
Theoretical Investigation of Geometries and Bonding of Indium Hydrides in the In2Hx and In3Hy (x = 0–4,6; y = 0–5) Series



Abstract
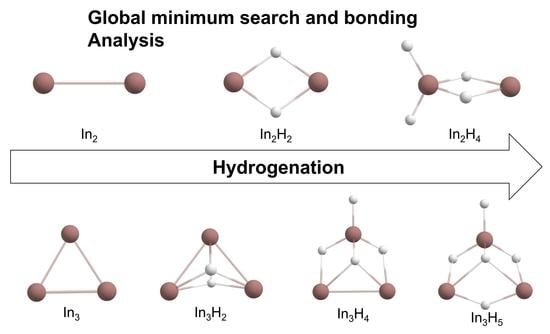
1. Introduction
2. Results and Discussions
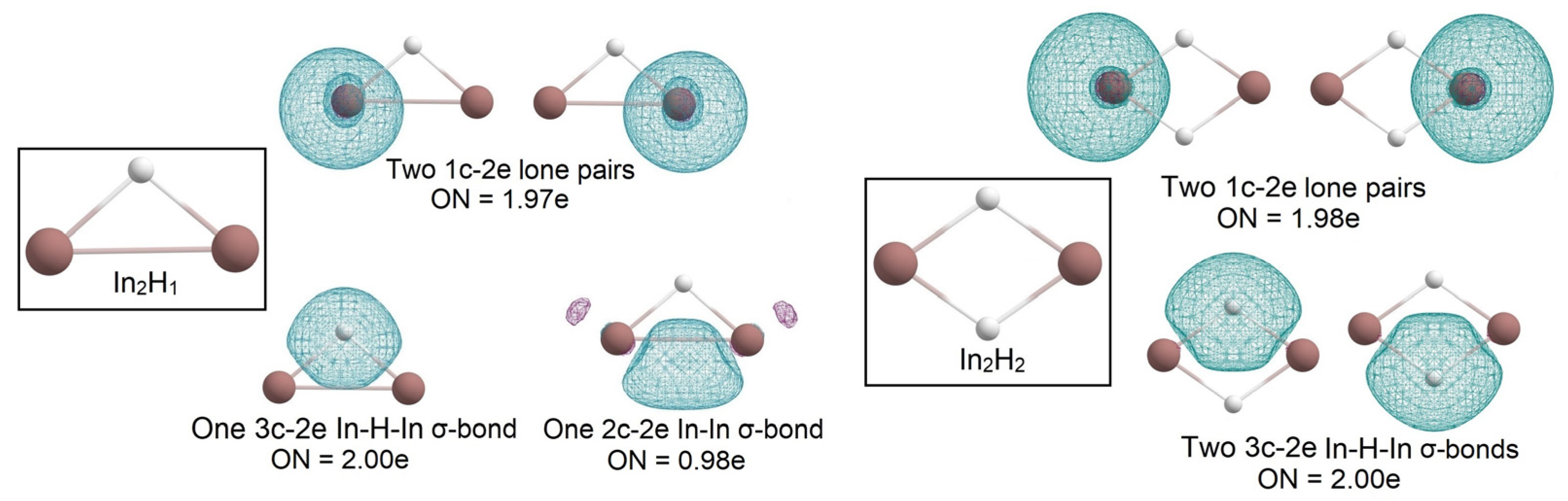
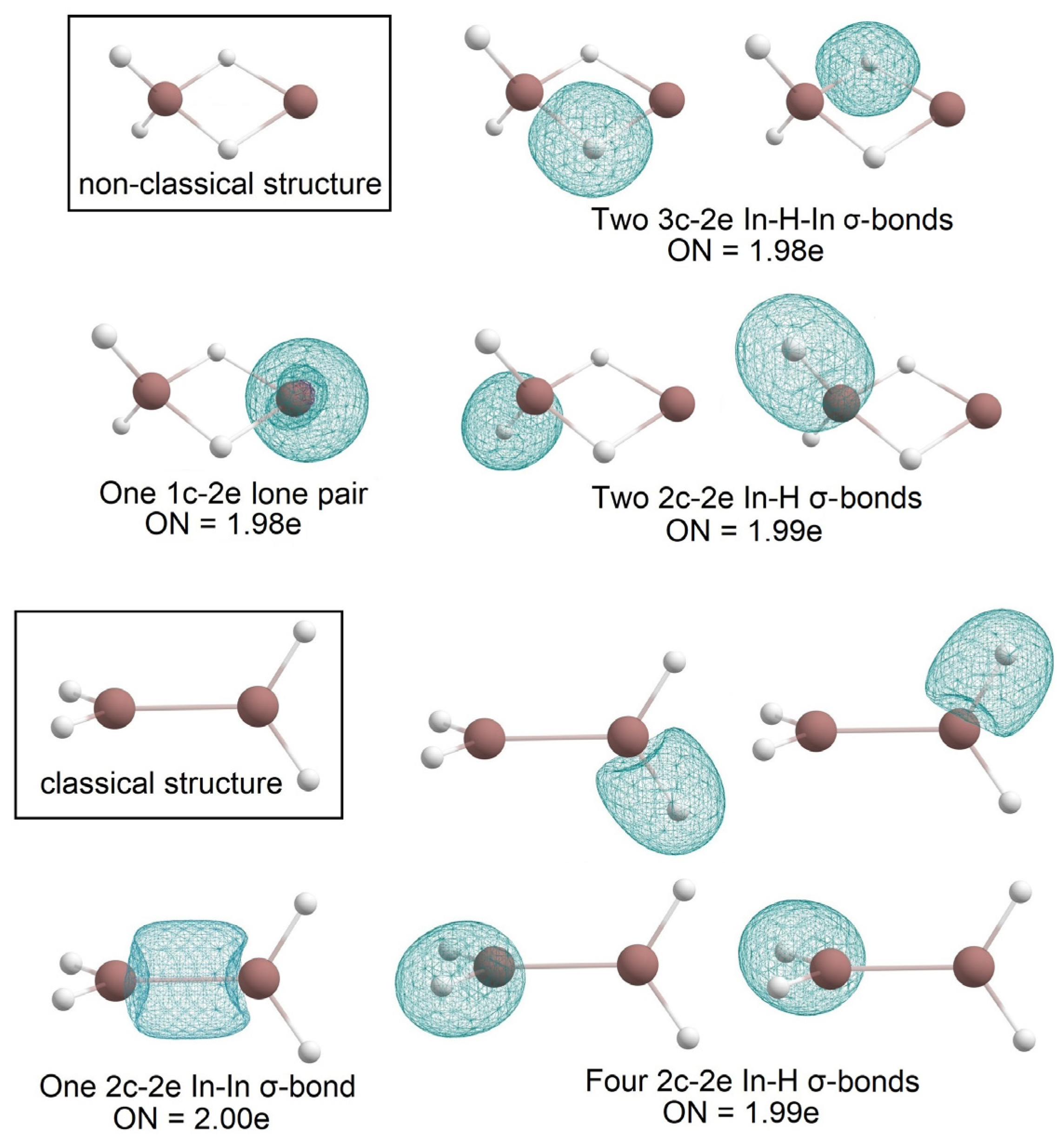
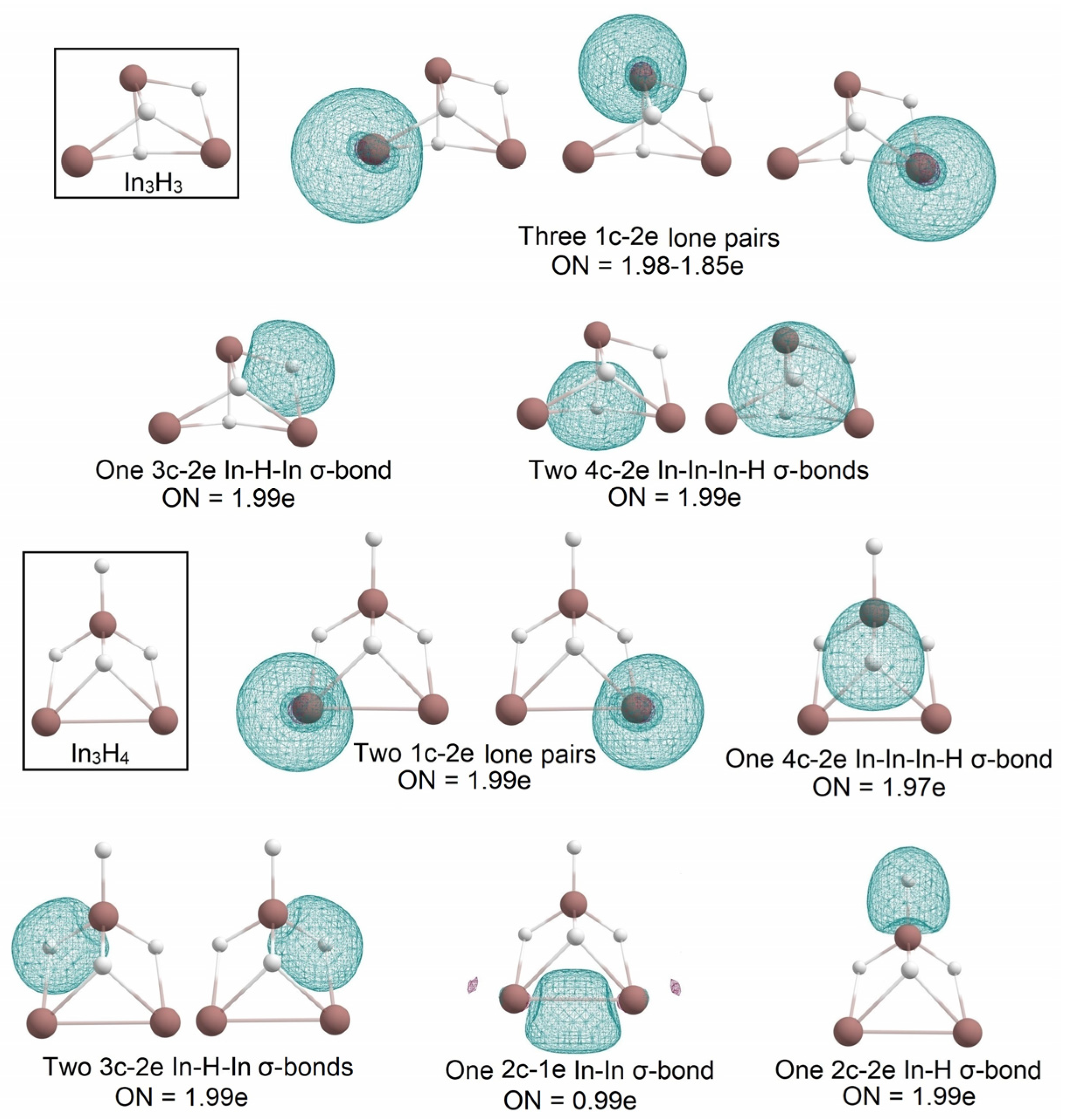
2.1. Global Geometry Optimization and Bonding Analysis
2.2. Thermodynamic Stability Observation
3. Theoretical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lipscomb, W.N. Boron Hydrides; Benjamin Inc.: New York, NY, USA, 1963. [Google Scholar]
- Muetterties, E.L. Boron Hydride Chemistry; Academic Press: New York, NY, USA, 1975. [Google Scholar]
- Pan, S.; Barroso, J.; Jalife, S.; Heine, T.; Asmis, K.R.; Merino, G. Fluxional Boron Clusters: From Theory to Reality. Acc. Chem. Res. 2019, 52, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
- Osorio, R.; Olson, J.K.; Tiznado, W.; Boldyrev, A.I. Analysis of Why Boron Avoids sp2 Hybridization and Classical Structures in the BnHn+2 Series. Eur. J. Chem. 2012, 18, 9677–9681. