
Nicotinic Acetylcholine Receptors and Microglia as Therapeutic and Imaging Targets in Alzheimer’s Disease

 ,
, 
Abstract
1. Introduction
2. nAChRs and AD Pathophysiology
2.1. Neuroprotection and nAChRs
2.2. Modulation of Neurotransmission through nAChRs by Aβ
2.3. Neuroinflammation and nAChRs
2.4. Regulation of nAChR Expression
3. Live Imaging of nAChRs
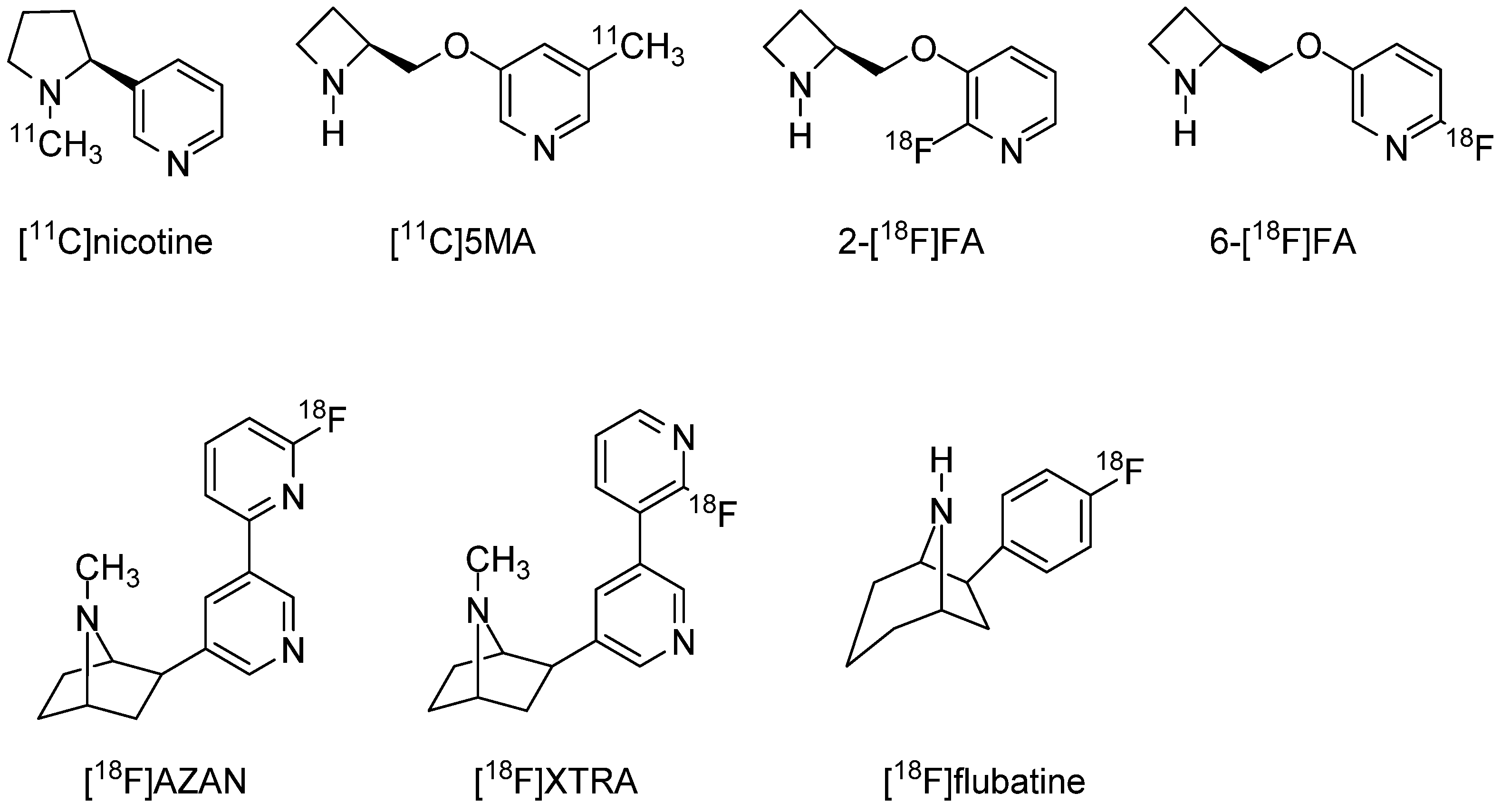
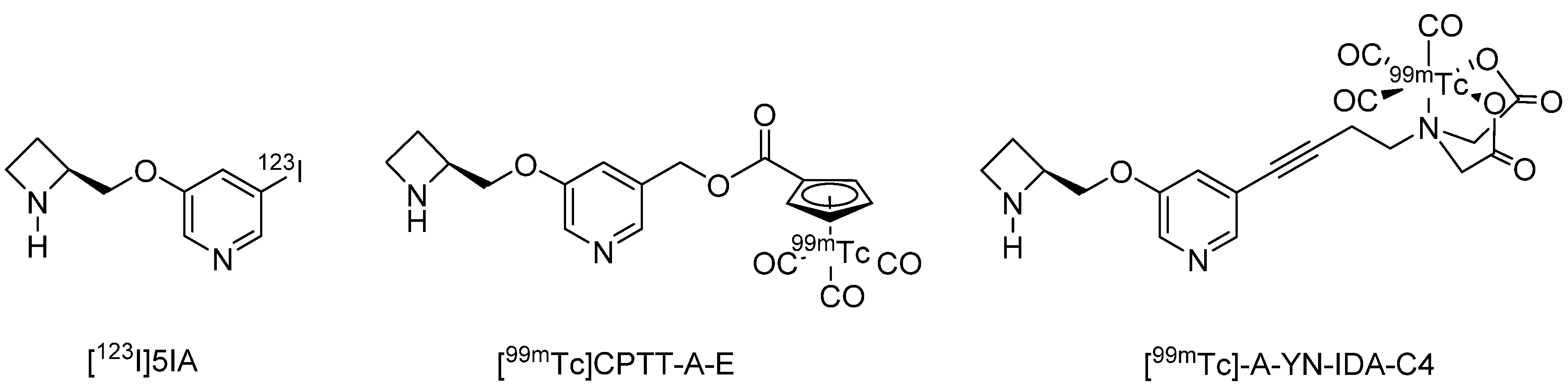
3.1. In Vivo PET and SPECT Imaging Probes for α4β2 nAChR
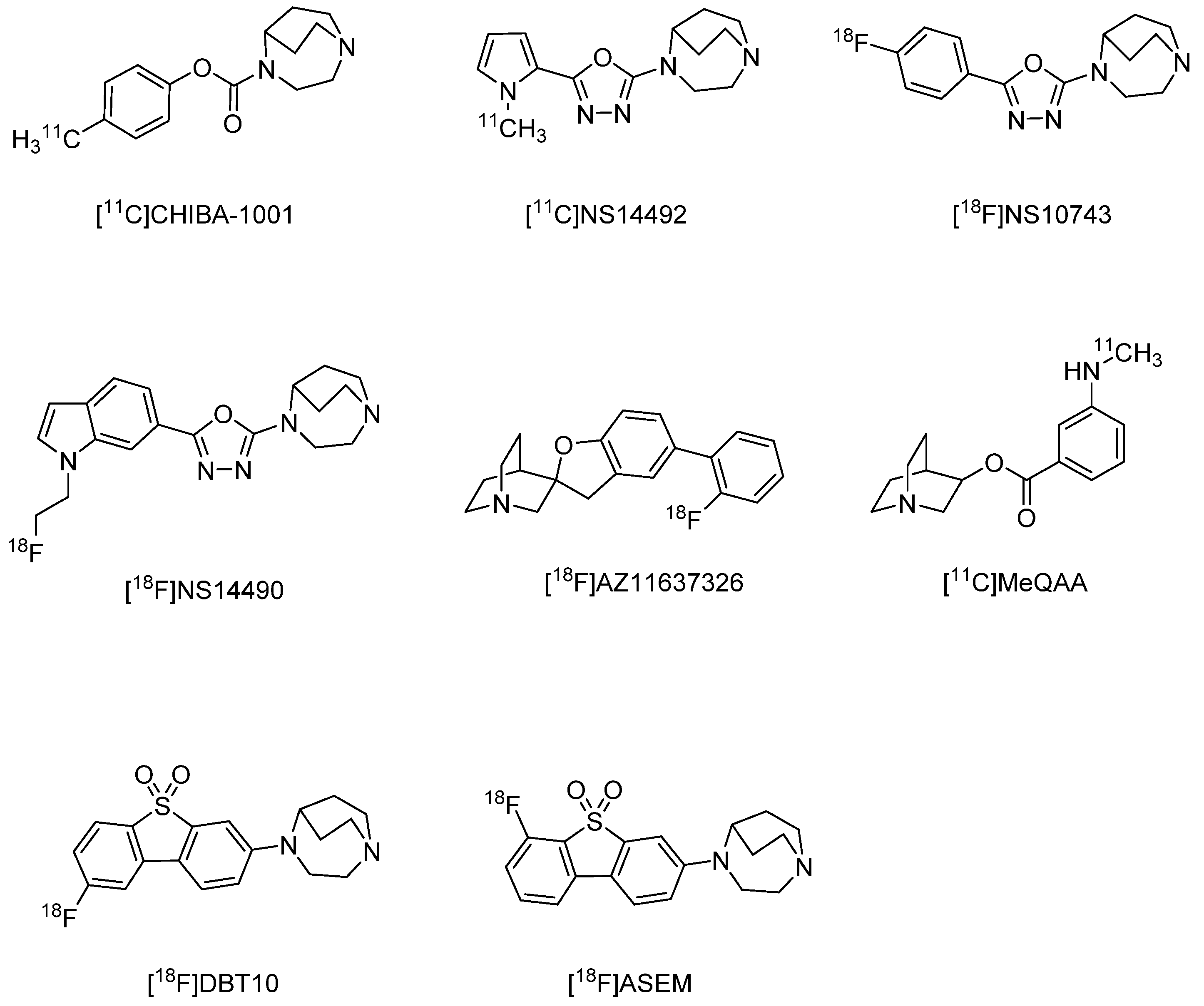
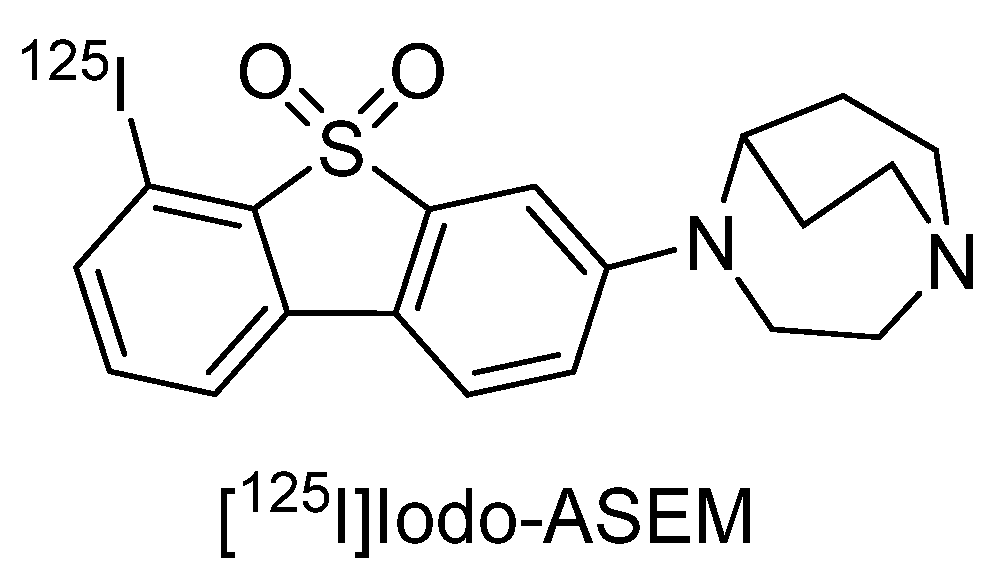
3.2. In Vivo PET and SPECT Imaging Probes for α7 nAChR
4. Microglia and AD Pathophysiology
5. Live Imaging of Microglia in AD
5.1. PK11195 and Its Targeting Protein TSPO (PBR)
5.2. Allelic Variances of TSPO
5.3. Distribution and Cell Origins of TSPO in the Brain
5.4. Recent Findings in Biological Function of TSPO
5.5. TSPO Expression in Pro-Inflammatory Activated Microglia as Detected by PET Imaging
5.6. TSPO Targeting Radioactive Imaging in AD Model Animals and AD Patients
5.7. Beyond Microglia Imaging by PET and SPECT
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| AChE | Acetylcholinesterase |
| AD | Alzheimer’s Disease |
| ago-PAM | Allosteric Agonist And Positive Allosteric Modulator |
| ALS | Amyotrophic Lateral Sclerosis |
| AMPARs | α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid-Type Glutamate Receptors |
| Arc | Activity-Regulated Cytoskeleton-Associated Protein |
| ASC | Apoptosis-Associated Speck-Like Protein Containing A Caspase Recruitment Domain |
| Aβ | Amyloid-β |
| BAMs | Border-Associated Macrophages |
| BDNF | Brain-Derived Neurotrophic Factor |
| CaM | Calmodulin |
| CaMKII | CaM-Binding Protein Kinase II |
| ChAT | Choline Acetyltransferase |
| CNS | Central Nervous System |
| CREB | cAMP Responsive Element-Binding Protein |
| CSF1R | Colony-Stimulating Factor 1 Receptor |
| Cytob558 | Cytochrome B558 |
| DAM | Disease-Associated Microglia |
| DAMPs | Damage-Associated Molecular Patterns |
| EAE | Experimental Autoimmune Encephalomyelitis |
| ER | Endoplasmic Reticulum |
| Erk1/2 | Extracellular Signal-Regulated Kinase 1/2 |
| FACS | Fluorescence-Activated Cell Sorting |
| GSDMD | Gasdermin D |
| HABs | High-Affinity Binders |
| HDAC1 | Histone Deacetylase 1 |
| HO-1 | Heme Oxygenase-1 |
| Iba-1 | Ionized Calcium-Binding Adaptor Molecule-1 |
| IFN-γ | Interferon-γ |
| IGF-1 | Insulin-Like Growth Factor-1 |
| IL | Interleukin |
| iNOS | Inducible Nitric Oxide Synthase |
| KD | Knockdown |
| KO | Knockout |
| LABs | Low-Affinity Binders |
| LPS | Lipopolysaccharide |
| LSP | Lipopolysaccharide |
| LTP | Long-Term Potentiation |
| MABs | Mixed-Affinity Binders |
| MAPKs | Mitogen-Activated Protein Kinases |
| miRNA | microRNA |
| MPTP | Mitochondrial Permeability Transition Pore |
| MRI | Magnetic Resonance Imaging |
| MS | Multiple Sclerosis |
| nAChRs | Nicotinic Acetylcholine Receptors |
| NAMPs | Neurodegeneration-Associated Molecular Patterns |
| NLRP3 | NOD-, LRR-, And Pyrin Domain Containing 3 |
| NOX2 | NADPH Oxidase 2 |
| NQO1 | NAD(P)H: Quinone Oxidoreductase-1 |
| P2Y12R | P2Y12 Receptor |
| PAMs | Positive Allosteric Modulators |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PBR | Peripheral-Type Benzodiazepine Receptor |
| PD | Parkinson’s Disease |
| PET | Positron Emission Tomography |
| PI3K | Phosphatidylinositol 3 Kinase |
| PRRs | Pattern Recognition Receptors |
| Rac1 | Ras-Related C3 Botulinum Toxin Substrate 1 |
| RIC-3 | Resistance To Inhibitors Of Cholinesterase-3 |
| ROS | Reactive Oxygen Species |
| scRNA-seq | Single Cell RNA-Sequencing |
| SIRT1 | Sirtuin 1 |
| SNP | Single Nucleotide Polymorphism |
| snRNA-seq | Single Nucleus RNA- Sequencing |
| SPECT | Single Photon Emission Computed Tomography |
| TNF | Tumor Necrosis Factor |
| TSPO | Translocator Protein |
| TXNRD1 | Thioredoxin Reductase |
| WAVE | Wiskott–Aldrich Syndrome Protein Family Verprolin-Homologous Protein |
| σ1-R | σ1 Receptor |
References
- Jorm, A.F.; Jolley, D. The incidence of dementia: A meta-analysis. Neurology 1998, 51, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Corrada, M.M.; Brookmeyer, R.; Paganini-Hill, A.; Berlau, D.; Kawas, C.H. Dementia incidence continues to increase with age in the oldest old: The 90+ study. Ann. Neurol. 2010, 67, 114–121. [Google Scholar] [CrossRef] [PubMed]
- 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022, 18, 700–789. [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergstrom, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Shimohama, S.; Taniguchi, T.; Fujiwara, M.; Kameyama, M. Changes in nicotinic and muscarinic cholinergic receptors in Alzheimer-type dementia. J. Neurochem. 1986, 46, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Flynn, D.D.; Mash, D.C. Characterization of L-[3H]nicotine binding in human cerebral cortex: Comparison between Alzheimer’s disease and the normal. J. Neurochem. 1986, 47, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Winblad, B. Reduced number of [3H]nicotine and [3H]acetylcholine binding sites in the frontal cortex of Alzheimer brains. Neurosci. Lett. 1986, 72, 115–119. [Google Scholar] [CrossRef]
- Mulugeta, E.; Karlsson, E.; Islam, A.; Kalaria, R.; Mangat, H.; Winblad, B.; Adem, A. Loss of muscarinic M4 receptors in hippocampus of Alzheimer patients. Brain Res. 2003, 960, 259–262. [Google Scholar] [CrossRef]
- Svensson, A.L.; Alafuzoff, I.; Nordberg, A. Characterization of muscarinic receptor subtypes in Alzheimer and control brain cortices by selective muscarinic antagonists. Brain Res. 1992, 596, 142–148. [Google Scholar] [CrossRef]
- Li, D.D.; Zhang, Y.H.; Zhang, W.; Zhao, P. Meta-Analysis of Randomized Controlled Trials on the Efficacy and Safety of Donepezil, Galantamine, Rivastigmine, and Memantine for the Treatment of Alzheimer’s Disease. Front. Neurosci. 2019, 13, 472. [Google Scholar] [CrossRef]
- Buckingham, S.D.; Jones, A.K.; Brown, L.A.; Sattelle, D.B. Nicotinic Acetylcholine Receptor Signalling: Roles in Alzheimer’s Disease and Amyloid Neuroprotection. Pharmacol. Rev. 2009, 61, 39–61. [Google Scholar] [CrossRef]
- Akaike, A.; Tamura, Y.; Yokota, T.; Shimohama, S.; Kimura, J. Nicotine-induced protection of cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res. 1994, 644, 181–187. [Google Scholar] [CrossRef]
- Kihara, T.; Shimohama, S.; Sawada, H.; Kimura, J.; Kume, T.; Kochiyama, H.; Maeda, T.; Akaike, A. Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann. Neurol. 1997, 42, 159–163. [Google Scholar] [CrossRef]
