
Small Molecules as Modulators of Voltage-Gated Calcium Channels in Neurological Disorders: State of the Art and Perspectives


Abstract
1. Introduction
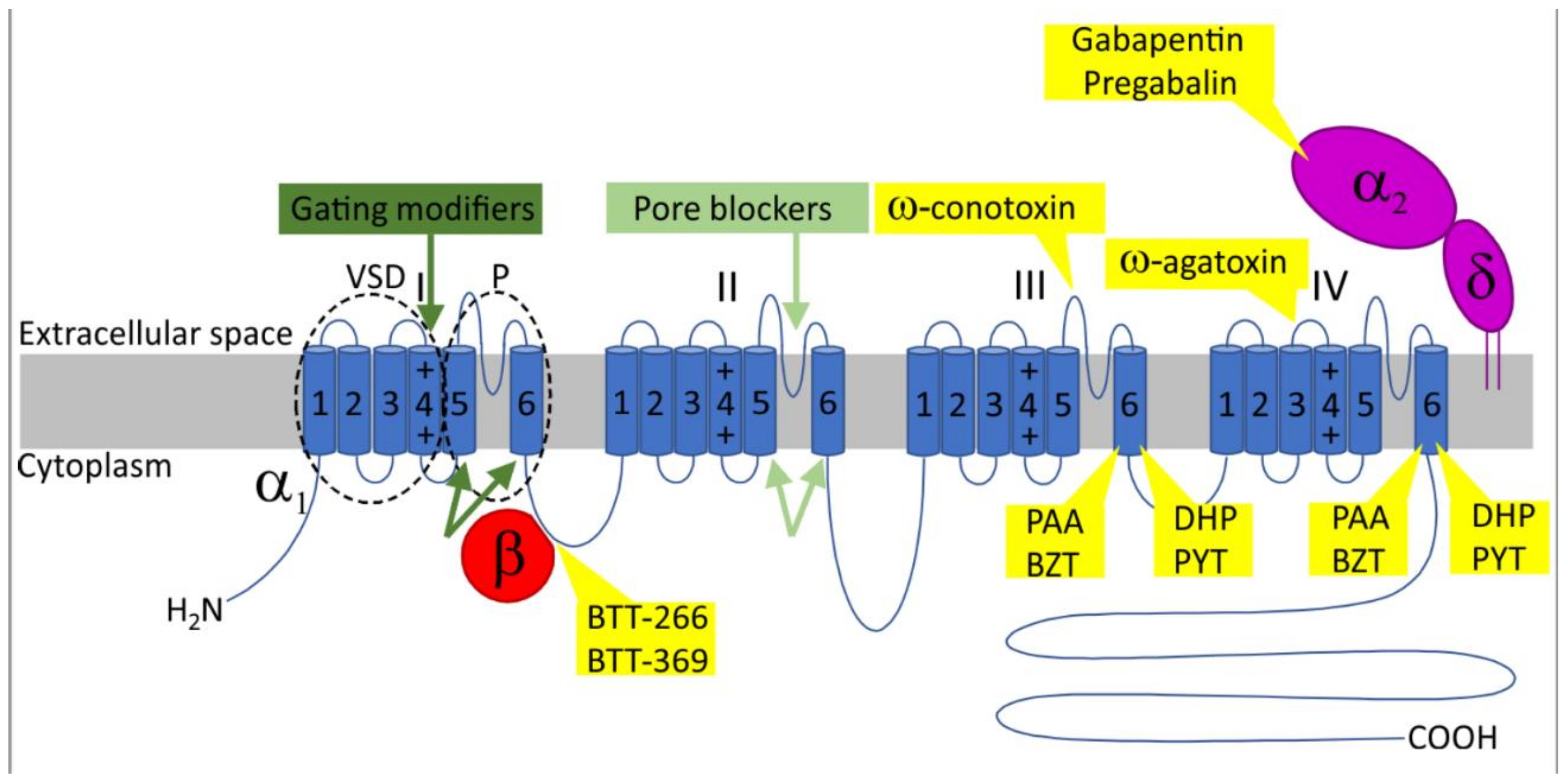
2. Voltage-Gated Calcium Channels
3. Physiological Roles of VGCCs in the Nervous System
4. L-type VGCCs in Psychiatric Disorders
5. VGCC Inhibitors in the Treatment of Parkinson’s Disease
6. The Potential of Pyrimidine-2,4,6-Triones (PYT) as CaV1.3 Selective Inhibitors
7. VGCCs Inhibitors in Pain Treatment
8. VGCCs in Seizure Disorders
9. VGCCs in Migraine
10. VGCCs in the Aging Brain
11. Summary
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.B. Channelopathies. Korean J. Pediatr. 2014, 57, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed]
- Heyes, S.; Pratt, W.S.; Rees, E.; Dahimene, S.; Ferron, L.; Owen, M.J.; Dolphin, A.C. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog. Neurobiol. 2015, 134, 36–54. [Google Scholar] [CrossRef]
- Flucher, B.E. Skeletal muscle CaV1.1 channelopathies. Pflugers Arch. 2020, 472, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Ablinger, C.; Geisler, S.M.; Stanika, R.I.; Klein, C.T.; Obermair, G.J. Neuronal alpha2delta proteins and brain disorders. Pflugers Arch. 2020, 472, 845–863. [Google Scholar] [CrossRef] [PubMed]
- Marcantoni, A.; Calorio, C.; Hidisoglu, E.; Chiantia, G.; Carbone, E. Cav1.2 channelopathies causing autism: New hallmarks on Timothy syndrome. Pflugers Arch. 2020, 472, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Discov. 2016, 15, 19–34. [Google Scholar] [CrossRef]
- Striessnig, J. Voltage-Gated Ca(2+)-Channel alpha1-Subunit de novo Missense Mutations: Gain or Loss of Function—Implications for Potential Therapies. Front. Synaptic Neurosci. 2021, 13, 634760. [Google Scholar] [CrossRef] [PubMed]
- Kabir, Z.D.; Martinez-Rivera, A.; Rajadhyaksha, A.M. From Gene to Behavior: L-Type Calcium Channel Mechanisms Underlying Neuropsychiatric Symptoms. Neurotherapeutics 2017, 14, 588–613. [Google Scholar] [CrossRef] [PubMed]
- Lory, P.; Nicole, S.; Monteil, A. Neuronal Cav3 channelopathies: Recent progress and perspectives. Pflugers Arch. 2020, 472, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Zamponi, G.W. Genetic T-type calcium channelopathies. J. Med. Genet. 2020, 57, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C. Voltage-gated calcium channels: Their discovery, function and importance as drug targets. Brain Neurosci. Adv. 2018, 2, 2398212818794805. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.W.; Lipscombe, D.; Madison, D.V.; Bley, K.R.; Fox, A.P. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988, 11, 431–438. [Google Scholar] [CrossRef]
- Nowycky, M.C.; Fox, A.P.; Tsien, R.W. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 1985, 316, 440–443. [Google Scholar] [CrossRef]
- Carbone, E.; Lux, H.D. A low voltage-activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature 1984, 310, 501–502. [Google Scholar] [CrossRef]
- Boland, L.M.; Morrill, J.A.; Bean, B.P. Omega-Conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. J. Neurosci. 1994, 14, 5011–5027. [Google Scholar] [CrossRef] [PubMed]
- Plummer, M.R.; Logothetis, D.E.; Hess, P. Elementary properties and pharmacological sensitivities of calcium channels in mammalian peripheral neurons. Neuron 1989, 2, 1453–1463. [Google Scholar] [CrossRef]
- Mintz, I.M.; Venema, V.J.; Swiderek, K.M.; Lee, T.D.; Bean, B.P.; Adams, M.E. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature 1992, 355, 827–829. [Google Scholar] [CrossRef]
- Llinas, R.; Sugimori, M.; Lin, J.W.; Cherksey, B. Blocking and isolation of a calcium channel from neurons in mammals and cephalopods utilizing a toxin fraction (FTX) from funnel-web spider poison. Proc. Natl. Acad. Sci. USA 1989, 86, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Randall, A.; Tsien, R.W. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J. Neurosci. 1995, 15, 2995–3012. [Google Scholar] [CrossRef]
- Newcomb, R.; Szoke, B.; Palma, A.; Wang, G.; Chen, X.; Hopkins, W.; Cong, R.; Miller, J.; Urge, L.; Tarczy-Hornoch, K.; et al. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry 1998, 37, 15353–15362. [Google Scholar] [CrossRef] [PubMed]
- Kimm, T.; Bean, B.P. Inhibition of A-type potassium current by the peptide toxin SNX-482. J. Neurosci. 2014, 34, 9182–9189. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Decher, N.; Kumar, P.; Sachse, F.B.; Beggs, A.H.; Sanguinetti, M.C.; Keating, M.T. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 8089–8096; discussion 8086–8088. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Yarotskyy, V.; Gao, G.; Du, L.; Ganapathi, S.B.; Peterson, B.Z.; Elmslie, K.S. Roscovitine binds to novel L-channel (CaV1.2) sites that separately affect activation and inactivation. J. Biol. Chem. 2010, 285, 43–53. [Google Scholar] [CrossRef]
- Yarotskyy, V.; Elmslie, K.S. Roscovitine, a cyclin-dependent kinase inhibitor, affects several gating mechanisms to inhibit cardiac L-type (Ca(V)1.2) calcium channels. Br. J. Pharmacol. 2007, 152, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.P.; Portmann, T.; Voineagu, I.; Yazawa, M.; Shcheglovitov, A.; Pasca, A.M.; Cord, B.; Palmer, T.D.; Chikahisa, S.; Nishino, S.; et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011, 17, 1657–1662. [Google Scholar] [CrossRef]
- Ferreira, M.A.; O’Donovan, M.C.; Meng, Y.A.; Jones, I.R.; Ruderfer, D.M.; Jones, L.; Fan, J.; Kirov, G.; Perlis, R.H.; Green, E.K.; et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 2008, 40, 1056–1058. [Google Scholar] [CrossRef] [PubMed]
- Sklar, P.; Smoller, J.W.; Fan, J.; Ferreira, M.A.; Perlis, R.H.; Chambert, K.; Nimgaonkar, V.L.; McQueen, M.B.; Faraone, S.V.; Kirby, A.; et al. Whole-genome association study of bipolar disorder. Mol. Psychiatry 2008, 13, 558–569. [Google Scholar] [CrossRef]
