
Sorption of Organic Contaminants by Stable Organic Matter Fraction in Soil



Abstract
1. Introduction
2. Results and Discussion
2.1. Spectroscopic Characterization of the S-SOM Fractions
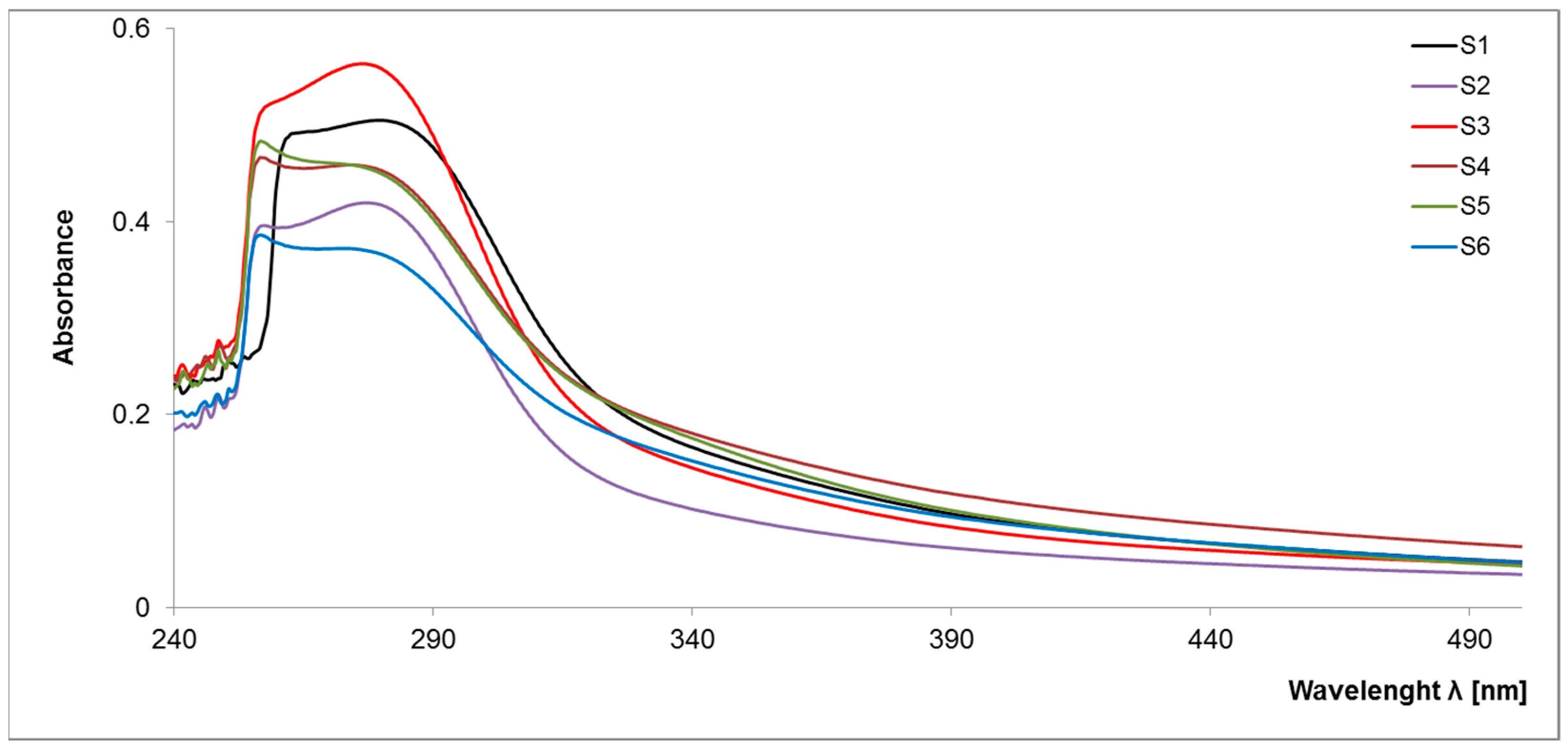
2.1.1. UV–VIS Spectroscopy
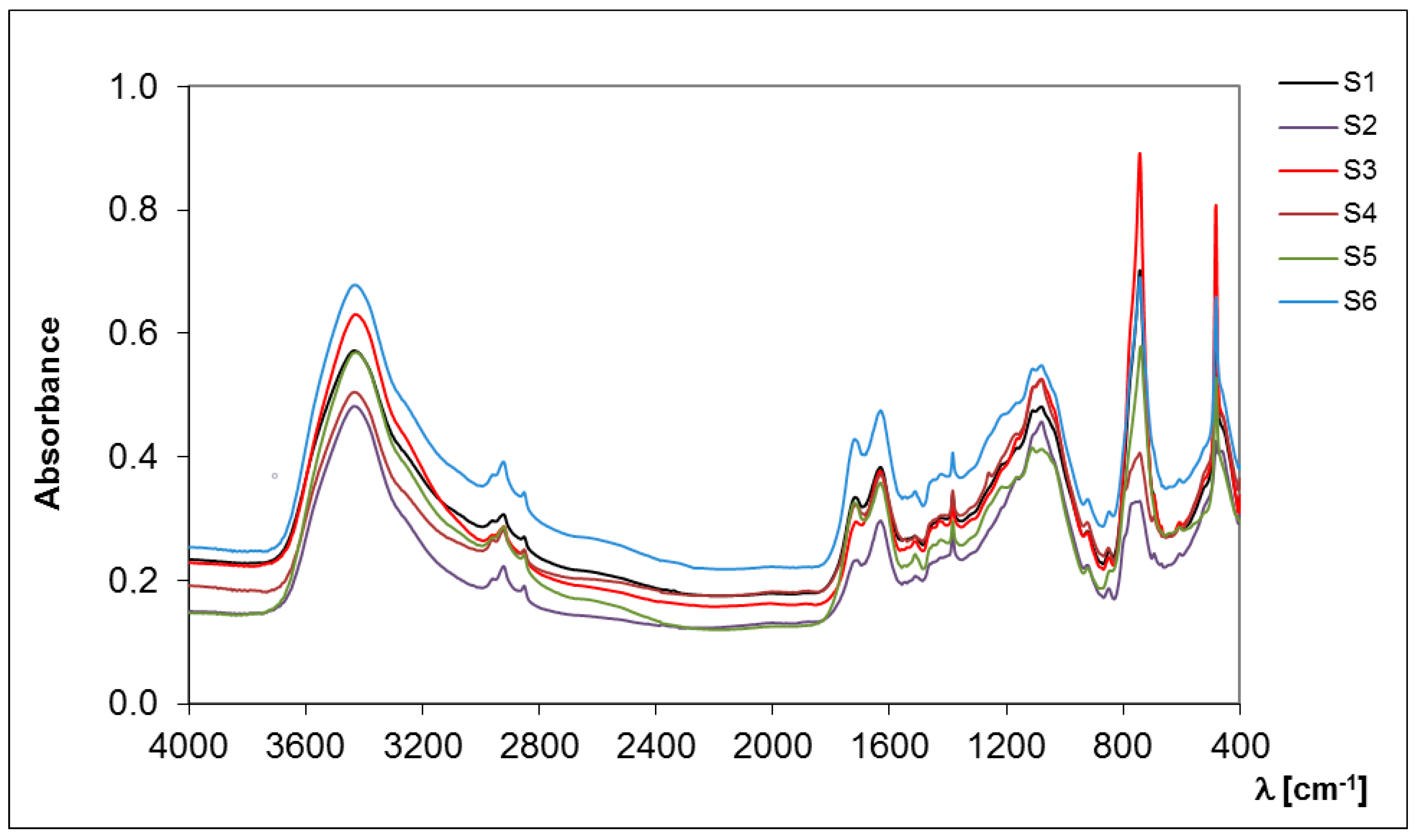
2.1.2. FT-IR Spectroscopy
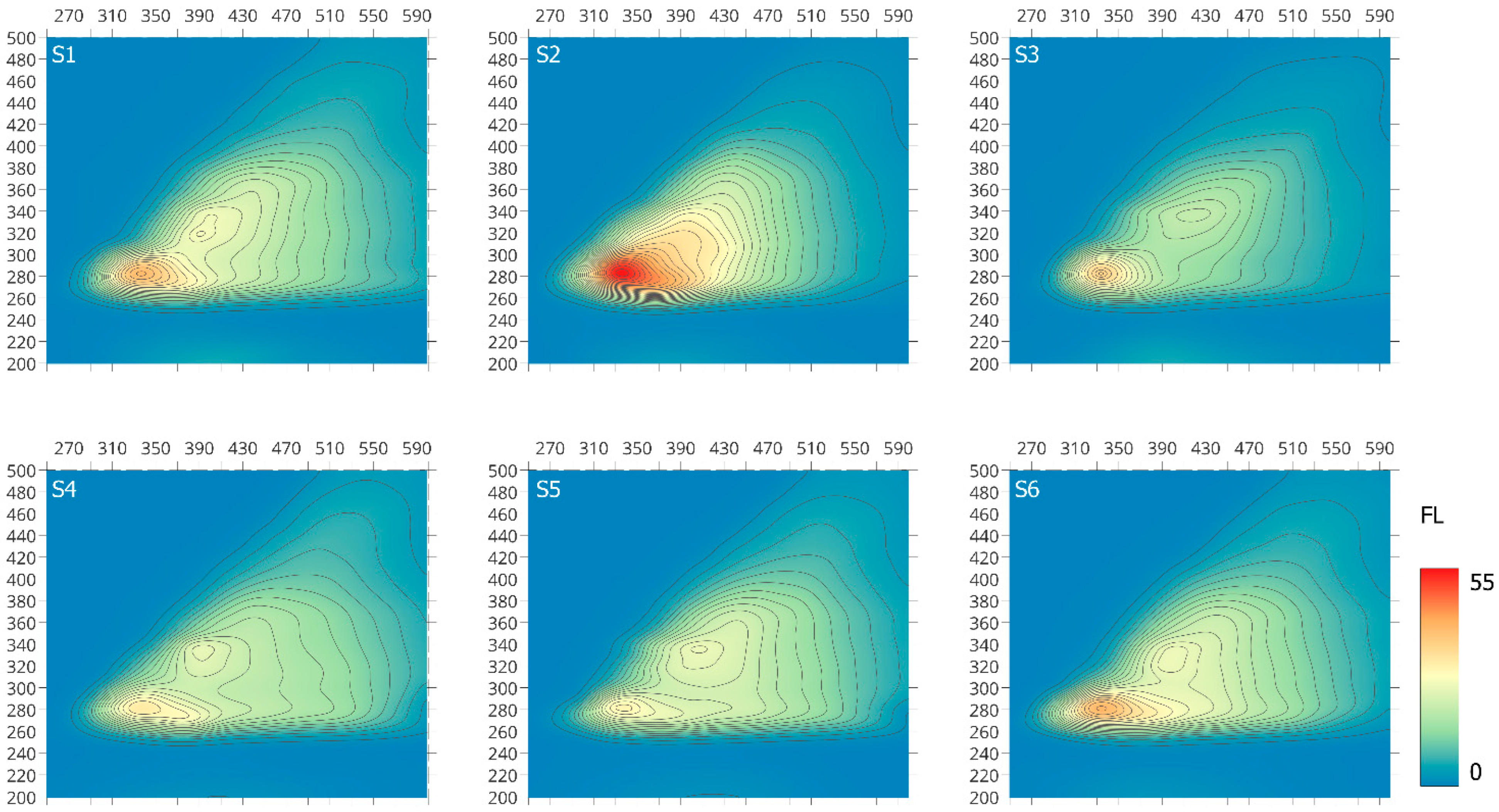
2.1.3. EEM Spectroscopy
2.2. Sorption of OCs to S-SOM
2.3. Influence of S-SOM Molecular Properties on OCs Sorption Affinity
3. Materials and Methods
3.1. Soil Sampling
3.2. Chemical and Physical Analysis
3.2.1. Soil Physicochemical Analysis
3.2.2. Extraction Procedure of Stable Organic Matter
3.2.3. Characteristics of the Isolated Stable Organic Matter Fraction
The UV–VIS Spectroscopy
The FT-IR Spectroscopy