- Kihara, T.; Shimohama, S.; Sawada, H.; Honda, K.; Nakamizo, T.; Shibasaki, H.; Kume, T.; Akaike, A. Alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J. Biol. Chem. 2001, 276, 13541–13546. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Ginhoux, F.; Shimohama, S. Roles of microglia in Alzheimer’s disease and impact of new findings on microglial heterogeneity as a target for therapeutic intervention. Biochem. Pharmacol. 2021, 192, 114754. [Google Scholar] [CrossRef] [PubMed]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Streit, W.J. Microglia and neuroprotection: Implications for Alzheimer’s disease. Brain Res. Brain Res. Rev. 2005, 48, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, P.; Oller, G.; Hoeffel, G.; Pont-Lezica, L.; Rostaing, P.; Low, D.; Bessis, A.; Ginhoux, F.; Garel, S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014, 8, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290 e1217. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef]
- Shimohama, S.; Kawamata, J. Roles of Nicotinic Acetylcholine Receptors in the Pathology and Treatment of Alzheimer’s and Parkinson’s Diseases; Springer: Singapore, 2018; pp. 137–158. [Google Scholar]
- Kume, T.; Takada-Takatori, Y. Nicotinic Acetylcholine Receptor Signaling: Roles in Neuroprotection; Springer: Singapore, 2018; pp. 59–71. [Google Scholar]
- Papke, R.L.; Horenstein, N.A. Therapeutic Targeting of alpha7 Nicotinic Acetylcholine Receptors. Pharmacol. Rev. 2021, 73, 1118–1149. [Google Scholar] [CrossRef]
- Guo, J.; Yang, G.; He, Y.; Xu, H.; Fan, H.; An, J.; Zhang, L.; Zhang, R.; Cao, G.; Hao, D.; et al. Involvement of α7nAChR in the Protective Effects of Genistein Against β-Amyloid-Induced Oxidative Stress in Neurons via a PI3K/Akt/Nrf2 Pathway-Related Mechanism. Cell. Mol. Neurobiol. 2021, 41, 377–393. [Google Scholar] [CrossRef]
- Fonar, G.; Polis, B.; Sams, D.S.; Levi, A.; Malka, A.; Bal, N.; Maltsev, A.; Elliott, E.; Samson, A.O. Modified Snake alpha-Neurotoxin Averts beta-Amyloid Binding to alpha7 Nicotinic Acetylcholine Receptor and Reverses Cognitive Deficits in Alzheimer’s Disease Mice. Mol. Neurobiol. 2021, 58, 2322–2341. [Google Scholar] [CrossRef]
- Chang, K.-W.; Zong, H.-F.; Ma, K.-G.; Zhai, W.-Y.; Yang, W.-N.; Hu, X.-D.; Xu, J.-H.; Chen, X.-L.; Ji, S.-F.; Qian, Y.-H. Activation of α7 nicotinic acetylcholine receptor alleviates Aβ1–42-induced neurotoxicity via downregulation of p38 and JNK MAPK signaling pathways. Neurochem. Int. 2018, 120, 238–250. [Google Scholar] [CrossRef]
- Li, L.; Liu, Z.; Jiang, Y.Y.; Shen, W.X.; Peng, Y.P.; Qiu, Y.H. Acetylcholine suppresses microglial inflammatory response via alpha7nAChR to protect hippocampal neurons. J. Integr. Neurosci. 2019, 18, 51–56. [Google Scholar] [PubMed]
- Cao, M.; MacDonald, J.W.; Liu, H.L.; Weaver, M.; Cortes, M.; Durosier, L.D.; Burns, P.; Fecteau, G.; Desrochers, A.; Schulkin, J.; et al. alpha7 Nicotinic Acetylcholine Receptor Signaling Modulates Ovine Fetal Brain Astrocytes Transcriptome in Response to Endotoxin. Front. Immunol. 2019, 10, 1063. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-W.; Zong, H.-F.; Wang, M.; Rizvi, M.Y.; Neha, S.I.; Yang, W.-N.; Ji, S.-F.; Ma, Y.-B.; Qian, Y.-H. PNU282987 alleviates Aβ-induced anxiety and depressive-like behaviors through upregulation of α7nAChR by ERK-serotonin receptors pathway. Neurosci. Lett. 2020, 731, 135118. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-L.; Deng, Y.-X.; Gao, Y.-M.; Dong, Y.-T.; Wang, F.; Guan, Z.-Z.; Hong, W.; Qi, X.-L. Activation of α7 nAChR by PNU-282987 improves synaptic and cognitive functions through restoring the expression of synaptic-associated proteins and the CaM-CaMKII-CREB signaling pathway. Aging 2020, 12, 543–570. [Google Scholar] [CrossRef] [PubMed]
- Potasiewicz, A.; Faron-Gorecka, A.; Popik, P.; Nikiforuk, A. Repeated treatment with alpha 7 nicotinic acetylcholine receptor ligands enhances cognitive processes and stimulates Erk1/2 and Arc genes in rats. Behav. Brain Res. 2021, 409, 113338. [Google Scholar] [CrossRef]
- Takata, K.; Amamiya, T.; Mizoguchi, H.; Kawanishi, S.; Kuroda, E.; Kitamura, R.; Ito, A.; Saito, Y.; Tawa, M.; Nagasawa, T.; et al. Alpha7 nicotinic acetylcholine receptor-specific agonist DMXBA (GTS-21) attenuates Abeta accumulation through suppression of neuronal gamma-secretase activity and promotion of microglial amyloid-beta phagocytosis and ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 62, 197–209. [Google Scholar]
- Patel, H.; McIntire, J.; Ryan, S.; Dunah, A.; Loring, R. Anti-inflammatory effects of astroglial α7 nicotinic acetylcholine receptors are mediated by inhibition of the NF-κB pathway and activation of the Nrf2 pathway. J. Neuroinflamm. 2017, 14, 192. [Google Scholar] [CrossRef]
- Li, H.; Gao, J.; Chang, Y.; Li, K.; Wang, L.; Ju, C.; Zhang, F. JWX-A0108, a positive allosteric modulator of alpha7 nAChR, attenuates cognitive deficits in APP/PS1 mice by suppressing NF-kappaB-mediated inflammation. Int. Immunopharmacol. 2021, 96, 107726. [Google Scholar] [CrossRef]
- Mizrachi, T.; Marsha, O.; Brusin, K.; Ben-David, Y.; Thakur, G.A.; Vaknin-Dembinsky, A.; Treinin, M.; Brenner, T. Suppression of neuroinflammation by an allosteric agonist and positive allosteric modulator of the α7 nicotinic acetylcholine receptor GAT107. J. Neuroinflamm. 2021, 18, 99. [Google Scholar] [CrossRef]
- Lin, M.-W.; Chen, Y.-H.; Yang, H.-B.; Lin, C.C.; Hung, S.-Y. Galantamine Inhibits Aβ1–42-Induced Neurotoxicity by Enhancing α7nAChR Expression as a Cargo Carrier for LC3 Binding and Aβ1–42 Engulfment During Autophagic Degradation. Neurotherapeutics 2020, 17, 676–689. [Google Scholar] [CrossRef]
- Takata, K.; Kitamura, Y.; Saeki, M.; Terada, M.; Kagitani, S.; Kitamura, R.; Fujikawa, Y.; Maelicke, A.; Tomimoto, H.; Taniguchi, T.; et al. Galantamine-induced amyloid-{beta} clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J. Biol. Chem. 2010, 285, 40180–40191. [Google Scholar] [CrossRef] [PubMed]
- Chalon, S.; Vetel, S.; Foucault-Fruchard, L.; Tronel, C.; Buron, F.; Vergote, J.; Bodard, S.; Routier, S.; Sérrière, S. Neuroprotective and anti-inflammatory effects of a therapy combining agonists of nicotinic α7 and σ1 receptors in a rat model of Parkinson’s disease. Neural Regen. Res. 2021, 16, 1099. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T. Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments. Behav. Brain Res. 2016, 296, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.P.; Stokoe, S.A.; Sathler, M.F.; Nichols, R.A.; Kim, S. Selective coactivation of α7- and α4β2-nicotinic acetylcholine receptors reverses beta-amyloid–induced synaptic dysfunction. J. Biol. Chem. 2021, 296, 100402. [Google Scholar] [CrossRef]
- Cao, K.; Dong, Y.-T.; Xiang, J.; Xu, Y.; Li, Y.; Song, H.; Yu, W.-F.; Qi, X.-L.; Guan, Z.-Z. The neuroprotective effects of SIRT1 in mice carrying the APP/PS1 double-transgenic mutation and in SH-SY5Y cells over-expressing human APP670/671 may involve elevated levels of α7 nicotinic acetylcholine receptors. Aging 2020, 12, 1792–1807. [Google Scholar] [CrossRef]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2012, 13, 93–110. [Google Scholar] [CrossRef]
- Adams, J.P.; Sweatt, J.D. Molecular psychology: Roles for the ERK MAP kinase cascade in memory. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 135–163. [Google Scholar] [CrossRef]