- Casamassima, F.; Huang, J.; Fava, M.; Sachs, G.S.; Smoller, J.W.; Cassano, G.B.; Lattanzi, L.; Fagerness, J.; Stange, J.P.; Perlis, R.H. Phenotypic effects of a bipolar liability gene among individuals with major depressive disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Green, E.K.; Grozeva, D.; Jones, I.; Jones, L.; Kirov, G.; Caesar, S.; Gordon-Smith, K.; Fraser, C.; Forty, L.; Russell, E.; et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol. Psychiatry 2010, 15, 1016–1022. [Google Scholar] [CrossRef]
- Shi, J.; Potash, J.B.; Knowles, J.A.; Weissman, M.M.; Coryell, W.; Scheftner, W.A.; Lawson, W.B.; DePaulo, J.R., Jr.; Gejman, P.V.; Sanders, A.R.; et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol. Psychiatry 2011, 16, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Nyegaard, M.; Demontis, D.; Foldager, L.; Hedemand, A.; Flint, T.J.; Sorensen, K.M.; Andersen, P.S.; Nordentoft, M.; Werge, T.; Pedersen, C.B.; et al. CACNA1C (rs1006737) is associated with schizophrenia. Mol. Psychiatry 2010, 15, 119–121. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 2013, 381, 1371–1379. [Google Scholar] [CrossRef]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Hori, H.; Yamamoto, N.; Fujii, T.; Teraishi, T.; Sasayama, D.; Matsuo, J.; Kawamoto, Y.; Kinoshita, Y.; Ota, M.; Hattori, K.; et al. Effects of the CACNA1C risk allele on neurocognition in patients with schizophrenia and healthy individuals. Sci. Rep. 2012, 2, 634. [Google Scholar] [CrossRef]
- Lencz, T.; Malhotra, A.K. Targeting the schizophrenia genome: A fast track strategy from GWAS to clinic. Mol. Psychiatry 2015, 20, 820–826. [Google Scholar] [CrossRef]
- Krzyzewska, I.M.; Ensink, J.B.M.; Nawijn, L.; Mul, A.N.; Koch, S.B.; Venema, A.; Shankar, V.; Frijling, J.L.; Veltman, D.J.; Lindauer, R.J.L.; et al. Genetic variant in CACNA1C is associated with PTSD in traumatized police officers. Eur. J. Hum. Genet. 2018, 26, 247–257. [Google Scholar] [CrossRef]
- Bavley, C.C.; Kabir, Z.D.; Walsh, A.P.; Kosovsky, M.; Hackett, J.; Sun, H.; Vazquez-Rosa, E.; Cintron-Perez, C.J.; Miller, E.; Koh, Y.; et al. Dopamine D1R-neuron cacna1c deficiency: A new model of extinction therapy-resistant post-traumatic stress. Mol. Psychiatry 2021, 26, 2286–2298. [Google Scholar] [CrossRef]
- Baig, S.M.; Koschak, A.; Lieb, A.; Gebhart, M.; Dafinger, C.; Nurnberg, G.; Ali, A.; Ahmad, I.; Sinnegger-Brauns, M.J.; Brandt, N.; et al. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 2011, 14, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Platzer, J.; Engel, J.; Schrott-Fischer, A.; Stephan, K.; Bova, S.; Chen, H.; Zheng, H.; Striessnig, J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 2000, 102, 89–97. [Google Scholar] [CrossRef]
- Ortner, N.J.; Kaserer, T.; Copeland, J.N.; Striessnig, J. De novo CACNA1D Ca(2+) channelopathies: Clinical phenotypes and molecular mechanism. Pflugers Arch. 2020, 472, 755–773. [Google Scholar] [CrossRef]
- Ament, S.A.; Szelinger, S.; Glusman, G.; Ashworth, J.; Hou, L.; Akula, N.; Shekhtman, T.; Badner, J.A.; Brunkow, M.E.; Mauldin, D.E.; et al. Rare variants in neuronal excitability genes influence risk for bipolar disorder. Proc. Natl. Acad. Sci. USA 2015, 112, 3576–3581. [Google Scholar] [CrossRef]
- Ross, J.; Gedvilaite, E.; Badner, J.A.; Erdman, C.; Baird, L.; Matsunami, N.; Leppert, M.; Xing, J.; Byerley, W. A Rare Variant in CACNA1D Segregates with 7 Bipolar I Disorder Cases in a Large Pedigree. Mol. Neuropsychiatry 2016, 2, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, L.; Kilicarslan, I.; Netzer, M.; Babai, N.; Seitter, H.; Koschak, A. Function of cone and cone-related pathways in CaV1.4 IT mice. Sci. Rep. 2021, 11, 2732. [Google Scholar] [CrossRef] [PubMed]
- Lodha, N.; Loucks, C.M.; Beaulieu, C.; Parboosingh, J.S.; Bech-Hansen, N.T. Congenital stationary night blindness: Mutation update and clinical variability. Adv. Exp. Med. Biol. 2012, 723, 371–379. [Google Scholar]
- Inagaki, A.; Frank, C.A.; Usachev, Y.M.; Benveniste, M.; Lee, A. Pharmacological correction of gating defects in the voltage-gated Ca(v)2.1 Ca(2)(+) channel due to a familial hemiplegic migraine mutation. Neuron 2014, 81, 91–102. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tottene, A.; Pivotto, F.; Fellin, T.; Cesetti, T.; van den Maagdenberg, A.M.; Pietrobon, D. Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J. Biol. Chem. 2005, 280, 17678–17686. [Google Scholar] [CrossRef]
- Waxman, S.G.; Zamponi, G.W. Regulating excitability of peripheral afferents: Emerging ion channel targets. Nat. Neurosci. 2014, 17, 153–163. [Google Scholar] [CrossRef]
- Marger, F.; Gelot, A.; Alloui, A.; Matricon, J.; Ferrer, J.F.; Barrere, C.; Pizzoccaro, A.; Muller, E.; Nargeot, J.; Snutch, T.P.; et al. T-type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 11268–11273. [Google Scholar] [CrossRef]
- Cizkova, D.; Marsala, J.; Lukacova, N.; Marsala, M.; Jergova, S.; Orendacova, J.; Yaksh, T.L. Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp. Brain Res. 2002, 147, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Jagodic, M.M.; Pathirathna, S.; Joksovic, P.M.; Lee, W.; Nelson, M.T.; Naik, A.K.; Su, P.; Jevtovic-Todorovic, V.; Todorovic, S.M. Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J. Neurophysiol. 2008, 99, 3151–3156. [Google Scholar] [CrossRef]
- Jiang, Y.Q.; Andrade, A.; Lipscombe, D. Spinal morphine but not ziconotide or gabapentin analgesia is affected by alternative splicing of voltage-gated calcium channel CaV2.2 pre-mRNA. Mol. Pain 2013, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Tedford, H.W.; Zamponi, G.W. Direct G protein modulation of Cav2 calcium channels. Pharmacol. Rev. 2006, 58, 837–862. [Google Scholar] [CrossRef]
- Bell, T.J.; Thaler, C.; Castiglioni, A.J.; Helton, T.D.; Lipscombe, D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron 2004, 41, 127–138. [Google Scholar] [CrossRef]