The EEM Spectroscopy
3.3. Experimental Design
3.3.1. Tested Compounds
3.3.2. Method for OCs Determination
3.3.3. Sorption Experiment
3.4. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zhang, D.; Hou, L.; Zhu, D.; Chen, W. Synergistic role of different soil components in slow sorption kinetics of polar organic contaminants. Environ. Pollut. 2014, 184, 123–130. [Google Scholar] [CrossRef]
- Ren, X.; Zeng, G.; Tang, L.; Wang, J.; Wan, J.; Liu, Y.; Yu, J.; Yi, H.; Ye, S.; Deng, R. Sorption, transport and biodegradation—An insight into bioavailability of persistent organic pollutants in soil. Sci. Total Environ. 2018, 610–611, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Owino Gudda, F.; Wu, C.; Ling, W.; El-Ramady, H.; Mosa, A.; Wang, J. Contributions of partition and adsorption to polycyclic aromatic hydrocarbons sorption by fractionated soil at different particle sizes. Chemosphere 2022, 301, 134715. [Google Scholar] [CrossRef] [PubMed]
- Neira-Albornoz, A.; Fuentesc, E.; Cáceres-Jensen, L. Connecting the evidence about organic pollutant sorption on soils with environmental regulation and decision-making: A scoping review. Chemosphere 2022, 308, 136164. [Google Scholar] [CrossRef] [PubMed]
- COM(2021)699 Final. Communication from the Commission to the European Parliament, the European Council, the Council, the European Economic and Social Committee and the Committee of the Regions. Brussels, 17.11.2021. Available online: https://www.eea.europa.eu/policy-documents/communication-from-the-commission-to-1 (accessed on 14 November 2012).