- Tzingounis, A.V.; Nicoll, R.A. Arc/Arg3.1: Linking Gene Expression to Synaptic Plasticity and Memory. Neuron 2006, 52, 403–407. [Google Scholar] [CrossRef]
- Malinow, R.; Malenka, R.C. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [Google Scholar] [CrossRef]
- Giocomo, L.M.; Hasselmo, M.E. Nicotinic modulation of glutamatergic synaptic transmission in region CA3 of the hippocampus. Eur. J. Neurosci. 2005, 22, 1349–1356. [Google Scholar] [CrossRef]
- Cheng, Q.; Yakel, J.L. The effect of alpha7 nicotinic receptor activation on glutamatergic transmission in the hippocampus. Biochem. Pharmacol. 2015, 97, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Kawai, H.; Berg, D.K. Beta-Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 4734–4739. [Google Scholar] [CrossRef] [PubMed]
- Gulisano, W.; Melone, M.; Ripoli, C.; Tropea, M.R.; Li Puma, D.D.; Giunta, S.; Cocco, S.; Marcotulli, D.; Origlia, N.; Palmeri, A.; et al. Neuromodulatory Action of Picomolar Extracellular Abeta42 Oligomers on Presynaptic and Postsynaptic Mechanisms Underlying Synaptic Function and Memory. J. Neurosci. 2019, 39, 5986–6000. [Google Scholar] [CrossRef] [PubMed]
- Tropea, M.R.; Li Puma, D.D.; Melone, M.; Gulisano, W.; Arancio, O.; Grassi, C.; Conti, F.; Puzzo, D. Genetic deletion of alpha7 nicotinic acetylcholine receptors induces an age-dependent Alzheimer’s disease-like pathology. Prog. Neurobiol. 2021, 206, 102154. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rahman, M.S.; Uddin, M.J.; Mamum-Or-Rashid, A.N.M.; Pang, M.-G.; Rhim, H. Emerging risk of environmental factors: Insight mechanisms of Alzheimer’s diseases. Environ. Sci. Pollut. Res. 2020, 27, 44659–44672. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS β-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdes-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019, 216, 60–70. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nistico, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef] [PubMed]
- Habbas, S.; Santello, M.; Becker, D.; Stubbe, H.; Zappia, G.; Liaudet, N.; Federica; Kollias, G.; Fontana, A.; Christopher; et al. Neuroinflammatory TNFα Impairs Memory via Astrocyte Signaling. Cell 2015, 163, 1730–1741. [Google Scholar] [CrossRef]
- Sun, L.-L.; Yang, T.-Y.; Wei, N.-N.; Lu, W.; Jiao, W.-X.; Zhou, Q.-Q.; Miao, Y.-Z.; Gao, Q.; Wang, X.-T.; Sun, Q.; et al. Pharmacological characterization of JWX-A0108 as a novel type I positive allosteric modulator of α7 nAChR that can reverse acoustic gating deficits in a mouse prepulse inhibition model. Acta Pharmacol. Sin. 2019, 40, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Pabst, M.; Braganza, O.; Dannenberg, H.; Hu, W.; Pothmann, L.; Rosen, J.; Mody, I.; Karen; Deisseroth, K.; Albert; et al. Astrocyte Intermediaries of Septal Cholinergic Modulation in the Hippocampus. Neuron 2016, 90, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.M.; Zhang, S.L.; Wang, X.L.; Guan, Z.Z.; Qi, X.L. Expression levels of the alpha7 nicotinic acetylcholine receptor in the brains of patients with Alzheimer’s disease and their effect on synaptic proteins in SH-SY5Y cells. Mol. Med. Rep. 2020, 22, 2063–2075. [Google Scholar] [CrossRef]
- Guan, Z.Z.; Zhang, X.; Ravid, R.; Nordberg, A. Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer’s disease. J. Neurochem. 2000, 74, 237–243. [Google Scholar] [CrossRef]
- Nakaizumi, K.; Ouchi, Y.; Terada, T.; Yoshikawa, E.; Kakimoto, A.; Isobe, T.; Bunai, T.; Yokokura, M.; Suzuki, K.; Magata, Y. In vivo Depiction of alpha7 Nicotinic Receptor Loss for Cognitive Decline in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 61, 1355–1365. [Google Scholar] [CrossRef]
- Song, C.; Shi, J.; Xu, J.; Zhao, L.; Zhang, Y.; Huang, W.; Qiu, Y.; Zhang, R.; Chen, H.; Wang, H. Post-transcriptional regulation of alpha7 nAChR expression by miR-98-5p modulates cognition and neuroinflammation in an animal model of Alzheimer’s disease. FASEB J. 2021, 35, e21658. [Google Scholar] [CrossRef]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef]
- Tan, L.; Yu, J.T.; Tan, M.S.; Liu, Q.Y.; Wang, H.F.; Zhang, W.; Jiang, T.; Tan, L. Genome-wide serum microRNA expression profiling identifies serum biomarkers for Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 40, 1017–1027. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, L.; Wang, Y.; Huang, S. Integrated analysis of miRNA and mRNA expression in the blood of patients with Alzheimer’s disease. Mol. Med. Rep. 2020, 22, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, N.; Asle-Rousta, M.; Rahnema, M.; Amini, R. Morin attenuates memory deficits in a rat model of Alzheimer’s disease by ameliorating oxidative stress and neuroinflammation. Eur. J. Pharmacol. 2021, 910, 174506. [Google Scholar] [CrossRef] [PubMed]
- Halevi, S. The C.elegansric-3 gene is required for maturation of nicotinic acetylcholine receptors. EMBO J. 2002, 21, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Mizrachi, T.; Vaknin-Dembinsky, A.; Brenner, T.; Treinin, M. Neuroinflammation Modulation via alpha7 Nicotinic Acetylcholine Receptor and Its Chaperone, RIC-3. Molecules 2021, 26, 6139. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Jose; Lord, B.; Anthony, S.; Weston, D. Brain α7 Nicotinic Acetylcholine Receptor Assembly Requires NACHO. Neuron 2016, 89, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Matta, J.A.; Gu, S.; Davini, W.B.; Lord, B.; Siuda, E.R.; Harrington, A.W.; Bredt, D.S. NACHO Mediates Nicotinic Acetylcholine Receptor Function throughout the Brain. Cell Rep. 2017, 19, 688–696. [Google Scholar] [CrossRef]
- Wu, M.; Liu, C.Z.; Barrall, E.A.; Rissman, R.A.; Joiner, W.J. Unbalanced Regulation of α7 nAChRs by Ly6h and NACHO Contributes to Neurotoxicity in Alzheimer’s Disease. J. Neurosci. 2021, 41, 8461–8474. [Google Scholar] [CrossRef]
- Posadas, I.; Lopez-Hernandez, B.; Cena, V. Nicotinic Receptors in Neurodegeneration. Curr. Neuropharmacol. 2013, 11, 298–314. [Google Scholar] [CrossRef]
- Wilens, T.E.; Decker, M.W. Neuronal nicotinic receptor agonists for the treatment of attention-deficit/hyperactivity disorder: Focus on cognition. Biochem. Pharmacol. 2007, 74, 1212–1223. [Google Scholar] [CrossRef]
- Sanberg, P.R.; Silver, A.A.; Shytle, R.D.; Philipp, M.K.; Cahill, D.W.; Fogelson, H.M.; McConville, B.J. Nicotine for the treatment of Tourette’s syndrome. Pharmacol. Ther. 1997, 74, 21–25. [Google Scholar] [CrossRef]
- Philip, N.S.; Carpenter, L.L.; Tyrka, A.R.; Price, L.H. Nicotinic acetylcholine receptors and depression: A review of the preclinical and clinical literature. Psychopharmacology 2010, 212, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Saji, H.; Magata, Y.; Yamada, Y.; Tajima, K.; Yonekura, Y.; Konishi, J.; Ohmomo, Y.; Yokoyama, A. Synthesis of (S)-N-[methyl-11C]nicotine and its regional distribution in the mouse brain: A potential tracer for visualization of brain nicotinic receptors by positron emission tomography. Chem. Pharm. Bull. 1992, 40, 734–736. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iida, Y.; Ogawa, M.; Ueda, M.; Tominaga, A.; Kawashima, H.; Magata, Y.; Nishiyama, S.; Tsukada, H.; Mukai, T.; Saji, H. Evaluation of 5-(11)C-methyl-A-85380 as an imaging agent for PET investigations of brain nicotinic acetylcholine receptors. J. Nucl. Med. 2004, 45, 878–884. [Google Scholar] [PubMed]