- Altier, C.; Dale, C.S.; Kisilevsky, A.E.; Chapman, K.; Castiglioni, A.J.; Matthews, E.A.; Evans, R.M.; Dickenson, A.H.; Lipscombe, D.; Vergnolle, N.; et al. Differential role of N-type calcium channel splice isoforms in pain. J. Neurosci. 2007, 27, 6363–6373. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Rostro, M.; Ramgoolam, K.; Pratt, W.S.; Kulik, A.; Dolphin, A.C. Ablation of alpha2delta-1 inhibits cell-surface trafficking of endogenous N-type calcium channels in the pain pathway in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E12043–E12052. [Google Scholar] [CrossRef]
- Luo, Z.D.; Chaplan, S.R.; Higuera, E.S.; Sorkin, L.S.; Stauderman, K.A.; Williams, M.E.; Yaksh, T.L. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J. Neurosci. 2001, 21, 1868–1875. [Google Scholar] [CrossRef]
- Bauer, C.S.; Nieto-Rostro, M.; Rahman, W.; Tran-Van-Minh, A.; Ferron, L.; Douglas, L.; Kadurin, I.; Sri Ranjan, Y.; Fernandez-Alacid, L.; Millar, N.S.; et al. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J. Neurosci. 2009, 29, 4076–4088. [Google Scholar] [CrossRef]
- Iyer, A.; Marson, A. Pharmacotherapy of focal epilepsy. Expert Opin. Pharm. 2014, 15, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, A.H.; McNaughton, N.C.; Pereverzev, A.; Schneider, T.; Randall, A.D. Actions of sipatrigine, 202W92 and lamotrigine on R-type and T-type Ca2+ channel currents. Eur. J. Pharmacol. 2003, 467, 77–80. [Google Scholar] [CrossRef]
- Dibue, M.; Kamp, M.A.; Alpdogan, S.; Tevoufouet, E.E.; Neiss, W.F.; Hescheler, J.; Schneider, T. Cav 2.3 (R-type) calcium channels are critical for mediating anticonvulsive and neuroprotective properties of lamotrigine in vivo. Epilepsia 2013, 54, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Kuzmiski, J.B.; Barr, W.; Zamponi, G.W.; MacVicar, B.A. Topiramate inhibits the initiation of plateau potentials in CA1 neurons by depressing R-type calcium channels. Epilepsia 2005, 46, 481–489. [Google Scholar] [CrossRef]
- Ernst, W.L.; Zhang, Y.; Yoo, J.W.; Ernst, S.J.; Noebels, J.L. Genetic enhancement of thalamocortical network activity by elevating alpha 1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J. Neurosci. 2009, 29, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Talley, E.M.; Solorzano, G.; Depaulis, A.; Perez-Reyes, E.; Bayliss, D.A. Low-voltage-activated calcium channel subunit expression in a genetic model of absence epilepsy in the rat. Brain Res. Mol. Brain Res. 2000, 75, 159–165. [Google Scholar] [CrossRef]
- Powell, K.L.; Cain, S.M.; Ng, C.; Sirdesai, S.; David, L.S.; Kyi, M.; Garcia, E.; Tyson, J.R.; Reid, C.A.; Bahlo, M.; et al. A Cav3.2 T-type calcium channel point mutation has splice-variant-specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J. Neurosci. 2009, 29, 371–380. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Lory, P.; Perez-Reyes, E. Role of voltage-gated calcium channels in epilepsy. Pflugers Arch. 2010, 460, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Khosravani, H.; Varela, D.; Bladen, C.; Williams, T.C.; Newman, M.R.; Scheffer, I.E.; Berkovic, S.F.; Mulley, J.C.; Zamponi, G.W. Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann. Neurol. 2007, 62, 560–568. [Google Scholar] [CrossRef]
- Vitko, I.; Bidaud, I.; Arias, J.M.; Mezghrani, A.; Lory, P.; Perez-Reyes, E. The I-II loop controls plasma membrane expression and gating of Ca(v)3.2 T-type Ca2+ channels: A paradigm for childhood absence epilepsy mutations. J. Neurosci. 2007, 27, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Khosravani, H.; Altier, C.; Simms, B.; Hamming, K.S.; Snutch, T.P.; Mezeyova, J.; McRory, J.E.; Zamponi, G.W. Gating effects of mutations in the Cav3.2 T-type calcium channel associated with childhood absence epilepsy. J. Biol. Chem. 2004, 279, 9681–9684. [Google Scholar] [CrossRef]
- Splawski, I.; Yoo, D.S.; Stotz, S.C.; Cherry, A.; Clapham, D.E.; Keating, M.T. CACNA1H mutations in Autism. spectrum disorders. J. Biol. Chem. 2006, 281, 22085–22091. [Google Scholar] [CrossRef] [PubMed]
- El Ghaleb, Y.; Schneeberger, P.E.; Fernandez-Quintero, M.L.; Geisler, S.M.; Pelizzari, S.; Polstra, A.M.; van Hagen, J.M.; Denecke, J.; Campiglio, M.; Liedl, K.R.; et al. CACNA1I gain-of-function mutations differentially affect channel gating and cause neurodevelopmental disorders. Brain 2021, 144, 2092–2106. [Google Scholar] [CrossRef]
- Dolphin, A.C. Voltage-gated calcium channels and their auxiliary subunits: Physiology and pathophysiology and pharmacology. J. Physiol. 2016, 594, 5369–5390. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C. Calcium channel auxiliary alpha2delta and beta subunits: Trafficking and one step beyond. Nat. Rev. Neurosci. 2012, 13, 542–555. [Google Scholar] [CrossRef]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of voltage-gated calcium channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef]
- Dolphin, A.C.; Lee, A. Presynaptic calcium channels: Specialized control of synaptic neurotransmitter release. Nat. Rev. Neurosci. 2020, 21, 213–229. [Google Scholar] [CrossRef]
- Dai, S.; Hall, D.D.; Hell, J.W. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 2009, 89, 411–452. [Google Scholar] [CrossRef]
- Harvey, R.D.; Hell, J.W. CaV1.2 signaling complexes in the heart. J. Mol. Cell Cardiol. 2013, 58, 143–152. [Google Scholar] [CrossRef]
- Patriarchi, T.; Buonarati, O.R.; Hell, J.W. Postsynaptic localization and regulation of AMPA receptors and Cav1.2 by beta2 adrenergic receptor/PKA and Ca(2+)/CaMKII signaling. EMBO J. 2018, 37, e99771. [Google Scholar] [CrossRef] [PubMed]
- Wild, A.R.; Dell’Acqua, M.L. Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol. Ther. 2018, 185, 99–121. [Google Scholar] [CrossRef]
- Calin-Jageman, I.; Lee, A. Ca(v)1 L-type Ca2+ channel signaling complexes in neurons. J. Neurochem. 2008, 105, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Nanou, E.; Catterall, W.A. Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron 2018, 98, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Hell, J.W.; Westenbroek, R.E.; Warner, C.; Ahlijanian, M.K.; Prystay, W.; Gilbert, M.M.; Snutch, T.P.; Catterall, W.A. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell Biol. 1993, 123, 949–962. [Google Scholar] [CrossRef]
