
Anticancer Activity of (±)-Kusunokinin Derivatives towards Cholangiocarcinoma Cells


Abstract

1. Introduction
2. Results
2.1. The Cytotoxic Effect of (±)-TTPG-A and (±)-TTPG-B on Four Types of Cancer and Normal Cells
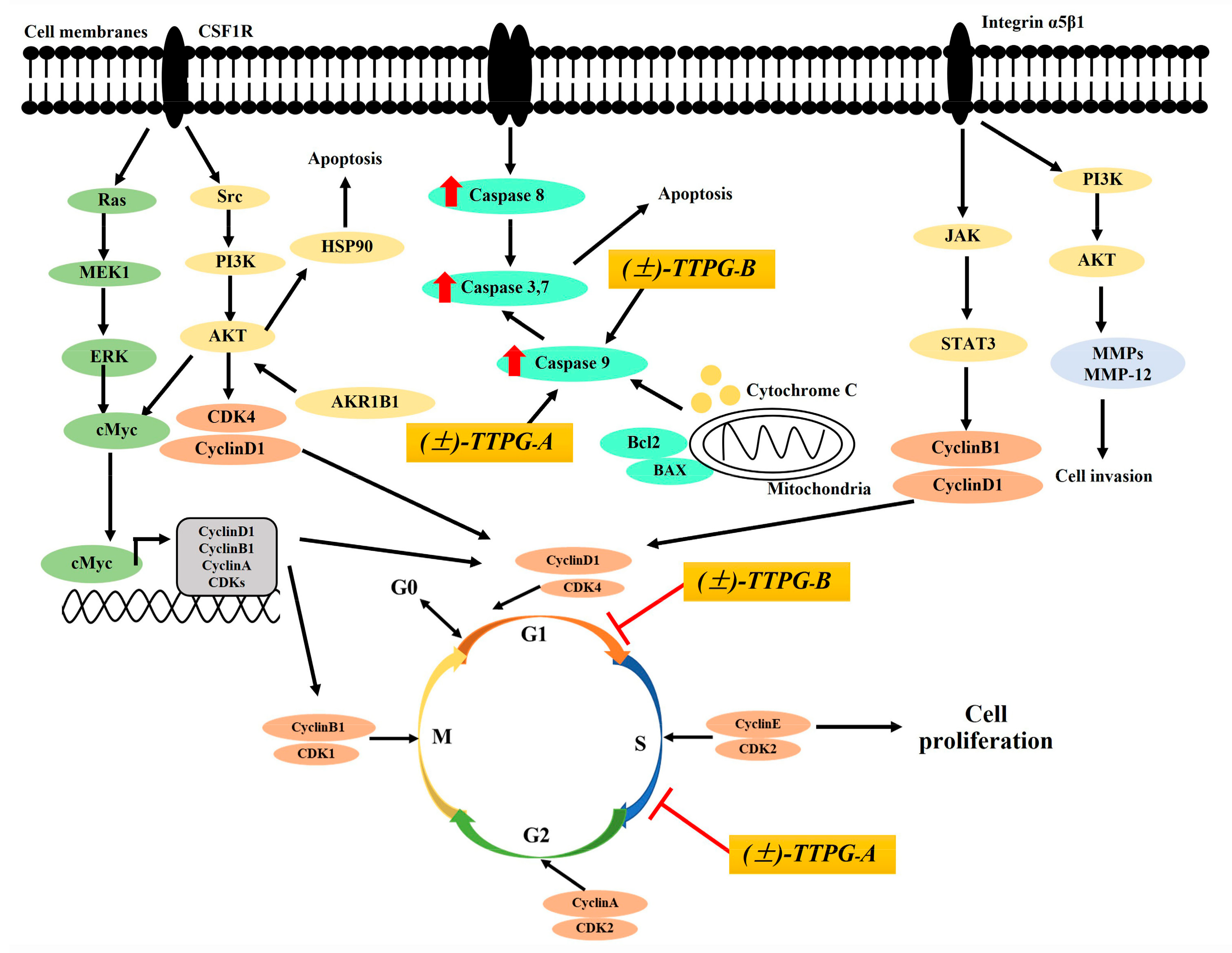
2.2. (±)-TTPG-A and (±)-TTPG-B Exerted Cell-Cycle Arrest
2.3. (±)-TTPG-A and (±)-TTPG-B Induced Apoptotic Cells
2.4. (±)-TTPG-A and (±)-TTPG-B Increased Multi-Caspase Activity
3. Discussion
4. Materials and Methods
4.1. Synthesis of (±)-Kusunokinin, (±)-TTPG-A and (±)-TTPG-B Compound
4.1.1. Synthesis of (±)-Kusunokinin
4.1.2. Synthesis (±)-TTPG-A
Preparation of 3,4-Dimethoxybenzaldehyde
Preparation of 4-(3,4-Dimethoxyphenyl)-3-(methoxycarbonyl)but-3-enoic Acid
Preparation of 3-(3,4-Dimethoxybenzyl)-4-methoxy-4-oxobutanoic Acid
Preparation of 4-(3,4-Dimethoxybenzyl)dihydrofuran-2(3H)-one
Preparation of 4-(Benzyloxy)-3-methoxybenzaldehyde
Preparation of 3-((4-(Benzyloxy)-3-methoxyphenyl)(hydroxy)methyl)-4-(3,4-dimethoxybenzyl)dihydrofuran-2(3H)-one
Preparation of 4-(3,4-Dimethoxybenzyl)-3-(4-hydroxy-3-methoxybenzyl)dihydrofuran-2(3H)-one ((±)-TTPG-A)
4.1.3. Synthesis (±)-TTPG-B
Preparation of 4-Butoxy-3-methoxybenzaldehyde
Preparation of 4-(4-Butoxy-3-methoxyphenyl)-3-(methoxycarbonyl)but-3-enoic Acid
Preparation of 3-(3,4-Dimethoxybenzyl)-4-methoxy-4-oxobutanoic Acid
Preparation of 4-(4-Butoxy-3-methoxybenzyl)dihydro-furan-2(3H)-one
Preparation of 3-((4-(Benzyloxy)-3-methoxyphenyl)(hydroxy)methyl)-4-(4-butoxy-3-methoxybenzyl)dihydrofuran-2(3H)-one
Preparation of 4-(4-Butoxy-3-methoxybenzyl)-3-(4-hydroxy-3-methoxybenzyl)dihydrofuran-2(3H)-one ((±)-TTPG-B)
4.2. Cell Culture
4.3. Cytotoxicity Assay
4.4. Cell Cycle Analysis Assay
4.5. Apoptosis Assay
4.6. Multi-Caspase Activity Assay
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, Y.; Liu, S. The new insight of treatment in Cholangiocarcinoma. J. Cancer 2022, 13, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Maughan, K.L.; Lutterbie, M.A.; Ham, P.S. Treatment of breast cancer. Am. Fam. Physician 2010, 81, 1339–1346. [Google Scholar] [PubMed]
- Grisold, W.; Briani, C.; Vass, A. Malignant cell infiltration in the peripheral nervous system. Handb. Clin. Neurol. 2013, 115, 685–712. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.S.; Ansari, J.; Spooner, D.; Hussain, S.A. Chemotherapy for breast cancer (Review). Oncol. Rep. 2010, 24, 1121–1131. [Google Scholar] [CrossRef]