- COM/2019/640 Final. Communication from the Commission to the European Parliament, the European Council, the Council, the European Economic and Social Committee and the Committee of the Regions. The European Green Deal. Brussels, 11.12.2019. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=COM%3A2019%3A640%3AFIN (accessed on 11 December 2019).
- Ukalska-Jaruga, A.; Smreczak, B.; Siebielec, G. Assessment of Pesticide Residue Content in Polish Agricultural Soils. Molecules 2020, 25, 587. [Google Scholar] [CrossRef] [PubMed]
- Ukalska-Jaruga, A.; Smreczak, B. The impact of organic matter on polycyclic aromatic hydrocarbon (PAH) availability and persistence in soils. Molecules 2020, 25, 2470. [Google Scholar] [CrossRef]
- Mordechay, E.; Mordehay, V.; Tarchitzky, J.; Chefetz, B. Fate of contaminants of emerging concern in the reclaimed wastewater-soil-plant continuum. Sci. Total Environ. 2022, 822, 153574. [Google Scholar] [CrossRef]
- Maliszewska-Kordybach, B.; Smreczak, B.; Klimkowicz-Pawlas, A. Concentrations, sources, and spatial distribution of individual polycyclic aromatic hydrocarbons (PAHs) in agricultural soils in the Eastern part of the EU: Poland as a case study. Sci. Total Environ. 2009, 407, 3746–3753. [Google Scholar] [CrossRef]
- Hayes, M.; Mylotte, R.; Swift, R. Humin: Its Composition and Importance in Soil Organic Matter. In Advances in Agronomy; Donald, L.S., Ed.; Academic Press: Burlington, UK, 2017; Volume 143, pp. 47–138. ISBN 978-0-12-812421-5. [Google Scholar]
- Pignatello, J. Dynamic interactions of natural organic matter and organic compounds. J. Soil Sediment 2012, 12, 1241–1256. [Google Scholar] [CrossRef]
- Kim, P.-G.; Tarafdar, A.; Kwon, J.-H. Effects of soil pH on the sorption capacity of soil organic matter for polycyclic aromatic hydrocarbons in unsaturated soils. Pedosphere, 2022; in press. [Google Scholar] [CrossRef]
- Schaumann, G. Soil organic matter beyond molecular structure Part I: Macromolecular and supramolecular characteristics. J. Plant Nutr. Soil Sci. 2006, 169, 145–156. [Google Scholar] [CrossRef]
- Schaumann, G. Soil organic matter beyond molecular structure Part II: Amorphous nature and physical aging. J. Plant Nutr. Soil Sci. 2006, 169, 157–167. [Google Scholar] [CrossRef]
- Ukalska-Jaruga, A.; Smreczak, B.; Klimkowicz-Pawlas, A. Soil organic matter composition as a factor affecting the accumulation of polycyclic aromatic hydrocarbons. J. Soils Sediments 2019, 19, 1890–1900. [Google Scholar] [CrossRef]
- Senesi, N. Humic Substances as Natural Nanoparticles Ubiquitous in the Environment. In Molecular Environmental Soil Science at the Interfaces in the Earth’s Critical Zone; Xu, J., Huang, P.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Zhang, F.; Zhang, W.; Wu, S.; Fu, X.; Li, S.; Yue, S. Analysis of UV–VIS spectral characteristics and content estimation of soil DOM under mulching practices. Ecol. Indic. 2022, 138, 108869. [Google Scholar] [CrossRef]
- Enev, V.; Pospisilova, L.; Klucakova, M.; Liptaj, T.; Doskocil, L. Spectral characterization of selected humic substances. Soil Water Res. 2014, 9, 9–17. [Google Scholar] [CrossRef]