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.T. Recent development and improvement for Boron hydride-based catalytic asymmetric reduction of unsymmetrical ketones. Chem. Soc. Rev. 2009, 38, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Sivaev, I.B.; Bregadze, V.V. Polyhedral Boranes for Medical Applications: Current Status and prospects. Eur. J. Inorg. Chem. 2009, 2009, 1433–1450. [Google Scholar] [CrossRef]
- Fakioğlu, E.; Yürüm, Y.; Veziroğlu, T.N. A review of hydrogen storage systems based on Boron and its compounds. Int. J. Hydrogen Energy 2004, 29, 1371–1376. [Google Scholar] [CrossRef]
- Sergeeva, A.P.; Popov, I.A.; Piazza, Z.A.; Li, W.L.; Romanescu, C.; Wang, L.S.; Boldyrev, A.I. Understanding Boron through Size-Selected Clusters: Structure, Chemical Bonding, and Fluxionality. Acc. Chem. Res. 2014, 47, 1349–1358. [Google Scholar] [CrossRef]
- Jian, T.; Chen, X.; Li, S.D.; Boldyrev, A.I.; Li, J.; Wang, L.S. Probing the structures and bonding of size-selected Boron and doped-Boron clusters. Chem. Soc. Rev. 2019, 48, 3550–3591. [Google Scholar] [CrossRef]
- Wiberg, E.; Schmidt, M.Z. Uber Wasserstoff-Verbindungen des Indiums. Z. Nat. B 1957, 12, 54. [Google Scholar] [CrossRef]
- Miyai, T.; Inoue, K.; Yasuda, M.; Shibata, I.; Baba, A. Preparation of a novel Indium hydride and application to practical organic synthesis. Tetrahedron Lett. 1998, 39, 1929–1932. [Google Scholar] [CrossRef]