- Tudoran, O.; Soritau, O.; Balacescu, L.; Visan, S.; Barbos, O.; Cojocneanu-Petric, R.; Balacescu, O.; Berindan-Neagoe, I. Regulation of stem cells-related signaling pathways in response to doxorubicin treatment in Hs578T triple-negative breast cancer cells. Mol. Cell. Biochem. 2015, 409, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Twelves, C.; Scheithauer, W.; McKendrick, J.; Seitz, J.F.; Van Hazel, G.; Wong, A.; Díaz-Rubio, E.; Gilberg, F.; Cassidy, J. Capecitabine versus 5-fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: Final results from the X-ACT trial with analysis by age and preliminary evidence of a pharmacodynamic marker of efficacy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23, 1190–1197. [Google Scholar] [CrossRef]
- Eckmann, K.R.; Patel, D.K.; Landgraf, A.; Slade, J.H.; Lin, E.; Kaur, H.; Loyer, E.; Weatherly, J.M.; Javle, M. Chemotherapy outcomes for the treatment of unresectable intrahepatic and hilar cholangiocarcinoma: A retrospective analysis. Gastrointest. Cancer Res. 2011, 4, 155–160. [Google Scholar]
- Ramírez-Merino, N.; Aix, S.P.; Cortés-Funes, H. Chemotherapy for cholangiocarcinoma: An update. World J. Gastrointest. Oncol. 2013, 5, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A. First-line treatment of ovarian cancer: Questions and controversies to address. Ther. Adv. Med. Oncol. 2018, 10, 1758835918768232. [Google Scholar] [CrossRef]
- Kayl, A.E.; Meyers, C.A. Side-effects of chemotherapy and quality of life in ovarian and breast cancer patients. Curr. Opin. Obstet. Gynecol. 2006, 18, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Aung, T.N.; Qu, Z.; Kortschak, R.D.; Adelson, D.L. Understanding the Effectiveness of Natural Compound Mixtures in Cancer through Their Molecular Mode of Action. Int. J. Mol. Sci. 2017, 18, 656. [Google Scholar] [CrossRef]
- Kreidieh, F.Y.; Moukadem, H.A.; El Saghir, N.S. Overview, prevention and management of chemotherapy extravasation. World J. Clin. Oncol. 2016, 7, 87–97. [Google Scholar] [CrossRef]
- Amawi, H.; Ashby, C.R., Jr.; Tiwari, A.K. Cancer chemoprevention through dietary flavonoids: What’s limiting? Chin. J. Cancer 2017, 36, 50. [Google Scholar] [CrossRef]
- Thoppil, R.J.; Bishayee, A. Terpenoids as potential chemopreventive and therapeutic agents in liver cancer. World J. Hepatol. 2011, 3, 228–249. [Google Scholar] [CrossRef]
- Saloustros, E.; Mavroudis, D.; Georgoulias, V. Paclitaxel and docetaxel in the treatment of breast cancer. Expert Opin. Pharmacother. 2008, 9, 2603–2616. [Google Scholar] [CrossRef]
- Sriwiriyajan, S.; Sukpondma, Y.; Srisawat, T.; Madla, S.; Graidist, P. (−)-Kusunokinin and piperloguminine from Piper nigrum: An alternative option to treat breast cancer. Biomed. Pharmacother. 2017, 92, 732–743. [Google Scholar] [CrossRef]