- Wild, A.R.; Sinnen, B.L.; Dittmer, P.J.; Kennedy, M.J.; Sather, W.A.; Dell’Acqua, M.L. Synapse-to-Nucleus Communication through NFAT Is Mediated by L-type Ca(2+) Channel Ca(2+) Spike Propagation to the Soma. Cell Rep. 2019, 26, 3537–3550.e4. [Google Scholar] [CrossRef] [PubMed]
- Oliveria, S.F.; Dell’Acqua, M.L.; Sather, W.A. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 2007, 55, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.G.; Groth, R.D.; Ma, H.; Barrett, C.F.; Owen, S.F.; Safa, P.; Tsien, R.W. Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 2012, 149, 1112–1124. [Google Scholar] [CrossRef]
- Patriarchi, T.; Qian, H.; Di Biase, V.; Malik, Z.A.; Chowdhury, D.; Price, J.L.; Hammes, E.A.; Buonarati, O.R.; Westenbroek, R.E.; Catterall, W.A.; et al. Phosphorylation of Cav1.2 on S1928 uncouples the L-type Ca2+ channel from the beta2 adrenergic receptor. EMBO J. 2016, 35, 1330–1345. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, P.S.; Lu, Y.; Rice, R.C.; Pieper, A.A.; Rajadhyaksha, A.M. Loss of Cav1.2 channels impairs hippocampal theta burst stimulation-induced long-term potentiation. Channels 2020, 14, 287–293. [Google Scholar] [CrossRef]
- Berkefeld, H.; Sailer, C.A.; Bildl, W.; Rohde, V.; Thumfart, J.O.; Eble, S.; Klugbauer, N.; Reisinger, E.; Bischofberger, J.; Oliver, D.; et al. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 2006, 314, 615–620. [Google Scholar] [CrossRef]
- Stanika, R.; Campiglio, M.; Pinggera, A.; Lee, A.; Striessnig, J.; Flucher, B.E.; Obermair, G.J. Splice variants of the CaV1.3 L-type calcium channel regulate dendritic spine morphology. Sci. Rep. 2016, 6, 34528. [Google Scholar] [CrossRef]
- Olson, P.A.; Tkatch, T.; Hernandez-Lopez, S.; Ulrich, S.; Ilijic, E.; Mugnaini, E.; Zhang, H.; Bezprozvanny, I.; Surmeier, D.J. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J. Neurosci. 2005, 25, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.L.; Shen, W.; Rafalovich, I.; Sebel, L.E.; Day, M.; Chan, C.S.; Surmeier, D.J. Regulation of dendritic calcium release in striatal spiny projection neurons. J. Neurophysiol. 2013, 110, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Pangrsic, T.; Singer, J.H.; Koschak, A. Voltage-Gated Calcium Channels: Key Players in Sensory Coding in the Retina and the Inner Ear. Physiol. Rev. 2018, 98, 2063–2096. [Google Scholar] [CrossRef] [PubMed]
- Brandt, A.; Khimich, D.; Moser, T. Few CaV1.3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J. Neurosci. 2005, 25, 11577–11585. [Google Scholar] [CrossRef]
- Lodha, N.; Bonfield, S.; Orton, N.C.; Doering, C.J.; McRory, J.E.; Mema, S.C.; Rehak, R.; Sauve, Y.; Tobias, R.; Stell, W.K.; et al. Congenital stationary night blindness in mice—A tale of two Cacna1f mutants. Adv. Exp. Med. Biol. 2010, 664, 549–558. [Google Scholar]
- Sudhof, T.C. The presynaptic active zone. Neuron 2012, 75, 11–25. [Google Scholar] [CrossRef]
- Wheeler, D.B.; Randall, A.; Tsien, R.W. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 1994, 264, 107–111. [Google Scholar] [CrossRef]
- Lazarevic, V.; Pothula, S.; Andres-Alonso, M.; Fejtova, A. Molecular mechanisms driving homeostatic plasticity of neurotransmitter release. Front. Cell Neurosci. 2013, 7, 244. [Google Scholar] [CrossRef]
- Cao, Y.Q.; Piedras-Renteria, E.S.; Smith, G.B.; Chen, G.; Harata, N.C.; Tsien, R.W. Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron 2004, 43, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Sutton, K.G.; McRory, J.E.; Guthrie, H.; Murphy, T.H.; Snutch, T.P. P/Q-type calcium channels mediate the activity-dependent feedback of syntaxin-1A. Nature 1999, 401, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Vecchia, D.; Tottene, A.; van den Maagdenberg, A.M.; Pietrobon, D. Abnormal cortical synaptic transmission in CaV2.1 knockin mice with the S218L missense mutation which causes a severe familial hemiplegic migraine syndrome in humans. Front. Cell Neurosci. 2015, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Perez-Reyes, E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar] [PubMed]
- Molineux, M.L.; McRory, J.E.; McKay, B.E.; Hamid, J.; Mehaffey, W.H.; Rehak, R.; Snutch, T.P.; Zamponi, G.W.; Turner, R.W. Specific T-type calcium channel isoforms are associated with distinct burst phenotypes in deep cerebellar nuclear neurons. Proc. Natl. Acad. Sci. USA 2006, 103, 5555–5560. [Google Scholar] [CrossRef]
- Chemin, J.; Monteil, A.; Bourinet, E.; Nargeot, J.; Lory, P. Alternatively spliced alpha(1G) (Ca(V)3.1) intracellular loops promote specific T-type Ca(2+) channel gating properties. Biophys. J. 2001, 80, 1238–1250. [Google Scholar] [CrossRef]
- Bigos, K.L.; Mattay, V.S.; Callicott, J.H.; Straub, R.E.; Vakkalanka, R.; Kolachana, B.; Hyde, T.M.; Lipska, B.K.; Kleinman, J.E.; Weinberger, D.R. Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch. Gen. Psychiatry 2010, 67, 939–945. [Google Scholar] [CrossRef]
- Thimm, M.; Kircher, T.; Kellermann, T.; Markov, V.; Krach, S.; Jansen, A.; Zerres, K.; Eggermann, T.; Stocker, T.; Shah, N.J.; et al. Effects of a CACNA1C genotype on attention networks in healthy individuals. Psychol Med. 2011, 41, 1551–1561. [Google Scholar] [CrossRef]
- Krug, A.; Nieratschker, V.; Markov, V.; Krach, S.; Jansen, A.; Zerres, K.; Eggermann, T.; Stocker, T.; Shah, N.J.; Treutlein, J.; et al. Effect of CACNA1C rs1006737 on neural correlates of verbal fluency in healthy individuals. Neuroimage 2010, 49, 1831–1836. [Google Scholar] [CrossRef]
- Vahdani, B.; Armani Kian, A.; Esmaeilzadeh, A.; Zenoozian, S.; Yousefi, V.; Mazloomzadeh, S. Adjunctive Raloxifene and Isradipine Improve Cognitive Functioning in Patients With Schizophrenia: A Pilot Study. J. Clin. PsychoPharmacol. 2020, 40, 457–463. [Google Scholar] [CrossRef]