- Horti, A.G.; Scheffel, U.; Koren, A.O.; Ravert, H.T.; Mathews, W.B.; Musachio, J.L.; Finley, P.A.; London, E.D.; Dannals, R.F. 2-[18F]Fluoro-A-85380, an in vivo tracer for the nicotinic acetylcholine receptors. Nucl. Med. Biol. 1998, 25, 599–603. [Google Scholar] [CrossRef]
- Valette, H.; Bottlaender, M.; Dolle, F.; Guenther, I.; Fuseau, C.; Coulon, C.; Ottaviani, M.; Crouzel, C. Imaging central nicotinic acetylcholine receptors in baboons with [18F]fluoro-A-85380. J. Nucl. Med. 1999, 40, 1374–1380. [Google Scholar] [PubMed]
- Doll, F.; Dolci, L.; Valette, H.; Hinnen, F.; Vaufrey, F.; Guenther, I.; Fuseau, C.; Coulon, C.; Bottlaender, M.; Crouzel, C. Synthesis and nicotinic acetylcholine receptor in vivo binding properties of 2-fluoro-3-[2(S)-2-azetidinylmethoxy]pyridine: A new positron emission tomography ligand for nicotinic receptors. J. Med. Chem. 1999, 42, 2251–2259. [Google Scholar] [CrossRef]
- Koren, A.O.; Horti, A.G.; Mukhin, A.G.; Gundisch, D.; Kimes, A.S.; Dannals, R.F.; London, E.D. 2-, 5-, and 6-Halo-3-(2(S)-azetidinylmethoxy)pyridines: Synthesis, affinity for nicotinic acetylcholine receptors, and molecular modeling. J. Med. Chem. 1998, 41, 3690–3698. [Google Scholar] [CrossRef]
- Wong, D.F.; Kuwabara, H.; Kim, J.; Brasic, J.R.; Chamroonrat, W.; Gao, Y.; Valentine, H.; Willis, W.; Mathur, A.; McCaul, M.E.; et al. PET imaging of high-affinity alpha4beta2 nicotinic acetylcholine receptors in humans with 18F-AZAN, a radioligand with optimal brain kinetics. J. Nucl. Med. 2013, 54, 1308–1314. [Google Scholar] [CrossRef]
- Kuwabara, H.; Wong, D.F.; Gao, Y.; Valentine, H.; Holt, D.P.; Ravert, H.T.; Dannals, R.F.; Horti, A.G. PET Imaging of nicotinic acetylcholine receptors in baboons with 18F-AZAN, a radioligand with improved brain kinetics. J. Nucl. Med. 2012, 53, 121–129. [Google Scholar] [CrossRef]
- Gao, Y.; Kuwabara, H.; Spivak, C.E.; Xiao, Y.; Kellar, K.; Ravert, H.T.; Kumar, A.; Alexander, M.; Hilton, J.; Wong, D.F.; et al. Discovery of (-)-7-methyl-2-exo-[3′-(6-[18F]fluoropyridin-2-yl)-5′-pyridinyl]-7-azabicyclo[2.2.1]heptane, a radiolabeled antagonist for cerebral nicotinic acetylcholine receptor (alpha4beta2-nAChR) with optimal positron emission tomography imaging properties. J. Med. Chem. 2008, 51, 4751–4764. [Google Scholar]
- Sattler, B.; Kranz, M.; Starke, A.; Wilke, S.; Donat, C.K.; Deuther-Conrad, W.; Patt, M.; Schildan, A.; Patt, J.; Smits, R.; et al. Internal dose assessment of (-)-18F-flubatine, comparing animal model datasets of mice and piglets with first-in-human results. J. Nucl. Med. 2014, 55, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Hillmer, A.T.; Esterlis, I.; Gallezot, J.D.; Bois, F.; Zheng, M.Q.; Nabulsi, N.; Lin, S.F.; Papke, R.L.; Huang, Y.; Sabri, O.; et al. Imaging of cerebral alpha4beta2* nicotinic acetylcholine receptors with (-)-[(18)F]Flubatine PET: Implementation of bolus plus constant infusion and sensitivity to acetylcholine in human brain. Neuroimage 2016, 141, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Deuther-Conrad, W.; Patt, J.T.; Lockman, P.R.; Allen, D.D.; Patt, M.; Schildan, A.; Ganapathy, V.; Steinbach, J.; Sabri, O.; Brust, P. Norchloro-fluoro-homoepibatidine (NCFHEB)—A promising radioligand for neuroimaging nicotinic acetylcholine receptors with PET. Eur. Neuropsychopharmacol. 2008, 18, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Brašić, J.R.; Cascella, N.; Kumar, A.; Zhou, Y.; Hilton, J.; Raymont, V.; Crabb, A.; Guevara, M.R.; Horti, A.G.; Wong, D.F. Positron emission tomography experience with 2-[18F]fluoro-3-(2(s)-azetidinylmethoxy)pyridine (2-[18F]fa) in the living human brain of smokers with paranoid schizophrenia. Synapse 2012, 66, 352–368. [Google Scholar] [CrossRef]
- Saji, H.; Ogawa, M.; Ueda, M.; Iida, Y.; Magata, Y.; Tominaga, A.; Kawashima, H.; Kitamura, Y.; Nakagawa, M.; Kiyono, Y.; et al. Evaluation of radioiodinated 5-iodo-3-(2(S)-azetidinylmethoxy)pyridine as a ligand for SPECT investigations of brain nicotinic acetylcholine receptors. Ann. Nucl. Med. 2002, 16, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Ichise, M.; van Dyck, C.H.; Zoghbi, S.S.; Tamagnan, G.; Mukhin, A.G.; Bozkurt, A.; Seneca, N.; Tipre, D.; DeNucci, C.C.; et al. Quantification of nicotinic acetylcholine receptors in human brain using [123I]5-I-A-85380 SPET. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Ueda, M.; Kawashima, H.; Arimitsu, K.; Yagi, Y.; Saji, H. Synthesis and biological evaluation of Tc-99m-cyclopentadienyltricarbonyl-technetium-labeled A-85380: An imaging probe for single-photon emission computed tomography investigation of nicotinic acetylcholine receptors in the brain. Bioorg. Med. Chem. 2019, 27, 2245–2252. [Google Scholar] [CrossRef]
- Mori, D.; Kimura, H.; Kawashima, H.; Yagi, Y.; Arimitsu, K.; Ono, M.; Saji, H. Development of (99m)Tc radiolabeled A85380 derivatives targeting cerebral nicotinic acetylcholine receptor: Novel radiopharmaceutical ligand (99m)Tc-A-YN-IDA-C4. Bioorg. Med. Chem. 2019, 27, 4200–4210. [Google Scholar] [CrossRef]
- Mathew, S.V.; Law, A.J.; Lipska, B.K.; Dávila-García, M.I.; Zamora, E.D.; Mitkus, S.N.; Vakkalanka, R.; Straub, R.E.; Weinberger, D.R.; Kleinman, J.E.; et al. α7 nicotinic acetylcholine receptor mRNA expression and binding in postmortem human brain are associated with genetic variation in neuregulin 1. Hum. Mol. Genet. 2007, 16, 2921–2932. [Google Scholar] [CrossRef][Green Version]
- Dani, J.A. Neuronal Nicotinic Acetylcholine Receptor Structure and Function and Response to Nicotine; Elsevier: Amsterdam, The Netherlands, 2015; pp. 3–19. [Google Scholar]
- Davies, A.R.; Hardick, D.J.; Blagbrough, I.S.; Potter, B.V.; Wolstenholme, A.J.; Wonnacott, S. Characterisation of the binding of [3H]methyllycaconitine: A new radioligand for labelling alpha 7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology 1999, 38, 679–690. [Google Scholar] [CrossRef]
- Toyohara, J.; Sakata, M.; Wu, J.; Ishikawa, M.; Oda, K.; Ishii, K.; Iyo, M.; Hashimoto, K.; Ishiwata, K. Preclinical and the first clinical studies on [11C]CHIBA-1001 for mapping alpha7 nicotinic receptors by positron emission tomography. Ann. Nucl. Med. 2009, 23, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Ettrup, A.; Mikkelsen, J.D.; Lehel, S.; Madsen, J.; Nielsen, E.O.; Palner, M.; Timmermann, D.B.; Peters, D.; Knudsen, G.M. 11C-NS14492 as a novel PET radioligand for imaging cerebral alpha7 nicotinic acetylcholine receptors: In vivo evaluation and drug occupancy measurements. J. Nucl. Med. 2011, 52, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Deuther-Conrad, W.; Fischer, S.; Hiller, A.; Nielsen, E.O.; Timmermann, D.B.; Steinbach, J.; Sabri, O.; Peters, D.; Brust, P. Molecular imaging of alpha7 nicotinic acetylcholine receptors: Design and evaluation of the potent radioligand [18F]NS10743. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Rotering, S.; Scheunemann, M.; Fischer, S.; Hiller, A.; Peters, D.; Deuther-Conrad, W.; Brust, P. Radiosynthesis and first evaluation in mice of [(18)F]NS14490 for molecular imaging of alpha7 nicotinic acetylcholine receptors. Bioorg. Med. Chem. 2013, 21, 2635–2642. [Google Scholar] [CrossRef]
