Bridelia balansae Tutcher" Molecules 28, no. 16: 6165. https://doi.org/10.3390/molecules28166165
APA StyleZhao, L., Xie, W.-J., Du, Y.-X., Xia, Y.-X., Liu, K.-L., Ku, C. F., Ou, Z., Wang, M.-Z., & Zhang, H.-J. (2023). Isolation and Anticancer Progression Evaluation of the Chemical Constituents from Bridelia balansae Tutcher. Molecules, 28(16), 6165. https://doi.org/10.3390/molecules28166165