- Clark, M.B.; Wrzesinski, T.; Garcia, A.B.; Hall, N.A.L.; Kleinman, J.E.; Hyde, T.; Weinberger, D.R.; Harrison, P.J.; Haerty, W.; Tunbridge, E.M. Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain. Mol. Psychiatry 2020, 25, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Seitter, H.; Koschak, A. Relevance of tissue specific subunit expression in channelopathies. Neuropharmacology 2018, 132, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Ostacher, M.J.; Iosifescu, D.V.; Hay, A.; Blumenthal, S.R.; Sklar, P.; Perlis, R.H. Pilot investigation of isradipine in the treatment of bipolar depression motivated by genome-wide association. Bipolar Disord 2014, 16, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Yarotskyy, V.; Gao, G.; Peterson, B.Z.; Elmslie, K.S. The Timothy syndrome mutation of cardiac CaV1.2 (L-type) channels: Multiple altered gating mechanisms and pharmacological restoration of inactivation. J. Physiol. 2009, 587, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tadross, M.R.; Tsien, R.W. Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 2016, 351, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Hofer, N.T.; Tuluc, P.; Ortner, N.J.; Nikonishyna, Y.V.; Fernandes-Quintero, M.L.; Liedl, K.R.; Flucher, B.E.; Cox, H.; Striessnig, J. Biophysical classification of a CACNA1D de novo mutation as a high-risk mutation for a severe neurodevelopmental disorder. Mol. Autism. 2020, 11, 4. [Google Scholar]
- Lu, A.T.; Dai, X.; Martinez-Agosto, J.A.; Cantor, R.M. Support for calcium channel gene defects in Autism. spectrum disorders. Mol. Autism. 2012, 3, 18. [Google Scholar] [CrossRef]
- Strom, S.P.; Stone, J.L.; Ten Bosch, J.R.; Merriman, B.; Cantor, R.M.; Geschwind, D.H.; Nelson, S.F. High-density SNP association study of the 17q21 chromosomal region linked to Autism. identifies CACNA1G as a novel candidate gene. Mol. Psychiatry 2010, 15, 996–1005. [Google Scholar] [CrossRef]
- Strawn, J.R.; Geracioti, T.D., Jr. The treatment of generalized anxiety disorder with pregabalin, an atypical anxiolytic. Neuropsychiatr. Dis. Treat. 2007, 3, 237–243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duval, E.R.; Javanbakht, A.; Liberzon, I. Neural circuits in anxiety and stress disorders: A focused review. Ther. Clin. Risk Manag. 2015, 11, 115–126. [Google Scholar]
- Shinnick-Gallagher, P.; McKernan, M.G.; Xie, J.; Zinebi, F. L-type voltage-gated calcium channels are involved in the in vivo and in vitro expression of fear conditioning. Ann. N. Y. Acad. Sci. 2003, 985, 135–149. [Google Scholar] [PubMed]
- Fulga, I.G.; Stroescu, V. Experimental reseArch. on the effect of calcium channel blockers nifedipine and verapamil on the anxiety in mice. Rom. J. Physiol. 1997, 34, 127–136. [Google Scholar] [PubMed]
- Busquet, P.; Nguyen, N.K.; Schmid, E.; Tanimoto, N.; Seeliger, M.W.; Ben-Yosef, T.; Mizuno, F.; Akopian, A.; Striessnig, J.; Singewald, N. CaV1.3 L-type Ca2+ channels modulate depression-like behaviour in mice independent of deaf phenotype. Int. J. Neuropsychopharmacol. 2010, 13, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Saegusa, H.; Kurihara, T.; Zong, S.; Kazuno, A.; Matsuda, Y.; Nonaka, T.; Han, W.; Toriyama, H.; Tanabe, T. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J. 2001, 20, 2349–2356. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013, 28, 715–724. [Google Scholar] [CrossRef]
- Zahodne, L.B.; Fernandez, H.H. Pathophysiology and treatment of psychosis in Parkinson’s disease: A review. Drugs Aging 2008, 25, 665–682. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef]
- Abdel-Salam, O.M. The paths to neurodegeneration in genetic Parkinson’s disease. CNS Neurol. Disord.-Drug Targets 2014, 13, 1485–1512. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Jick, S.S.; Meier, C.R. Use of antihypertensives and the risk of Parkinson disease. Neurology 2008, 70, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, B.; Svanstrom, H.; Nielsen, N.M.; Fugger, L.; Melbye, M.; Hviid, A. Use of calcium channel blockers and Parkinson’sdisease. Am. J. Epidemiol. 2012, 175, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.; Rhodes, S.L.; Qian, L.; Schernhammer, E.; Olsen, J.H.; Friis, S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol. 2010, 67, 600–606. [Google Scholar] [PubMed]
- Liss, B.; Striessnig, J. The Potential of L-Type Calcium Channels as a Drug Target for Neuroprotective Therapy in Parkinson’s Disease. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 263–289. [Google Scholar] [CrossRef] [PubMed]
- Putzier, I.; Kullmann, P.H.; Horn, J.P.; Levitan, E.S. Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J. Neurosci. 2009, 29, 15414–15419. [Google Scholar] [CrossRef]
- Chan, C.S.; Gertler, T.S.; Surmeier, D.J. A molecular basis for the increased vulnerability of substantia nigra dopamine neurons in aging and Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. 1), S63–S70. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Hurley, M.J.; Brandon, B.; Gentleman, S.M.; Dexter, D.T. Parkinson’s disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain 2013, 136, 2077–2097. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Lipscombe, D. Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 2001, 21, 5944–5951. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, S.; Harvey, K.E.; Salyer, A.E.; Li, T.A.; Rantz, E.K.; Lill, M.A.; Hockerman, G.H. Molecular Determinants of the Differential Modulation of Cav1.2 and Cav1.3 by Nifedipine and FPL 64176. Mol. Pharmacol. 2018, 94, 973–983. [Google Scholar] [CrossRef]
- Lipscombe, D.; Helton, T.D.; Xu, W. L-type calcium channels: The low down. J. Neurophysiol. 2004, 92, 2633–2641. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study, G. Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson’s disease (STEADY-PD). Mov. Disord. 2013, 28, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group STEADY-PD III Investigators. Isradipine Versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann. Intern. Med. 2020, 172, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Venuto, C.S.; Yang, L.; Javidnia, M.; Oakes, D.; James Surmeier, D.; Simuni, T. Isradipine plasma pharmacokinetics and exposure-response in early Parkinson’s disease. Ann. Clin. Transl. Neurol. 2021, 8, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.H.; Yang, Y.C.; Pan, M.K.; Huang, C.S.; Kuo, C.C. Modulation of subthalamic T-type Ca(2+) channels remedies locomotor deficits in a rat model of Parkinson disease. J. Clin. Investig. 2011, 121, 3289–3305. [Google Scholar] [CrossRef] [PubMed]