- Ravert, H.T.; Dorff, P.; Foss, C.A.; Mease, R.C.; Fan, H.; Holmquist, C.R.; Phillips, E.; McCarthy, D.J.; Heys, J.R.; Holt, D.P.; et al. Radiochemical synthesis and in vivo evaluation of [18F]AZ11637326: An agonist probe for the alpha7 nicotinic acetylcholine receptor. Nucl. Med. Biol. 2013, 40, 731–739. [Google Scholar] [CrossRef]
- Ogawa, M.; Nishiyama, S.; Tsukada, H.; Hatano, K.; Fuchigami, T.; Yamaguchi, H.; Matsushima, Y.; Ito, K.; Magata, Y. Synthesis and evaluation of new imaging agent for central nicotinic acetylcholine receptor alpha7 subtype. Nucl. Med. Biol. 2010, 37, 347–355. [Google Scholar] [CrossRef]
- Wong, D.F.; Kuwabara, H.; Pomper, M.; Holt, D.P.; Brasic, J.R.; George, N.; Frolov, B.; Willis, W.; Gao, Y.; Valentine, H.; et al. Human Brain Imaging of α7 nAChR with [18F]ASEM: A New PET Radiotracer for Neuropsychiatry and Determination of Drug Occupancy. Mol. Imaging Biol. 2014, 16, 730–738. [Google Scholar] [CrossRef]
- Gao, Y.; Kellar, K.J.; Yasuda, R.P.; Tran, T.; Xiao, Y.; Dannals, R.F.; Horti, A.G. Derivatives of dibenzothiophene for positron emission tomography imaging of alpha7-nicotinic acetylcholine receptors. J. Med. Chem. 2013, 56, 7574–7589. [Google Scholar] [CrossRef]
- Hillmer, A.T.; Li, S.; Zheng, M.Q.; Scheunemann, M.; Lin, S.F.; Nabulsi, N.; Holden, D.; Pracitto, R.; Labaree, D.; Ropchan, J.; et al. PET imaging of alpha7 nicotinic acetylcholine receptors: A comparative study of [(18)F]ASEM and [(18)F]DBT-10 in nonhuman primates, and further evaluation of [(18)F]ASEM in humans. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1042–1050. [Google Scholar] [CrossRef]
- Gao, Y.; Mease, R.C.; Olson, T.T.; Kellar, K.J.; Dannals, R.F.; Pomper, M.G.; Horti, A.G. [(125)I]Iodo-ASEM, a specific in vivo radioligand for alpha7-nAChR. Nucl. Med. Biol. 2015, 42, 488–493. [Google Scholar] [CrossRef]
- Donat, C.K.; Hansen, H.H.; Hansen, H.D.; Mease, R.C.; Horti, A.G.; Pomper, M.G.; L’Estrade, E.T.; Herth, M.M.; Peters, D.; Knudsen, G.M.; et al. In Vitro and In Vivo Characterization of Dibenzothiophene Derivatives [(125)I]Iodo-ASEM and [(18)F]ASEM as Radiotracers of Homo- and Heteromeric alpha7 Nicotinic Acetylcholine Receptors. Molecules 2020, 25, 1425. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef] [PubMed]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef] [PubMed]
- Tangestani Fard, M.; Stough, C. A Review and Hypothesized Model of the Mechanisms That Underpin the Relationship Between Inflammation and Cognition in the Elderly. Front. Aging Neurosci. 2019, 11, 56. [Google Scholar] [CrossRef]
- Ahmad, M.A.; Kareem, O.; Khushtar, M.; Akbar, M.; Haque, M.R.; Iqubal, A.; Haider, M.F.; Pottoo, F.H.; Abdulla, F.S.; Al-Haidar, M.B.; et al. Neuroinflammation: A Potential Risk for Dementia. Int. J. Mol. Sci. 2022, 23, 616. [Google Scholar] [CrossRef]
- Wright, A.L.; Zinn, R.; Hohensinn, B.; Konen, L.M.; Beynon, S.B.; Tan, R.P.; Clark, I.A.; Abdipranoto, A.; Vissel, B. Neuroinflammation and Neuronal Loss Precede Aβ Plaque Deposition in the hAPP-J20 Mouse Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e59586. [Google Scholar] [CrossRef]
- Van Zeller, M.; Dias, D.; Sebastião, A.M.; Valente, C.A. NLRP3 Inflammasome: A Starring Role in Amyloid-β- and Tau-Driven Pathological Events in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 83, 939–961. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Mejias, N.H.; Martinez, C.C.; Stephens, M.E.; de Rivero Vaccari, J.P. Contribution of the inflammasome to inflammaging. J. Inflamm. 2018, 15, 23. [Google Scholar] [CrossRef]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- Griffin, W.S.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflamm. 2006, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; O’Banion, M.K.; Terwel, D.; Kummer, M.P. Neuroinflammatory processes in Alzheimer’s disease. J. Neural Transm. 2010, 117, 919–947. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Puxeddu, I.; Napoletano, S.; Scala, E.; Melillo, D.; Manocchio, S.; Angiolillo, A.; Migliorini, P.; Boraschi, D.; Vitale, E.; et al. Circulating levels of IL-1 family cytokines and receptors in Alzheimer’s disease: New markers of disease progression? J. Neuroinflamm. 2018, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.-P.; Rivest, S. Bone Marrow-Derived Microglia Play a Critical Role in Restricting Senile Plaque Formation in Alzheimer’s Disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Lamkanfi, M. Vishva Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 Mediates Pathological Effects of Microglia on Tau Phosphorylation and on Synaptophysin Synthesis in Cortical Neurons through a p38-MAPK Pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef]
- Chan, A.H.; Schroder, K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 2020, 217, jem.20190314. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Martin-Sanchez, F.; Gomez, A.I.; Martinez, C.M.; Amores-Iniesta, J.; Compan, V.; Barbera-Cremades, M.; Yague, J.; Ruiz-Ortiz, E.; Anton, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef]
- Prada, I.; Gabrielli, M.; Turola, E.; Iorio, A.; D’Arrigo, G.; Parolisi, R.; De Luca, M.; Pacifici, M.; Bastoni, M.; Lombardi, M.; et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018, 135, 529–550. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Vergara, C.; Houben, S.; Suain, V.; Yilmaz, Z.; De Decker, R.; Vanden Dries, V.; Boom, A.; Mansour, S.; Leroy, K.; Ando, K.; et al. Amyloid-beta pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019, 137, 397–412. [Google Scholar] [CrossRef]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-beta plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sun, L.; Hashioka, S.; Yu, S.; Schwab, C.; Okada, R.; Hayashi, Y.; McGeer, P.L.; Nakanishi, H. Differential pathways for interleukin-1β production activated by chromogranin A and amyloid β in microglia. Neurobiol. Aging 2013, 34, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Grehan, B.; Lynch, M.A. Glial Uptake of Amyloid Beta Induces NLRP3 Inflammasome Formation via Cathepsin-Dependent Degradation of NLRP10. Neuromol. Med. 2014, 16, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada Gonzalez, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e210. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e256. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Bottcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Thion, M.S.; Ginhoux, F.; Garel, S. Microglia and early brain development: An intimate journey. Science 2018, 362, 185–189. [Google Scholar] [CrossRef]