- Korshin, G.V.; Benjamin, M.M. Monitoring the properties of natural organic matter through UV spectroscopy: A consistent theory. Water Res. 1997, 31, 1787–1795. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, Y.; Fan, Y.; Zhang, F.; Tan, W.; He, X.; Xi, B. Roles of bacterial community in the transformation of dissolved organic matter for the stability and safety of material during sludge composting. Bioresour. Technol. 2018, 267, 378–385. [Google Scholar] [CrossRef]
- Zheng, W.; Yang, Z.; Huang, L.; Chen, Y. Roles of organic matter transformation in the bioavailability of Cu and Zn during sepiolite-amended pig manure composting. J. Environ. Manag. 2022, 314, 115046. [Google Scholar] [CrossRef]
- Ukalska-Jaruga, A.; Bejger, R.; Debaene, G.; Smreczak, B. Characterization of soil organic matter individual fractions (fulvic acids, humic acids, and humins) by spectroscopic and electrochemical techniques in agricultural soils. Agronomy 2021, 11, 1067. [Google Scholar] [CrossRef]
- Tatzber, M.; Stemmer, M.; Spiegel, H.; Katzlberger, C.; Haberhauer, G.; Mentler, A.; Gerzabek, M.H. FTIR-spectroscopic characterization of humic acids and humin fractions obtained by advanced NaOH, Na4P2O7, and Na2CO3 extraction procedures. J. Plant Nutr. Soil Sci. 2007, 170, 522–529. [Google Scholar] [CrossRef]
- Helal, A.A.; Murad, G.A.; Helal, A.A. Characterization of different humic materials by various analytical techniques. Arab. J. Chem. 2011, 4, 51–54. [Google Scholar] [CrossRef]
- Capriel, P. Hydrophobicity of organic matter in arable soils: Influence of management. Eur. J. Soil Sci. 1997, 48, 457–462. [Google Scholar] [CrossRef]
- Souza, F.D.; Bragança, S.R. Extraction and characterization of humic acid from coal for the application as dispersant of ceramic powders. J. Mater. Res. Technol. 2019, 7, 254–260. [Google Scholar] [CrossRef]
- Mastrolonardo, G.; Certini, G.; Krebs, R.; Forte, C.; Egli, M. Effects of fire on soil organic matter quality along an altitudinal sequence on Mt. Etna, Sicily. Catena 2013, 110, 133–145. [Google Scholar] [CrossRef]
- Tan, K.H. Humic Matter in Soil and the Environment. Principles and Controversies; CRC Press Taylor & Francis Group: Boca Raton, FL, USA; London, UK; New York, NY, USA, 2014. [Google Scholar]
- Paul, E. The nature and dynamic of soil organic matter: Plant inputs, microbial transformations, and organic matter stabilization. Soil Biol. 2016, 98, 109–126. [Google Scholar] [CrossRef]
- Schulten, H.; Leinweber, P. New insights into organic-mineral particles: Composition, properties and models of molecular structure. Biol. Fert. Soils 2000, 30, 399–432. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Agblevor, F.A.; El-Zawawy, W.K. Isolation and characterization of cellulose and lignin from steam-exploded lignocellulosic biomass. BioResources 2010, 5, 397–418. [Google Scholar]
- Perez, J.; Munoz-Dorado, J.; de la Rubin, T.; Martinez, J. Biodegradation and biological treatments of cellulose, hemicelluloses and lignin: An overview. Int. Microbiol. 2002, 5, 53–63. [Google Scholar] [CrossRef]
- Mansfield, S.D. Solutions for dissolution-engineering cell walls for deconstruction. Curr. Opin. Biotechnol. 2009, 20, 286–294. [Google Scholar] [CrossRef]
- Hames, D.; Hooper, N. Biochemistry, 4th ed.; Garland Science: New York, NY, USA, 2011; pp. 27–47. [Google Scholar]