- Tabata, Y.; Imaizumi, Y.; Sugawara, M.; Andoh-Noda, T.; Banno, S.; Chai, M.; Sone, T.; Yamazaki, K.; Ito, M.; Tsukahara, K.; et al. T-type Calcium Channels Determine the Vulnerability of Dopaminergic Neurons to Mitochondrial Stress in Familial Parkinson Disease. Stem Cell Reports 2018, 11, 1171–1184. [Google Scholar] [CrossRef]
- Weiss, N.; Zamponi, G.W. T-Type Channel Druggability at a Crossroads. ACS Chem. Neurosci. 2019, 10, 1124–1126. [Google Scholar] [CrossRef]
- Weiss, N.; Zamponi, G.W. T-type calcium channels: From molecule to therapeutic opportunities. Int. J. BioChem. Cell Biol. 2019, 108, 34–39. [Google Scholar] [CrossRef]
- Ortner, N.J. Voltage-Gated Ca(2+) Channels in Dopaminergic Substantia Nigra Neurons: Therapeutic Targets for Neuroprotection in Parkinson’s Disease? Front. Synaptic Neurosci. 2021, 13, 636103. [Google Scholar] [CrossRef] [PubMed]
- Striessnig, J.; Bolz, H.J.; Koschak, A. Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflugers Arch. 2010, 460, 361–374. [Google Scholar] [CrossRef]
- Martinez-Rivera, A.; Hao, J.; Tropea, T.F.; Giordano, T.P.; Kosovsky, M.; Rice, R.C.; Lee, A.; Huganir, R.L.; Striessnig, J.; Addy, N.A.; et al. Enhancing VTA Cav1.3 L-type Ca(2+) channel activity promotes cocaine and mood-related behaviors via overlapping AMPA receptor mechanisms in the nucleus accumbens. Mol. Psychiatry 2017, 22, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zhang, Z.; Zhang, W.; Ding, Y.; Zhao, F.; Zhang, J.; Song, Y. Investigation of the Selectivity of L-Type Voltage-Gated Calcium Channels 1.3 for Pyrimidine-2,4,6-Triones Derivatives Based on Molecular Dynamics Simulation. Molecules 2020, 25, 5440. [Google Scholar] [CrossRef]
- Kang, S.; Cooper, G.; Dunne, S.F.; Dusel, B.; Luan, C.H.; Surmeier, D.J.; Silverman, R.B. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson’s disease. Nat. Commun. 2012, 3, 1146. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.; Kang, S.; Perez-Rosello, T.; Guzman, J.N.; Galtieri, D.; Xie, Z.; Kondapalli, J.; Mordell, J.; Silverman, R.B.; Surmeier, D.J. A Single Amino Acid Determines the Selectivity and Efficacy of Selective Negative Allosteric Modulators of CaV1.3 L-Type Calcium Channels. ACS Chem. Biol. 2020, 15, 2539–2550. [Google Scholar] [CrossRef]
- Huang, H.; Ng, C.Y.; Yu, D.; Zhai, J.; Lam, Y.; Soong, T.W. Modest CaV1.342-selective inhibition by compound 8 is beta-subunit dependent. Nat. Commun. 2014, 5, 4481. [Google Scholar] [CrossRef]
- Ortner, N.J.; Bock, G.; Vandael, D.H.; Mauersberger, R.; Draheim, H.J.; Gust, R.; Carbone, E.; Tuluc, P.; Striessnig, J. Pyrimidine-2,4,6-triones are a new class of voltage-gated L-type Ca2+ channel activators. Nat. Commun. 2014, 5, 3897. [Google Scholar] [CrossRef] [PubMed]
- Degoulet, M.; Stelly, C.E.; Ahn, K.C.; Morikawa, H. L-type Ca(2)(+) channel blockade with antihypertensive medication disrupts VTA synaptic plasticity and drug-associated contextual memory. Mol. Psychiatry 2016, 21, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Hendrich, J.; Bauer, C.S.; Dolphin, A.C. Chronic pregabalin inhibits synaptic transmission between rat dorsal root ganglion and dorsal horn neurons in culture. Channels 2012, 6, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Rauck, R.L.; Wallace, M.S.; Burton, A.W.; Kapural, L.; North, J.M. Intrathecal ziconotide for neuropathic pain: A review. Pain Pract. 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Staats, P.S.; Yearwood, T.; Charapata, S.G.; Presley, R.W.; Wallace, M.S.; Byas-Smith, M.; Fisher, R.; Bryce, D.A.; Mangieri, E.A.; Luther, R.R.; et al. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: A randomized controlled trial. JAMA 2004, 291, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Rauck, R.L.; Wallace, M.S.; Leong, M.S.; Minehart, M.; Webster, L.R.; Charapata, S.G.; Abraham, J.E.; Buffington, D.E.; Ellis, D.; Kartzinel, R.; et al. A randomized, double-blind, placebo-controlled study of intrathecal ziconotide in adults with severe chronic pain. J. Pain Symptom Manag. 2006, 31, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.S.; Charapata, S.G.; Fisher, R.; Byas-Smith, M.; Staats, P.S.; Mayo, M.; McGuire, D.; Ellis, D.; Ziconotide Nonmalignant Pain Study, G. Intrathecal ziconotide in the treatment of chronic nonmalignant pain: A randomized, double-blind, placebo-controlled clinical trial. Neuromodulation 2006, 9, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Feng, Z.P.; Zhang, L.; Pajouhesh, H.; Ding, Y.; Belardetti, F.; Pajouhesh, H.; Dolphin, D.; Mitscher, L.A.; Snutch, T.P. Scaffold-based design and synthesis of potent N-type calcium channel blockers. Bioorganic Med. Chem. Lett. 2009, 19, 6467–6472. [Google Scholar] [CrossRef]
- Swensen, A.M.; Herrington, J.; Bugianesi, R.M.; Dai, G.; Haedo, R.J.; Ratliff, K.S.; Smith, M.M.; Warren, V.A.; Arneric, S.P.; Eduljee, C.; et al. Characterization of the substituted N-triazole oxindole TROX-1, a small-molecule, state-dependent inhibitor of Ca(V)2 calcium channels. Mol. Pharmacol. 2012, 81, 488–497. [Google Scholar] [CrossRef]
- Mathela, C.S.; Chanotiya, C.S.; Sammal, S.S.; Pant, A.K.; Pandey, S. Compositional diversity of terpenoids in the Himalayan Valeriana genera. Chem. Biodivers 2005, 2, 1174–1182. [Google Scholar] [CrossRef]
- Jiang, H.H.; Dong, W.; Zhou, J.; Hu, J.M.; Yang, J.; Nian, Y. Ca(v)2.2 and Ca(v)3.1 calcium channel inhibitors from Valeriana jatamansi Jones. Rsc Adv. 2017, 7, 45878–45884. [Google Scholar] [CrossRef]