- Hunt, P.; Schwerdtfeger, P. Are the Compounds InH3 and TlH3 Stable Gas Phase or Solid State Species? Inorg. Chem. 1996, 35, 2085–2088. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Heath, G.A.; Dolg, M.; Bennett, M.A. Low Valencies and Periodic Trends in Heavy Element Chemistry. A Theoretical Study of Relativistic Effects and Electron Correlation Effects in Group 13 and Period 6 Hydrides and Halides. Am. Chem. Soc. 1992, 114, 7518–7527. [Google Scholar] [CrossRef]
- Liu, Y.; Duan, D.; Tian, F.; Liu, H.; Wang, C.; Huang, X.; Li, D.; Ma, Y.; Liu, B.; Cui, T. Pressure-Induced Structures and Properties in Indium Hydrides. Inorg. Chem. 2015, 54, 9924–9928. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.P.; Herzberg, G. Chapter IV Constants of Diatomic Molecules. In Molecular Spectra and Molecular Structure, 1st ed.; Springer: New York, NY, USA, 1979. [Google Scholar]
- Edlén, B.; Ölme, A.; Herzberg, G.; Johns, J.W.C. Ionization Potential of Boron, and the Isotopic and Fine Structure of 2s2p2.2D. J. Opt. Soc. Am. 1970, 60, 889. [Google Scholar] [CrossRef]
- Paschen, F.; Campbell, J.S. Das erste Funkenspektrum des Indiums in II. Ann. Phys. 1938, 423, 29–75. [Google Scholar] [CrossRef]