- Clapp, C.E.; Hayes, M.H.B.; Simpson, A.J.; Kingery, W.L. Chemistry of soil organic matter. In Chemical Processes in Soil; Tabatabai, M.A., Sparks, D.L., Eds.; Soil Science Society of America: Madison, WI, USA, 2005; pp. 1–150. [Google Scholar]
- Deshmukh, A.P.; Simpson, A.J.; Hadad, C.M.; Hatcher, P.G. Insights into the structure of cutin and cutan from Agave americana leaf cuticle using HRMAS NMR spectroscopy. Org. Geochem. 2005, 36, 1072–1085. [Google Scholar] [CrossRef]
- Turner, J.W.; Hartman, B.E.; Hatcher, P.G. Structural characterisation of suberan isolated from river birch (Betula nigra) bark. Org. Geochem. 2013, 57, 41–53. [Google Scholar] [CrossRef]
- Simpson, A.J.; Simpson, M.J. Nuclear magnetic resonance analysis of natural organic matter. In Biophysio-Chemical Processes Involving Natural Non-Living Organic Matter in Environmental Systems; Senesi, N., Xing, B., Huang, P.M., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 589–650. [Google Scholar]
- Skjemstad, J.O.; Reicosky, D.C.; Wilts, A.R.; McGowan, J.A. Charcoal carbon in U.S. agricultural soils. Soil Sci. Soc. Am. J. 2002, 66, 1249–1255. [Google Scholar] [CrossRef]
- De Souza Sierra, M.M.; Donard, O.F.X.; Lamotte, M.; Belin, C.; Ewald, M. Fluorescence spectroscopy of coastal and marine waters. Mar. Chem. 1994, 47, 127–144. [Google Scholar] [CrossRef]
- Cook, R.L.; Langford, C.H. Metal ion qenching of fulvic acid fluorescence intensities and lifetimes: Nonlineatities and a possible three-component model. Anal. Chem. 1995, 67, 174–180. [Google Scholar] [CrossRef]
- Fuentes, M.; González-Gaitano, G.; García-Mina, J.M. The usefulness of UV—Visible and fluorescence spectroscopies to study the chemical nature of humic substances from soils and composts. Org. Geochem. 2006, 37, 1949–1959. [Google Scholar] [CrossRef]
- Fernández-Romero, M.L.; Clark, J.M.; Collins, C.D.; Parras-Alcántara, L. Evaluation of optical techniques for characterizing soil organic matter quality in agricultural soils. Soil Tillage Res. 2016, 155, 450–460. [Google Scholar] [CrossRef]
- Mielnik, L.; Weber, J.; Podlasinski, M.; Kocowicz, A. Fluorescence properties of humic substances transformed in ectohumus horizons of Podzols affected by alkaline fly-ash. Land Degrad. Dev. 2021, 32, 3487–3497. [Google Scholar] [CrossRef]
- Senesi, N.; D’Orazio, V. Fluorescence spectroscopy. In Encyclopedia of Soils in the Environment; Hillel, D., Ed.; Academic Press: Cambridge, MA, USA; Elsevier Science: Amsterdam, The Netherlands, 2005; pp. 35–52. [Google Scholar]
- Yu, J.; Xiao, K.; Xue, W.; Shen, Y.-X.; Tan, J.; Liang, S.; Wang, Y.; Huang, X. Excitation-emission matrix (EEM) fluorescence spectroscopy for characterization of organic matter in membrane bioreactors: Principles, methods and applications. Front. Environ. Sci. Eng. 2020, 14, 31. [Google Scholar] [CrossRef]
- Chen, W.; Westerhoff, P.; Leenheer, J.A.; Booksh, K. Fluorescence Excitation-Emission Matrix Regional Integration to Quantify Spectra for Dissolved Organic Matter. Environ. Sci. Technol. 2003, 37, 5701–5710. [Google Scholar] [CrossRef]
- Boguta, P.; Pieczywek, P.M.; Sokołowska, Z. A comparative study of the application of fluorescence excitation-emission matricescombined with parallel factor analysis and nonnegative matrix factorization in the analysis of Zn complexation by humic acids. Sensors 2016, 16, 1760. [Google Scholar] [CrossRef]