- Subasinghe, N.L.; Wall, M.J.; Winters, M.P.; Qin, N.; Lubin, M.L.; Finley, M.F.; Brandt, M.R.; Neeper, M.P.; Schneider, C.R.; Colburn, R.W.; et al. A novel series of pyrazolylpiperidine N-type calcium channel blockers. Bioorganic Med. Chem. Lett. 2012, 22, 4080–4083. [Google Scholar] [CrossRef]
- Shao, P.P.; Ye, F.; Chakravarty, P.K.; Varughese, D.J.; Herrington, J.B.; Dai, G.; Bugianesi, R.M.; Haedo, R.J.; Swensen, A.M.; Warren, V.A.; et al. Aminopiperidine sulfonamide Cav2.2 channel inhibitors for the treatment of chronic pain. J. Med. Chem. 2012, 55, 9847–9855. [Google Scholar] [CrossRef]
- Chen, X.; Liu, D.; Zhou, D.; Si, Y.; Xu, D.; Stamatkin, C.W.; Ghozayel, M.K.; Ripsch, M.S.; Obukhov, A.G.; White, F.A.; et al. Small-molecule CaValpha1CaVbeta antagonist suppresses neuronal voltage-gated calcium-channel trafficking. Proc. Natl. Acad. Sci. USA 2018, 115, E10566–E10575. [Google Scholar] [CrossRef]
- Van Petegem, F.; Duderstadt, K.E.; Clark, K.A.; Wang, M.; Minor, D.L., Jr. Alanine-scanning mutagenesis defines a conserved energetic hotspot in the CaValpha1 AID-CaVbeta interaction site that is critical for channel modulation. Structure 2008, 16, 280–294. [Google Scholar] [CrossRef]
- Campiglio, M.; Coste de Bagneaux, P.; Ortner, N.J.; Tuluc, P.; Van Petegem, F.; Flucher, B.E. STAC proteins associate to the IQ domain of CaV1.2 and inhibit calcium-dependent inactivation. Proc. Natl. Acad. Sci. USA 2018, 115, 1376–1381. [Google Scholar] [CrossRef]
- Horstick, E.J.; Linsley, J.W.; Dowling, J.J.; Hauser, M.A.; McDonald, K.K.; Ashley-Koch, A.; Saint-Amant, L.; Satish, A.; Cui, W.W.; Zhou, W.; et al. Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat. Commun. 2013, 4, 1952. [Google Scholar] [CrossRef] [PubMed]
- Moshe, S.L.; Perucca, E.; Ryvlin, P.; Tomson, T. Epilepsy: New advances. Lancet 2015, 385, 884–898. [Google Scholar] [CrossRef]
- Herman, S.T. Epilepsy after brain insult: Targeting epileptogenesis. Neurology 2002, 59 (Suppl. S5), S21–S26. [Google Scholar] [CrossRef]
- Noebels, J.L. The biology of epilepsy genes. Annu. Rev. Neurosci. 2003, 26, 599–625. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Scheffer, I.E.; Berkovic, S.F.; Dibbens, L.M.; Mulley, J.C. Channelopathies in idiopathic epilepsy. Neurotherapeutics 2007, 4, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Khosravani, H.; Zamponi, G.W. Voltage-gated calcium channels and idiopathic generalized epilepsies. Physiol. Rev. 2006, 86, 941–966. [Google Scholar] [CrossRef] [PubMed]
- Meisel, C.; Loddenkemper, T. Seizure prediction and intervention. Neuropharmacology 2020, 172, 107898. [Google Scholar] [CrossRef]
- Perucca, E.; Tomson, T. The pharmacological treatment of epilepsy in adults. Lancet. Neurol. 2011, 10, 446–456. [Google Scholar] [CrossRef]
- Loscher, W.; Schmidt, D. Modern antiepileptic drug development has failed to deliver: Ways out of the current dilemma. Epilepsia 2011, 52, 657–678. [Google Scholar] [CrossRef] [PubMed]
- Brodie, M.J.; Barry, S.J.; Bamagous, G.A.; Norrie, J.D.; Kwan, P. Patterns of treatment response in newly diagnosed epilepsy. Neurology 2012, 78, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Ryvlin, P.; Cross, J.H.; Rheims, S. Epilepsy surgery in children and adults. Lancet Neurol. 2014, 13, 1114–1126. [Google Scholar] [CrossRef]
- Dalic, L.; Cook, M.J. Managing drug-resistant epilepsy: Challenges and solutions. Neuropsychiatr. Dis. Treat. 2016, 12, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Huguenard, J.R. Block of T -Type Ca(2+) Channels Is an Important Action of Succinimide Antiabsence Drugs. Epilepsy Curr. 2002, 2, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Gomora, J.C.; Daud, A.N.; Weiergraber, M.; Perez-Reyes, E. Block of cloned human T-type calcium channels by succinimide antiepileptic drugs. Mol. Pharmacol. 2001, 60, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Crunelli, V.; Leresche, N. Block of Thalamic T-Type Ca(2+) Channels by Ethosuximide Is Not the Whole Story. Epilepsy Curr. 2002, 2, 53–56. [Google Scholar] [CrossRef]
- Goren, M.Z.; Onat, F. Ethosuximide: From bench to bedside. CNS Drug Rev. 2007, 13, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Terzioglu, B.; Aypak, C.; Onat, F.Y.; Kucukibrahimoglu, E.; Ozkaynakci, A.E.; Goren, M.Z. The effects of ethosuximide on amino acids in genetic absence epilepsy rat model. J. Pharmacol. Sci. 2006, 100, 227–233. [Google Scholar] [CrossRef]
- Ziyatdinova, S.; Gurevicius, K.; Kutchiashvili, N.; Bolkvadze, T.; Nissinen, J.; Tanila, H.; Pitkanen, A. Spontaneous epileptiform discharges in a mouse model of Alzheimer’s disease are suppressed by antiepileptic drugs that block sodium channels. Epilepsy Res. 2011, 94, 75–85. [Google Scholar] [CrossRef]
- Matar, N.; Jin, W.; Wrubel, H.; Hescheler, J.; Schneider, T.; Weiergraber, M. Zonisamide block of cloned human T-type voltage-gated calcium channels. Epilepsy Res. 2009, 83, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Murakami, T.; Ono, H. Zonisamide suppresses pain symptoms of formalin-induced inflammatory and streptozotocin-induced diabetic neuropathy. J. Pharmacol. Sci. 2008, 107, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Tringham, E.; Powell, K.L.; Cain, S.M.; Kuplast, K.; Mezeyova, J.; Weerapura, M.; Eduljee, C.; Jiang, X.; Smith, P.; Morrison, J.L.; et al. T-type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci. Transl. Med. 2012, 4, 121ra19. [Google Scholar] [CrossRef]
- Johannessen Landmark, C.; Beiske, G.; Baftiu, A.; Burns, M.L.; Johannessen, S.I. Experience from therapeutic drug monitoring and gender aspects of gabapentin and pregabalin in clinical practice. Seizure 2015, 28, 88–91. [Google Scholar] [CrossRef]
- Radzicki, D.; Yau, H.J.; Pollema-Mays, S.L.; Mlsna, L.; Cho, K.; Koh, S.; Martina, M. Temperature-sensitive Cav1.2 calcium channels support intrinsic firing of pyramidal neurons and provide a target for the treatment of febrile seizures. J. Neurosci. 2013, 33, 9920–9931. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Nukada, T.; Namiki, Y.; Miyashita, Y.; Hatsuno, K.; Ueno, Y.; Yamakawa, T.; Isshiki, T. Five different profiles of dihydropyridines in blocking T-type Ca(2+) channel subtypes (Ca(v)3.1 (alpha(1G)), Ca(v)3.2 (alpha(1H)), and Ca(v)3.3 (alpha(1I))) expressed in Xenopus oocytes. Eur. J. Pharmacol. 2009, 613, 100–107. [Google Scholar] [CrossRef]