- Mulla, H.F.; Franke, P.R.; Sargenta, C.; Douberlya, G.E.; Turney, J.M.; Schaefer, H.F., III. Four isomers of In2H2: A careful comparison between theory and experiment. Mol. Phys. 2021, 119, e1979675. [Google Scholar] [CrossRef]
- Báez-Grezab, R.; Ruizc, L.; Pino-Rios, R.; Tiznado, W. Which NICS method is most consistent with ring current analysis? Assessment in simple monocycles. RSC Adv. 2018, 8, 13446–13453. [Google Scholar] [CrossRef]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.V.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef]
- Sergeeva, A.P.; Averkiev, B.B.; Zhai, H.-J.; Boldyrev, A.I.; Wang, L.S. All-Boron analogues of aromatic hydrocarbons: B17− and B18−. J. Chem. Phys. 2011, 134, 224304. [Google Scholar] [CrossRef]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.J.; Wang, L.S. All-Boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry. Coord. Chem. Rev. 2006, 250, 2811–2866. [Google Scholar] [CrossRef]
- Saunders, M. Stochastic search for isomers on a quantum mechanical surface. J. Comput. Chem. 2004, 25, 621–626. [Google Scholar] [CrossRef]
- Coalescence Modification to Saunders KICK Version. Available online: https://kick.science/KICK_Saunders_coalescence.html (accessed on 25 November 2022).