- Senesi, N.; Miano, T.M.; Provenzano, M.R. Fluorescence Spectroscopy as a Means of Distingiushing Fulvic and Humic Acids from Dissolved and Sedimentary Aquatic Sources and Terrestrial Sources. In Humic Substances in the Aquatic and Terrastrial Environment; Allard, B., Boren, H., Grimvall, A., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 63–73. [Google Scholar]
- Cory, R.M.; Mcknight, D.M. Fluorescence spectroscopy reveals ubiquitous presence of oxidized and reduced quinones in dissolved organic matter. Environ. Sci. Technol. 2005, 39, 8142–8149. [Google Scholar] [CrossRef] [PubMed]
- Venkanna, B.K.; Swati, B.V. Basic Thermodynamics; PHI Learning Pvt. Ltd.: New Dheli, India, 2010; ISBN 9788120341128. [Google Scholar]
- Eeshwarasinghe, D.; Loganathan, P.; Vigneswaran, S. Simultaneous removal of polycyclic aromatic hydrocarbons and heavy metals from water using granular activated carbon. Chemosphere 2019, 223, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Hayes, M.; Novotny, E.; Simpson, A. Isolation and fractionation of soil humin using alkaline urea and dimethylsulphoxide plus sulphuric acid. Naturwissenschaften 2011, 98, 7–13. [Google Scholar] [CrossRef]
- Matějková, S.; Šimon, T. Application of FTIR spectroscopy for evaluation of hydrophobic/hydrophilic organic components in arable soil. Plant Soil Environ. 2012, 58, 192–195. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S-SOM Sample | ε 280 | ε 465 | ε 665 | ε 280/465 | ε 280/665 | ε 465/665 |
|---|---|---|---|---|---|---|
| S1 | 498.5 | 55.9 | 22.5 | 8.92 | 22.16 | 2.48 |
| S2 | 417.4 | 40.1 | 19.9 | 10.40 | 20.98 | 2.02 |
| S3 | 558.8 | 52.2 | 26.3 | 10.70 | 21.27 | 1.99 |
| S4 | 453.3 | 75.6 | 31.7 | 6.00 | 14.30 | 2.38 |
| S5 | 449.9 | 54.7 | 17.7 | 8.23 | 25.45 | 3.09 |
| S6 | 366.4 | 57.5 | 22.9 | 6.38 | 16.03 | 2.51 |
| Sample of S-SOM | Hydrophobic (Intensity) | Hydrophilic (Intensity) | Hydrophobicity Index (HI) |
|---|---|---|---|
| S1 | 0.698 | 17.175 | 0.041 |
| S2 | 1.103 | 13.359 | 0.083 |
| S3 | 1.112 | 17.109 | 0.065 |
| S4 | 1.027 | 18.005 | 0.057 |
| S5 | 1.247 | 22.176 | 0.102 |
| S6 | 0.669 | 18.583 | 0.050 |
| Sample of S-SOM | IFl (I) [a.u.] | IFl (II) [a.u.] | IFl (I)/IFl (II) |
|---|---|---|---|
| S1 | 39.5 | 26.8 | 1.47 |
| S2 | 54.5 | n.d | - |
| S3 | 35.5 | 19.9 | 1.78 |
| S4 | 31.5 | 24.7 | 1.27 |
| S5 | 29.6 | 25.1 | 1.17 |
| S6 | 40.0 | 25.6 | 1.56 |
| Sample of S-SOM | Sorption Equation | Total Accumulated Amount | ||
|---|---|---|---|---|
| Atrazine | ||||
| S1 | y = 0.192e−0.028x | R2 = 0.986 | 99.93% | Aa |
| S2 | y = 0.074e−0.021x | R2 = 0.922 | 87.52% | Ba |
| S3 | y = 0.067e−0.016x | R2 = 0.923 | 99.91% | Aa |
| S4 | y = 0.099e−0.019x | R2 = 0.926 | 99.78% | Aa |
| S5 | y = 0.140e−0.027x | R2 = 0.932 | 99.84% | Aa |
| S6 | y = 0.264e−0.028x | R2 = 0.887 | 99.84% | Aa |
| DDT | ||||
| S1 | y = 10.02e−0.015x | R2 = 0.903 | 74.35% | Bb |
| S2 | y = 9.03e−0.007x | R2 = 0.876 | 75.50% | Ba |
| S3 | y = 9.26e−0.001x | R2 = 0.977 | 79.99% | ABb |
| S4 | y = 9.15e−0.003x | R2 = 0.989 | 64.67% | Cb |
| S5 | y = 9.50e−0.007x | R2 = 0.920 | 64.22% | Cb |
| S6 | y = 10.82e−0.012x | R2 = 0.956 | 81.64% | Ab |
| Chrysene | ||||
| S1 | y = 23.38e−0.002x | R2 = 0.935 | 79.82% | Ab |
| S2 | y = 17.94e−0.006x | R2 = 0.924 | 48.56% | Cc |
| S3 | y = 14.89e−0.006x | R2 = 0.946 | 45.24% | Cc |