- Bian, Z.; Gong, Y.; Huang, T.; Lee, C.Z.W.; Bian, L.; Bai, Z.; Shi, H.; Zeng, Y.; Liu, C.; He, J.; et al. Deciphering human macrophage development at single-cell resolution. Nature 2020, 582, 571–576. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Liang, P.; Hou, B.; Lu, X.; Ma, Q.; Yu, X.; Han, S.; Peng, B.; Chen, T.; Liu, W.; et al. The effect of dipeptidyl peptidase IV on disease-associated microglia phenotypic transformation in epilepsy. J. Neuroinflamm. 2021, 18, 112. [Google Scholar] [CrossRef]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Moller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e386. [Google Scholar] [CrossRef]
- McQuade, A.; Kang, Y.J.; Hasselmann, J.; Jairaman, A.; Sotelo, A.; Coburn, M.; Shabestari, S.K.; Chadarevian, J.P.; Fote, G.; Tu, C.H.; et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020, 11, 5370. [Google Scholar] [CrossRef]
- Cagnin, A.; Kassiou, M.; Meikle, S.R.; Banati, R.B. Positron emission tomography imaging of neuroinflammation. Neurotherapeutics 2007, 4, 443–452. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapere, J.J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef]
- Braestrup, C.; Squires, R.F. Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H)diazepam binding. Proc. Natl. Acad. Sci. USA 1977, 74, 3805–3809. [Google Scholar] [CrossRef] [PubMed]
- Schoemaker, H.; Bliss, M.; Yamamura, H.I. Specific high-affinity saturable binding of [3H] R05-4864 to benzodiazepine binding sites in the rat cerebral cortex. Eur. J. Pharmacol. 1981, 71, 173–175. [Google Scholar] [CrossRef]
- Marangos, P.J.; Patel, J.; Boulenger, J.P.; Clark-Rosenberg, R. Characterization of peripheral-type benzodiazepine binding sites in brain using [3H]Ro 5-4864. Mol. Pharmacol. 1982, 22, 26–32. [Google Scholar] [PubMed]
- Lacapere, J.J.; Papadopoulos, V. Peripheral-type benzodiazepine receptor: Structure and function of a cholesterol-binding protein in steroid and bile acid biosynthesis. Steroids 2003, 68, 569–585. [Google Scholar] [CrossRef]
- Taketani, S.; Kohno, H.; Okuda, M.; Furukawa, T.; Tokunaga, R. Induction of peripheral-type benzodiazepine receptors during differentiation of mouse erythroleukemia cells. A possible involvement of these receptors in heme biosynthesis. J. Biol. Chem. 1994, 269, 7527–7531. [Google Scholar] [CrossRef]
- Wright, G.; Reichenbecher, V. The effects of superoxide and the peripheral benzodiazepine receptor ligands on the mitochondrial processing of manganese-dependent superoxide dismutase. Exp. Cell Res. 1999, 246, 443–450. [Google Scholar] [CrossRef]
- McEnery, M.W.; Snowman, A.M.; Trifiletti, R.R.; Snyder, S.H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA 1992, 89, 3170–3174. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Amri, H.; Boujrad, N.; Cascio, C.; Culty, M.; Garnier, M.; Hardwick, M.; Li, H.; Vidic, B.; Brown, A.S.; et al. Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids 1997, 62, 21–28. [Google Scholar] [CrossRef]
- Banati, R.B.; Middleton, R.J.; Chan, R.; Hatty, C.R.; Kam, W.W.; Quin, C.; Graeber, M.B.; Parmar, A.; Zahra, D.; Callaghan, P.; et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 2014, 5, 5452. [Google Scholar] [CrossRef]
- Tu, L.N.; Morohaku, K.; Manna, P.R.; Pelton, S.H.; Butler, W.R.; Stocco, D.M.; Selvaraj, V. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J. Biol. Chem. 2014, 289, 27444–27454. [Google Scholar] [CrossRef]
- Wang, H.; Zhai, K.; Xue, Y.; Yang, J.; Yang, Q.; Fu, Y.; Hu, Y.; Liu, F.; Wang, W.; Cui, L.; et al. Global Deletion of TSPO Does Not Affect the Viability and Gene Expression Profile. PLoS ONE 2016, 11, e0167307. [Google Scholar] [CrossRef] [PubMed]
- Morohaku, K.; Pelton, S.H.; Daugherty, D.J.; Butler, W.R.; Deng, W.; Selvaraj, V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology 2014, 155, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Sileikyte, J.; Blachly-Dyson, E.; Sewell, R.; Carpi, A.; Menabo, R.; Di Lisa, F.; Ricchelli, F.; Bernardi, P.; Forte, M. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)). J. Biol. Chem. 2014, 289, 13769–13781. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, V.; Stocco, D.M. The changing landscape in translocator protein (TSPO) function. Trends Endocrinol. Metab. 2015, 26, 341–348. [Google Scholar] [CrossRef]
- Zhao, A.H.; Tu, L.N.; Mukai, C.; Sirivelu, M.P.; Pillai, V.V.; Morohaku, K.; Cohen, R.; Selvaraj, V. Mitochondrial Translocator Protein (TSPO) Function Is Not Essential for Heme Biosynthesis. J. Biol. Chem. 2016, 291, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Imaizumi, M.; Zoghbi, S.S.; Fujimura, Y.; Farris, A.G.; Suhara, T.; Hong, J.; Pike, V.W.; Innis, R.B. Kinetic analysis in healthy humans of a novel positron emission tomography radioligand to image the peripheral benzodiazepine receptor, a potential biomarker for inflammation. Neuroimage 2008, 40, 43–52. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Fujita, M.; Fujimura, Y.; Kimura, N.; Jenko, K.J.; Kannan, P.; Hong, J.; Morse, C.L.; Zoghbi, S.S.; Gladding, R.L.; et al. Comparison of [(11)C]-(R)-PK 11195 and [(11)C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: Implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage 2010, 49, 2924–2932. [Google Scholar] [CrossRef]
- Owen, D.R.; Howell, O.W.; Tang, S.P.; Wells, L.A.; Bennacef, I.; Bergstrom, M.; Gunn, R.N.; Rabiner, E.A.; Wilkins, M.R.; Reynolds, R.; et al. Two binding sites for [3H]PBR28 in human brain: Implications for TSPO PET imaging of neuroinflammation. J. Cereb. Blood Flow Metab. 2010, 30, 1608–1618. [Google Scholar] [CrossRef]
- Owen, D.R.; Gunn, R.N.; Rabiner, E.A.; Bennacef, I.; Fujita, M.; Kreisl, W.C.; Innis, R.B.; Pike, V.W.; Reynolds, R.; Matthews, P.M.; et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J. Nucl. Med. 2011, 52, 24–32. [Google Scholar] [CrossRef]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18-kDa Translocator Protein (TSPO) Polymorphism Explains Differences in Binding Affinity of the PET Radioligand PBR28. J. Cereb. Blood Flow Metab. 2012, 32, 1–5. [Google Scholar] [CrossRef]
- Betlazar, C.; Harrison-Brown, M.; Middleton, R.J.; Banati, R.; Liu, G.J. Cellular Sources and Regional Variations in the Expression of the Neuroinflammatory Marker Translocator Protein (TSPO) in the Normal Brain. Int. J. Mol. Sci. 2018, 19, 2707. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.B.; Tsartsalis, S.; Ceyzeriat, K.; Fraser, B.H.; Gregoire, M.C.; Kovari, E.; Millet, P. Astrocytic TSPO Upregulation Appears Before Microglial TSPO in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 77, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Marks, J.D.; Das, S.; Hyman, B.T.; Serrano-Pozo, A. Characterization of the 18 kDa translocator protein (TSPO) expression in post-mortem normal and Alzheimer’s disease brains. Brain Pathol. 2020, 30, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Hillmer, A.T.; Holden, D.; Fowles, K.; Nabulsi, N.; West, B.L.; Carson, R.E.; Cosgrove, K.P. Microglial depletion and activation: A [(11)C]PBR28 PET study in nonhuman primates. EJNMMI Res. 2017, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Bader, S.; Wolf, L.; Milenkovic, V.M.; Gruber, M.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. Differential effects of TSPO ligands on mitochondrial function in mouse microglia cells. Psychoneuroendocrinology 2019, 106, 65–76. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Fan, J.; Zirkin, B. Translocator protein (18 kDa): An update on its function in steroidogenesis. J. Neuroendocrinol. 2018, 30, e12500. [Google Scholar] [CrossRef]