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H.; Hay, P.J. Modern Theoretical Chemistry; Schaefer, H.F., III, Ed.; Plenum: New York, NY, USA, 1977; Volume 3, pp. 1–28. [Google Scholar]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 12, e1606. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms Boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Metz, B.; Stoll, H. Small-core multiconfiguration-Dirac–Hartree–Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563. [Google Scholar] [CrossRef]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13–15 elements. J. Chem. Phys. 2003, 119, 11099. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Stability analysis for solutions of the closed shell Kohn–Sham equation. J. Chem. Phys. 1996, 104, 9047. [Google Scholar] [CrossRef]
- Balasubramanian, K.; Li, J. Spectroscopic properties and potential energy surfaces of In2. J. Chem. Phys. 1998, 88, 4979. [Google Scholar] [CrossRef]
- Tkachenko, N.V.; Boldyrev, A.I. Chemical bonding analysis of excited states using the Adaptive Natural Density Partitioning method. Phys. Chem. Chem. Phys. 2019, 21, 9590–9596. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef] [PubMed]
- Chemcraft. Available online: http://www.chemcraftprog.com (accessed on 25 November 2022).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stoichiometry | Decomposition Product | Stability | Eproducts − Ereagents, kcal/mol |
|---|---|---|---|
| In2H2 | In2 + H2 | stable | 25.2 |
| In2H3 | In2H + H2 | unstable | −0.1 |
| In2H4 | In2H2 + H2In2 + 2H2 | stable stable | 4.8 30.0 |
| In2H6 | In2H4 + H2In2H2 + 2H2In2 + 3H2 | unstable stable stable | −2.1 2.7 27.9 |
| In3H2 | In3 + H2 | stable | 12.1 |
| In3H3 | In3H + H2 | stable | 12.2 |
| In3H4 | In3H2 + H2In3 + 2H2 | unstable stable | −0.5 11.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozdeev, A.S.; Rublev, P.; Scheiner, S.; Boldyrev, A.I. Theoretical Investigation of Geometries and Bonding of Indium Hydrides in the In2Hx and In3Hy (x = 0–4,6; y = 0–5) Series. Molecules 2023, 28, 183. https://doi.org/10.3390/molecules28010183
Pozdeev AS, Rublev P, Scheiner S, Boldyrev AI. Theoretical Investigation of Geometries and Bonding of Indium Hydrides in the In2Hx and In3Hy (x = 0–4,6; y = 0–5) Series. Molecules. 2023; 28(1):183. https://doi.org/10.3390/molecules28010183
Chicago/Turabian StylePozdeev, Anton S., Pavel Rublev, Steve Scheiner, and Alexander I. Boldyrev. 2023. "Theoretical Investigation of Geometries and Bonding of Indium Hydrides in the In2Hx and In3Hy (x = 0–4,6; y = 0–5) Series" Molecules 28, no. 1: 183. https://doi.org/10.3390/molecules28010183
APA StylePozdeev, A. S., Rublev, P., Scheiner, S., & Boldyrev, A. I. (2023). Theoretical Investigation of Geometries and Bonding of Indium Hydrides in the In2Hx and In3Hy (x = 0–4,6; y = 0–5) Series. Molecules, 28(1), 183. https://doi.org/10.3390/molecules28010183