| S4 | y = 21.05e−0.007x | R2 = 0.932 | 35.23% | Dc |
| S5 | y = 11.19e−0.001x | R2 = 0.929 | 49.15% | Cc |
| S6 | y = 14.85e−0.009x | R2 = 0.902 | 60.40% | Bc |
| Parameters | PCA 1 | PCA 2 | PCA 3 |
|---|---|---|---|
| ε 280/465 | 0.96 | 0.13 | −0.08 |
| ε 280/665 | 0.67 | 0.54 | −0.71 |
| ε 465/665 | 0.96 | 0.13 | −0.08 |
| IFl (I)/IFl (II) | 0.78 | −0.53 | 0.33 |
| HI | 0.26 | 0.90 | 0.13 |
| Atrazine sorption | −0.70 | −0.17 | −0.42 |
| DDT sorption | 0.71 | −0.78 | 0.07 |
| Chrysene sorption | 0.17 | −0.48 | −0.77 |
| % of variance | 45 | 30 | 18 |
| Cumulative % | 45 | 75 | 93 |
| Soil Properties | S1 | S2 | S3 | S4 | S5 | S6 |
|---|---|---|---|---|---|---|
| Clay (%) | 1 | 1 | 1 | 2 | 0 | 8 |
| Silt (%) | 66 | 7 | 70 | 17 | 0 | 33 |
| Sand (%) | 33 | 92 | 29 | 81 | 0 | 59 |
| WRB taxonomy | Fluvisols | Luviosols | Luviosols | Luviosols | Fluvisols | Leptosols |
| pH in KCl | 7.3 | 6.4 | 6.9 | 7.1 | 7.3 | 7.4 |
| TOC (g kg−1) | 19.2 | 19.3 | 16.9 | 15.0 | 58.7 | 29.4 |
| TN (g kg−1) | 2.2 | 1.9 | 1.8 | 1.4 | 6.6 | 2.9 |
| TC/TN | 10.8 | 11.4 | 10.2 | 14.8 | 11.4 | 13.4 |
| Atrazine | Chrysene | 44′DDT | |
|---|---|---|---|
| Molecule structure |  |  |  |
| Type of compound | Pesticide (herbicide) | Polycyclic aromatic hydrocarbons | Pesticide (insecticide) |
| Molecular weight (g mol−1) | 215.68 | 228.29 | 354.49 |
| Water solubility (mg l−1) | 35 | 0.0015 | 0.006 |
| log Ko/w | 2.7 | 5.91 | 6.91 |
| Vapor pressure (mPa) | 0.039 | 1.04·10−6 | 0.025 |
| Soil degradation, DT 50 (days) | 75 | 120 | 6200 |
| Bioconcentration factor (l kg−1) | 4.3 | 0.00033 | 3173 |
| Parameter | GC-MS/MS |
|---|---|
| Injection mode | Hot-splitless; MMI injection mode |
| Injection volume | 2 μL |
| Inlet temperature | 280 °C |
| Carrier gas | He, constant flow 1.00 mL min−1 (column 2 = 1.20 mL min−1) |
| Detector temperature | - |
| Makeup gas | - |
| Oven program | 70 °C for 2 min 25 °C/min to 150 °C for 0 min; 3 °C/min to 200 °C for 0 min; 8 °C/min to 280 °C for 10 min hold time |
| MS transfer line temperature | 280 °C |
| Backflush settings | 5 min during post-run / 310 °C |
| Aux EPC pressure | ~50 psi |
| Inlet pressure | ~2 psi |
| Column pressure | ~3 psi |
| Electron energy | 70 eV |
| MS1 and MS2 resolution | Wide |
| Collision cell | 1.5 mL min−1 N2 and 2.25 mL min−1 He |
| Source temperature | 300 °C |
| Quad temperatures | 150 °C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ukalska-Jaruga, A.; Bejger, R.; Smreczak, B.; Podlasiński, M. Sorption of Organic Contaminants by Stable Organic Matter Fraction in Soil. Molecules 2023, 28, 429. https://doi.org/10.3390/molecules28010429
Ukalska-Jaruga A, Bejger R, Smreczak B, Podlasiński M. Sorption of Organic Contaminants by Stable Organic Matter Fraction in Soil. Molecules. 2023; 28(1):429. https://doi.org/10.3390/molecules28010429
Chicago/Turabian StyleUkalska-Jaruga, Aleksandra, Romualda Bejger, Bożena Smreczak, and Marek Podlasiński. 2023. "Sorption of Organic Contaminants by Stable Organic Matter Fraction in Soil" Molecules 28, no. 1: 429. https://doi.org/10.3390/molecules28010429
APA StyleUkalska-Jaruga, A., Bejger, R., Smreczak, B., & Podlasiński, M. (2023). Sorption of Organic Contaminants by Stable Organic Matter Fraction in Soil. Molecules, 28(1), 429. https://doi.org/10.3390/molecules28010429