- Lejri, I.; Grimm, A.; Halle, F.; Abarghaz, M.; Klein, C.; Maitre, M.; Schmitt, M.; Bourguignon, J.J.; Mensah-Nyagan, A.G.; Bihel, F.; et al. TSPO Ligands Boost Mitochondrial Function and Pregnenolone Synthesis. J. Alzheimer’s Dis. 2019, 72, 1045–1058. [Google Scholar] [CrossRef]
- Owen, D.R.; Narayan, N.; Wells, L.; Healy, L.; Smyth, E.; Rabiner, E.A.; Galloway, D.; Williams, J.B.; Lehr, J.; Mandhair, H.; et al. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 2017, 37, 2679–2690. [Google Scholar] [CrossRef]
- Milenkovic, V.M.; Slim, D.; Bader, S.; Koch, V.; Heinl, E.S.; Alvarez-Carbonell, D.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. CRISPR-Cas9 Mediated TSPO Gene Knockout alters Respiration and Cellular Metabolism in Human Primary Microglia Cells. Int. J. Mol. Sci. 2019, 20, 3359. [Google Scholar] [CrossRef]
- Yao, R.; Pan, R.; Shang, C.; Li, X.; Cheng, J.; Xu, J.; Li, Y. Translocator Protein 18 kDa (TSPO) Deficiency Inhibits Microglial Activation and Impairs Mitochondrial Function. Front. Pharmacol. 2020, 11, 986. [Google Scholar] [CrossRef] [PubMed]
- Loth, M.K.; Guariglia, S.R.; Re, D.B.; Perez, J.; de Paiva, V.N.; Dziedzic, J.L.; Chambers, J.W.; Azzam, D.J.; Guilarte, T.R. A Novel Interaction of Translocator Protein 18 kDa (TSPO) with NADPH Oxidase in Microglia. Mol. Neurobiol. 2020, 57, 4467–4487. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Herb, M.; Schramm, M.; Langmann, T. The TSPO-NOX1 axis controls phagocyte-triggered pathological angiogenesis in the eye. Nat. Commun. 2020, 11, 2709. [Google Scholar] [CrossRef] [PubMed]
- Batarseh, A.; Papadopoulos, V. Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Mol. Cell. Endocrinol. 2010, 327, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef]
- Shimoyama, S.; Furukawa, T.; Ogata, Y.; Nikaido, Y.; Koga, K.; Sakamoto, Y.; Ueno, S.; Nakamura, K. Lipopolysaccharide induces mouse translocator protein (18 kDa) expression via the AP-1 complex in the microglial cell line, BV-2. PLoS ONE 2019, 14, e0222861. [Google Scholar] [CrossRef]
- Hannestad, J.; Gallezot, J.D.; Schafbauer, T.; Lim, K.; Kloczynski, T.; Morris, E.D.; Carson, R.E.; Ding, Y.S.; Cosgrove, K.P. Endotoxin-induced systemic inflammation activates microglia: [(1)(1)C]PBR28 positron emission tomography in nonhuman primates. Neuroimage 2012, 63, 232–239. [Google Scholar] [CrossRef]
- Woodcock, E.A.; Hillmer, A.T.; Sandiego, C.M.; Maruff, P.; Carson, R.E.; Cosgrove, K.P.; Pietrzak, R.H. Acute neuroimmune stimulation impairs verbal memory in adults: A PET brain imaging study. Brain. Behav. Immun. 2020, 91, 784–787. [Google Scholar] [CrossRef]
- Sandiego, C.M.; Gallezot, J.-D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.-F.; Matuskey, D.; Lee, J.-Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef]
- Tournier, B.B.; Tsartsalis, S.; Ceyzeriat, K.; Medina, Z.; Fraser, B.H.; Gregoire, M.C.; Kovari, E.; Millet, P. Fluorescence-activated cell sorting to reveal the cell origin of radioligand binding. J. Cereb. Blood Flow Metab. 2020, 40, 1242–1255. [Google Scholar] [CrossRef]
- Tournier, B.B.; Tsartsalis, S.; Ceyzériat, K.; Garibotto, V.; Millet, P. In Vivo TSPO Signal and Neuroinflammation in Alzheimer’s Disease. Cells 2020, 9, 1941. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.B.; Tsartsalis, S.; Rigaud, D.; Fossey, C.; Cailly, T.; Fabis, F.; Pham, T.; Gregoire, M.C.; Kovari, E.; Moulin-Sallanon, M.; et al. TSPO and amyloid deposits in sub-regions of the hippocampus in the 3xTgAD mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 121, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Hamelin, L.; Lagarde, J.; Dorothee, G.; Potier, M.C.; Corlier, F.; Kuhnast, B.; Caille, F.; Dubois, B.; Fillon, L.; Chupin, M.; et al. Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer’s disease. Brain 2018, 141, 1855–1870. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Okello, A.A.; Brooks, D.J.; Edison, P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain 2015, 138, 3685–3698. [Google Scholar] [CrossRef] [PubMed]
- Tuisku, J.; Plaven-Sigray, P.; Gaiser, E.C.; Airas, L.; Al-Abdulrasul, H.; Bruck, A.; Carson, R.E.; Chen, M.K.; Cosgrove, K.P.; Ekblad, L.; et al. Effects of age, BMI and sex on the glial cell marker TSPO—A multicentre [(11)C]PBR28 HRRT PET study. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2329–2338. [Google Scholar] [CrossRef]
- Lyoo, C.H.; Ikawa, M.; Liow, J.S.; Zoghbi, S.S.; Morse, C.L.; Pike, V.W.; Fujita, M.; Innis, R.B.; Kreisl, W.C. Cerebellum Can Serve As a Pseudo-Reference Region in Alzheimer Disease to Detect Neuroinflammation Measured with PET Radioligand Binding to Translocator Protein. J. Nucl. Med. 2015, 56, 701–706. [Google Scholar] [CrossRef]
- Golla, S.S.; Boellaard, R.; Oikonen, V.; Hoffmann, A.; van Berckel, B.N.; Windhorst, A.D.; Virta, J.; Haaparanta-Solin, M.; Luoto, P.; Savisto, N.; et al. Quantification of [18F]DPA-714 binding in the human brain: Initial studies in healthy controls and Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2015, 35, 766–772. [Google Scholar] [CrossRef]
- Gulyas, B.; Vas, A.; Toth, M.; Takano, A.; Varrone, A.; Cselenyi, Z.; Schain, M.; Mattsson, P.; Halldin, C. Age and disease related changes in the translocator protein (TSPO) system in the human brain: Positron emission tomography measurements with [11C]vinpocetine. Neuroimage 2011, 56, 1111–1121. [Google Scholar] [CrossRef]
- Schuitemaker, A.; Kropholler, M.A.; Boellaard, R.; van der Flier, W.M.; Kloet, R.W.; van der Doef, T.F.; Knol, D.L.; Windhorst, A.D.; Luurtsema, G.; Barkhof, F.; et al. Microglial activation in Alzheimer’s disease: An (R)-[(1)(1)C]PK11195 positron emission tomography study. Neurobiol. Aging 2013, 34, 128–136. [Google Scholar] [CrossRef]
- Zeineh, M.M.; Chen, Y.; Kitzler, H.H.; Hammond, R.; Vogel, H.; Rutt, B.K. Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol. Aging 2015, 36, 2483–2500. [Google Scholar] [CrossRef]
- Takata, K.; Kitamura, Y.; Yanagisawa, D.; Morikawa, S.; Morita, M.; Inubushi, T.; Tsuchiya, D.; Chishiro, S.; Saeki, M.; Taniguchi, T.; et al. Microglial transplantation increases amyloid-beta clearance in Alzheimer model rats. FEBS Lett. 2007, 581, 475–478. [Google Scholar] [CrossRef] [PubMed]