- Rattanaburee, T.; Thongpanchang, T.; Wongma, K.; Tedasen, A.; Sukpondma, Y.; Graidist, P. Anticancer activity of synthetic (±)-kusunokinin and its derivative (±)-bursehernin on human cancer cell lines. Biomed. Pharmacother. 2019, 117, 109115. [Google Scholar] [CrossRef]
- Tedasen, A.; Dokduang, S.; Sukpondma, Y.; Lailerd, N.; Madla, S.; Sriwiriyajan, S.; Rattanaburee, T.; Tipmanee, V.; Graidist, P. (−)-Kusunokinin inhibits breast cancer in N-nitrosomethylurea-induced mammary tumor rats. Eur. J. Pharmacol. 2020, 882, 173311. [Google Scholar] [CrossRef] [PubMed]
- Rattanaburee, T.; Tipmanee, V.; Tedasen, A.; Thongpanchang, T.; Graidist, P. Inhibition of CSF1R and AKT by (±)-kusunokinin hinders breast cancer cell proliferation. Biomed. Pharmacother. 2020, 129, 110361. [Google Scholar] [CrossRef] [PubMed]
- Tanawattanasuntorn, T.; Thongpanchang, T.; Rungrotmongkol, T.; Hanpaibool, C.; Graidist, P.; Tipmanee, V. (−)-Kusunokinin as a Potential Aldose Reductase Inhibitor: Equivalency Observed via AKR1B1 Dynamics Simulation. ACS Omega 2021, 6, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Mad-Adam, N.; Rattanaburee, T.; Tanawattanasuntorn, T.; Graidist, P. Effects of trans-(±)-kusunokinin on chemosensitive and chemoresistant ovarian cancer cells. Oncol. Lett. 2022, 23, 59. [Google Scholar] [CrossRef] [PubMed]
- Maphanao, P.; Thanan, R.; Loilome, W.; Chio-Srichan, S.; Wongwattanakul, M.; Sakonsinsiri, C. Synchrotron FTIR microspectroscopy revealed apoptosis-induced biomolecular changes of cholangiocarcinoma cells treated with ursolic acid. Biochim. Biophys. Acta Gen. Subj 2020, 1864, 129708. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, X.; Zhang, Y.; Qi, R.; Li, Z.; Zhang, K.; Liu, Z.; Yang, X. Lobaplatin induces apoptosis and arrests cell cycle progression in human cholangiocarcinoma cell line RBE. Biomed. Pharmacother. 2012, 66, 161–166. [Google Scholar] [CrossRef]
- Kotawong, K.; Chaijaroenkul, W.; Muhamad, P.; Na-Bangchang, K. Cytotoxic activities and effects of atractylodin and beta-eudesmol on the cell cycle arrest and apoptosis on cholangiocarcinoma cell line. J Pharm. Sci 2018, 136, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Samukawa, E.; Fujihara, S.; Oura, K.; Iwama, H.; Yamana, Y.; Tadokoro, T.; Chiyo, T.; Kobayashi, K.; Morishita, A.; Nakahara, M.; et al. Angiotensin receptor blocker telmisartan inhibits cell proliferation and tumor growth of cholangiocarcinoma through cell cycle arrest. Int. J. Oncol. 2017, 51, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Srijiwangsa, P.; Ponnikorn, S.; Na-Bangchang, K. Effect of beta-Eudesmol on NQO1 suppression-enhanced sensitivity of cholangiocarcinoma cells to chemotherapeutic agents. BMC Pharm. Toxicol. 2018, 19, 32. [Google Scholar] [CrossRef]