- Chang, S.Y.; Yong, T.F.; Yu, C.Y.; Liang, M.C.; Pletnikova, O.; Troncoso, J.; Burgunder, J.M.; Soong, T.W. Age and gender-dependent alternative splicing of P/Q-type calcium channel EF-hand. Neuroscience 2007, 145, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, I.E.; Abele-Henckels, K.; Zhang, W.K.; Lin, B.; Yu, Y.; Geyman, L.S.; Ehlers, M.D.; Pnevmatikakis, E.A.; Yang, J. Age-related homeostatic midchannel proteolysis of neuronal L-type voltage-gated Ca(2)(+) channels. Neuron 2014, 82, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Patriarchi, T.; Price, J.L.; Matt, L.; Lee, B.; Nieves-Cintron, M.; Buonarati, O.R.; Chowdhury, D.; Nanou, E.; Nystoriak, M.A.; et al. Phosphorylation of Ser1928 mediates the enhanced activity of the L-type Ca2+ channel Cav1.2 by the beta2-adrenergic receptor in neurons. Sci. Signal. 2017, 10, eaaf9647. [Google Scholar] [CrossRef] [PubMed]
- Davare, M.A.; Hell, J.W. Increased phosphorylation of the neuronal L-type Ca(2+) channel Ca(v)1.2 during aging. Proc. Natl. Acad. Sci. USA 2003, 100, 16018–16023. [Google Scholar] [CrossRef] [PubMed]
- Folci, A.; Steinberger, A.; Lee, B.; Stanika, R.; Scheruebel, S.; Campiglio, M.; Ramprecht, C.; Pelzmann, B.; Hell, J.W.; Obermair, G.J.; et al. Molecular mimicking of C-terminal phosphorylation tunes the surface dynamics of CaV1.2 calcium channels in hippocampal neurons. J. Biol. Chem. 2018, 293, 1040–1053. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Current Type | CaV Nomenclature | Specific Blocker | Gene | Main Physiological Role | Disease |
|---|---|---|---|---|---|
| L | CaV1.1 | DHP | CACNA1S | Excitation-contraction coupling in skeletal muscle, regulation of gene transcription | Hypokalemic periodic paralysis [5], normokalemic periodic paralysis; malignant hypothermia susceptibility [5] |
| CaV1.2 | DHP | CACNA1C | Excitation-contraction coupling in cardiac muscle, regulation of gene transcription, endocrine secretion, spine and dendritic calcium signaling in neurons | Timothy syndrome [25,26,27,28,29], bipolar disorder [30,31], depressive disorder [32,33,34], schizophrenia [33,35,36,37,38,39], post-traumatic stress syndrome [40,41], Brugada syndrome (# 611875), cardiac Long QT syndrome [# 618447] | |
| CaV1.3 | DHP | CACNA1D | Hearing, cardiac and neuronal pace-making activity, spine and dendritic calcium signaling in neurons | Deafness [42,43], autism [44], bipolar disorder [45,46], sinoatrial dysfunction (# 614896) | |
| CaV1.4 | DHP | CACNA1F | Retinal neurotransmission | Congenital stationary night blindness [47,48], X-linked Cone-Rode dystrophy (# 300476), Aland Island eye disease (# 300600) | |
| N | CaV2.1 | ω-conotoxin-GVIA | CACNA1A | Neurotransmitter release, somatodendritic calcium signaling | Familial hemiplegic migraine [49,50], ataxia (# 108500, # 183086) |
| P/Q | CaV2.2 | ω-agatoxin-IVA | CACNA1B | Pain [8,51,52,53,54,55,56,57,58,59,60,61], neurodevelopmental disorder # 618497 | |
| R | CaV2.3 | SNX-482 | CACNA1E | Neurotransmitter release, membrane excitability | Seizure [62,63,64,65], neurodevelopmental disorder(# 618497), encephalopathy (# 618285) |
| T | CaV3.1 | Ethosuximide Zonisamide | CACNA1G | Membrane excitability, pace-making, firing, subthreshold oscillations | Seizure [66], spinocerebellar ataxia (# 616795 and # 618087) |
| CaV3.2 | Ethosuximide Zonisamide | CACNA1H | Seizure [67,68,69,70,71,72], autism [73], pain [51,52,53,54], hyperaldosteronism (# 617027) | ||
| CaV3.3 | Ethosuximide Zonisamide | CACNA1I | Seizure and neurodevelopmental disorders [74] |
| Small Molecules | Approved Applications | Target | Potential Applications # |
|---|---|---|---|
| Isradipine | Hypertension | L-type channels | Autism [44,116], failed Phase-III trial for PD [116], dependency [151] |
| Nimpodipine | Hypertension | L-type channels | Anxiety [121], febrile seizures [195] |
| Roscovitine | NA | CaV1.2, L-type currents | Timothy syndrome [29,114] |
| Pregabalin | Pain and seizures | CaVα2δ | Anxiety [119] |
| Gabapentin | Pain and seizures | CaVα2δ | Anxiety [119] |
| NNC 55-0396 | NA | T-type currents | PD [145] |
| Valeriana jatamansi derived small molecules | NA | CaV2.2, CaV3.1 | Pain [167] |
| Ziconotide | Pain | CaV2.2 | NA |
| BTT-266, BTT-369 | NA | β binding domain on α1 | Pain [170] |
| Ethosuximide | Seizures | T-type channels | Pain [185] |
| Valproate | Seizures | T-type channels | PD [190,191] |
| Zonisamide | Seizures | T-type channels | Pain and PD [191,192] |
| NP118809 (or Z160) | NA | N-type channels | Pain [164,193] |
| Z944 | NA | T-type channels | Seizures, pain [193] |
| Lamotrigine | Seizures | R-type channels | Pain [62] |
| Benzohydroquinone | NA | CaV2.1 | Familial hemiplegic migraine 1 [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanzetti, S.; Di Biase, V. Small Molecules as Modulators of Voltage-Gated Calcium Channels in Neurological Disorders: State of the Art and Perspectives. Molecules 2022, 27, 1312. https://doi.org/10.3390/molecules27041312
Lanzetti S, Di Biase V. Small Molecules as Modulators of Voltage-Gated Calcium Channels in Neurological Disorders: State of the Art and Perspectives. Molecules. 2022; 27(4):1312. https://doi.org/10.3390/molecules27041312
Chicago/Turabian StyleLanzetti, Stefano, and Valentina Di Biase. 2022. "Small Molecules as Modulators of Voltage-Gated Calcium Channels in Neurological Disorders: State of the Art and Perspectives" Molecules 27, no. 4: 1312. https://doi.org/10.3390/molecules27041312
APA StyleLanzetti, S., & Di Biase, V. (2022). Small Molecules as Modulators of Voltage-Gated Calcium Channels in Neurological Disorders: State of the Art and Perspectives. Molecules, 27(4), 1312. https://doi.org/10.3390/molecules27041312