- McLarnon, J.G.; Ryu, J.K.; Walker, D.G.; Choi, H.B. Upregulated expression of purinergic P2X(7) receptor in Alzheimer disease and amyloid-beta peptide-treated microglia and in peptide-injected rat hippocampus. J. Neuropathol. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Francistiova, L.; Bianchi, C.; Di Lauro, C.; Sebastian-Serrano, A.; de Diego-Garcia, L.; Kobolak, J.; Dinnyes, A.; Diaz-Hernandez, M. The Role of P2X7 Receptor in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 94. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Won, S.M.; Gwag, B.J.; Lee, Y.B. Microglial P2X(7) receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Exp. Mol. Med. 2011, 43, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Dubyak, G.R. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am. J. Physiol. Cell Physiol. 2004, 286, C1100–C1108. [Google Scholar] [CrossRef]
- Chiozzi, P.; Sarti, A.C.; Sanz, J.M.; Giuliani, A.L.; Adinolfi, E.; Vultaggio-Poma, V.; Falzoni, S.; Di Virgilio, F. Amyloid beta-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci. Rep. 2019, 9, 6475. [Google Scholar] [CrossRef] [PubMed]
- Beaino, W.; Janssen, B.; Vugts, D.J.; de Vries, H.E.; Windhorst, A.D. Towards PET imaging of the dynamic phenotypes of microglia. Clin. Exp. Immunol. 2021, 206, 282–300. [Google Scholar] [CrossRef] [PubMed]
- Mildner, A.; Huang, H.; Radke, J.; Stenzel, W.; Priller, J. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 2017, 65, 375–387. [Google Scholar] [CrossRef]
- Villa, A.; Klein, B.; Janssen, B.; Pedragosa, J.; Pepe, G.; Zinnhardt, B.; Vugts, D.J.; Gelosa, P.; Sironi, L.; Beaino, W.; et al. Identification of new molecular targets for PET imaging of the microglial anti-inflammatory activation state. Theranostics 2018, 8, 5400–5418. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Bao, X.; Wang, R. Clinical PET Imaging of Microglial Activation: Implications for Microglial Therapeutics in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 314. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
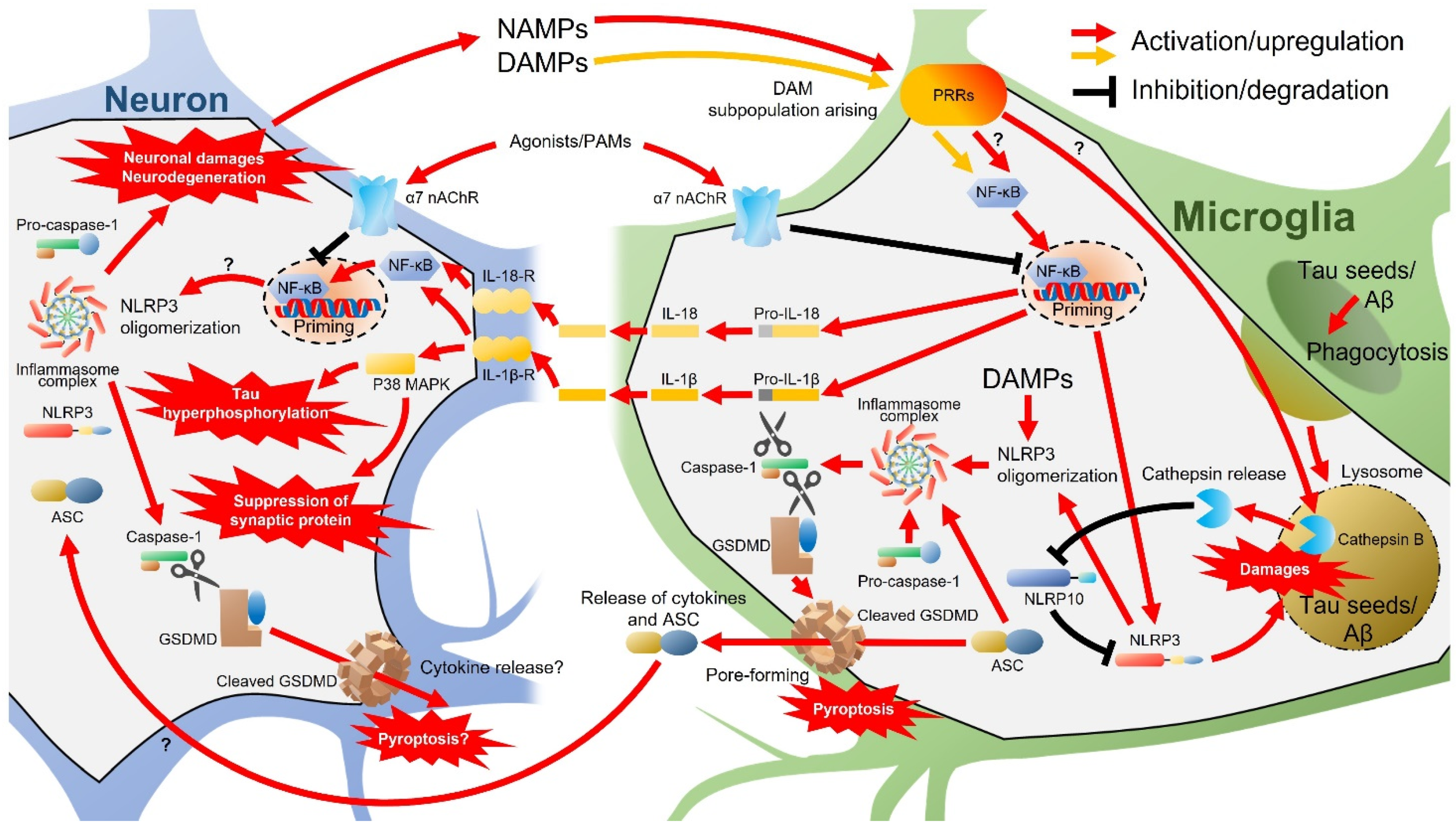{kind=link}
| Description | Agent | Action | Model | Ref. |
|---|---|---|---|---|
| Non-selective nAChR agonist | ACh | Activates PI3K/Akt, Nfr2/keap1 | Primary cultured mouse hippocampal neurons treated with Aβ25-35 | [40] |
| Activates Erk1/2 | AD model mice (3xTgAD) | [41] | ||
| Inhibits MAPKs (p38 MAPK, JNK) | Mice intrahippocampally-injected with Aβ1–42 | [42] | ||
| Inhibits phosphorylation of p44/42 and p38 MAPKs → Suppresses TNF-α | Primary cultured mouse microglia treated with LPS | [23] | ||
| Suppress iNOS, TNF-α, IL-1β → Restores IGF | Primary cultured mouse microglia treated with LPS | [43] | ||
| Selective α7 nAChR agonist | AR-R17779 | Reverses the pro-inflammatory phenotype | Primary cultured fetal sheep astrocytes | [44] |
| PNU-282987 | Inhibits Erk → Restores 5-HT1A, 2C → Improves anxiety and depressive-like behaviors | Aβ-injected mice | [45] | |
| Activates CaM-CaMKII-CREB → Improves learning and memory | AD model mice (APPswe/PSldE9) | [46] | ||
| Selective α7 nAChR partial agonist | A582941 | Increases Erk1/2, MAPKs, Arc → Behavior: pro-cognitive activity | Rats | [47] |
| DMXBA (GST-21) | Promotes microglial Aβ phagocytosis → Improves brain Aβ burden and memory Dysfunction → Suppresses γ-secretase activity | Primary cultured rat microglia Human neuroblastoma SH-SY5Y cells AD model mice (APdE9) | [48] | |
| Inhibits NF-κB → Suppresses IL-6 and TNF-α Activates Nfr2 → Increases HO-1, TXNRD1, NQO1 | Primary cultured mouse astrocytes treated with LPS | [49] | ||
| Selective α7 nAChR antagonist | α-bungarotoxin | Enhances the inflammatory phenotype | Primary cultured fetal sheep astrocytes | [44] |
| Type I PAM for α7 nAChR | CCMI | Increases Erk1/2, MAPKs, Arc → Behavior: pro-cognitive activity | Rats | [47] |
| JWX-A0108 | Inhibits NF-κB → Suppresses TNF-α, IL-1β, IL-6 | AD model mice (APP/PS1) | [50] | |
| Type II PAM for α7 nAChR | PNU-120596 | Increases: BDNF → Behavior pro-cognitive activity | Rats | [47] |
| Ago-PAM for α7 nAChR | GAT107 | Suppresses peripheral immune reactions, neuroinflammation | EAE mice | [51] |
| AChE inhibitor | Galantamine | Activates JNK → Increases α7 nAChRs Inhibits Akt → Induces autophagy → Promotes Aβ sequestration | Human neuroblastoma SH-SY5Y cells | [52] |
| Enhances nAChR sensitivity to choline → Activates CaM-CaMKII and CaM- Rac1-WAVE signaling → Promotes microglial Aβ phagocytosis | Primary cultured rat microglia AD model mice (APdE9) | [53] | ||
| Simultaneous stimulation Selective α7 nAChR agonist Selective σ1 receptor agonist | PHA-543613 PRE-084 | Modulates glial cells → Increases ACh by σ1-R stimulation | 6-OHDA rat model of PD | [54,55] |
| Simultaneous stimulation Selective α7 nAChR agonist Selective α4β2 nAChR agonist | PNU-282987 RJR-2403 oxalate | Inhibits dephosphorylation of AMPAR GluA1 subunit → Reduces AMPARs | Primary cultured mouse hippocampal neurons treated with Aβ1–42 oligomers | [56] |
| NAD-dependent deacetylase | SIRT1 | Activates Erk1/2 → Increases α7 nAChRs | AD model mice (APdE9) Human neuroblastoma SH-SY5Y cells | [57] |
| Effect | Agent | Action | Ref. |
|---|---|---|---|
| Downregulation of α7 nAChR | miR-98-5p | Negatively regulates the expression of α7 nAChRs | [80] |
| Upregulation of α7 nAChR | SIRT1 | Activates the Erk1/2 signaling pathway | [57] |
| Galantamine | Activates JNK signaling | [52] | |
| SP600125 | Inhibits JNK signaling | [42] | |
| SB202190 | Inhibits p38 MAPK signaling | [42] | |
| Morin | Restores decreased α7 nAChR mRNA expression | [84] | |
| RIC-3 | Promotes functional assembly of α7 nAChRs | [85,86] | |
| NACHO | Promotes functional assembly of α7 nAChRs | [87,88] | |
| Ly6h | Promotes functional assembly of α7 nAChRs | [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takata, K.; Kimura, H.; Yanagisawa, D.; Harada, K.; Nishimura, K.; Kitamura, Y.; Shimohama, S.; Tooyama, I. Nicotinic Acetylcholine Receptors and Microglia as Therapeutic and Imaging Targets in Alzheimer’s Disease. Molecules 2022, 27, 2780. https://doi.org/10.3390/molecules27092780
Takata K, Kimura H, Yanagisawa D, Harada K, Nishimura K, Kitamura Y, Shimohama S, Tooyama I. Nicotinic Acetylcholine Receptors and Microglia as Therapeutic and Imaging Targets in Alzheimer’s Disease. Molecules. 2022; 27(9):2780. https://doi.org/10.3390/molecules27092780
Chicago/Turabian StyleTakata, Kazuyuki, Hiroyuki Kimura, Daijiro Yanagisawa, Koki Harada, Kaneyasu Nishimura, Yoshihisa Kitamura, Shun Shimohama, and Ikuo Tooyama. 2022. "Nicotinic Acetylcholine Receptors and Microglia as Therapeutic and Imaging Targets in Alzheimer’s Disease" Molecules 27, no. 9: 2780. https://doi.org/10.3390/molecules27092780
APA StyleTakata, K., Kimura, H., Yanagisawa, D., Harada, K., Nishimura, K., Kitamura, Y., Shimohama, S., & Tooyama, I. (2022). Nicotinic Acetylcholine Receptors and Microglia as Therapeutic and Imaging Targets in Alzheimer’s Disease. Molecules, 27(9), 2780. https://doi.org/10.3390/molecules27092780