- Lee, K.; Ahn, J.H.; Lee, K.T.; Jang, D.S.; Choi, J.H. Deoxyschizandrin, Isolated from Schisandra Berries, Induces Cell Cycle Arrest in Ovarian Cancer Cells and Inhibits the Protumoural Activation of Tumour-Associated Macrophages. Nutrients 2018, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Ríos, J.L.; Giner, R.M.; Prieto, J.M. New findings on the bioactivity of lignans. In Studies in Natural Products Chemistry; Atta-ur, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2002; Volume 26, pp. 183–292. [Google Scholar]
- Utispan, K.; Sonongbua, J.; Thuwajit, P.; Chau-In, S.; Pairojkul, C.; Wongkham, S.; Thuwajit, C. Periostin activates integrin α5β1 through a PI3K/AKT-dependent pathway in invasion of cholangiocarcinoma. Int. J. Oncol. 2012, 41, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, K.; Block, K.; Nayak, B.K.; Livi, C.B.; Venkatachalam, M.A.; Sudarshan, S. PI3K regulation of the SKP-2/p27 axis through mTORC2. Oncogene 2013, 32, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Shiratori, Y.; Rengifo, W.; Igarashi, K.; Yamagata, M.; Kurokohchi, K.; Uchida, N.; Miyauchi, Y.; Yoshiji, H.; Watanabe, S.; et al. Cyclins and cyclin-dependent kinases: Comparative study of hepatocellular carcinoma versus cirrhosis. Hepatology 2003, 37, 534–543. [Google Scholar] [CrossRef]
- Thoma, O.-M.; Neurath, M.F.; Waldner, M.J. Cyclin-Dependent Kinase Inhibitors and Their Therapeutic Potential in Colorectal Cancer Treatment. Front. Pharmacol. 2021, 12, 757120. [Google Scholar] [CrossRef]
- Ke, F.; Wang, Z.; Song, X.; Ma, Q.; Hu, Y.; Jiang, L.; Zhang, Y.; Liu, Y.; Zhang, Y.; Gong, W. Cryptotanshinone induces cell cycle arrest and apoptosis through the JAK2/STAT3 and PI3K/Akt/NFκB pathways in cholangiocarcinoma cells. Drug Des. Dev. Ther. 2017, 11, 1753–1766. [Google Scholar] [CrossRef] [PubMed]
- Saranaruk, P.; Kariya, R.; Sittithumcharee, G.; Boueroy, P.; Boonmars, T.; Sawanyawisuth, K.; Wongkham, C.; Wongkham, S.; Okada, S.; Vaeteewoottacharn, K. Chromomycin A3 suppresses cholangiocarcinoma growth by induction of S phase cell cycle arrest and suppression of Sp1related antiapoptotic proteins. Int. J. Mol. Med. 2020, 45, 1005–1016. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Ganeshpure, P.A.; Stevenson, R. Synthesis of aryltetralin and dibenzylbutyrolactone lignans:(±)-lintetralin,(±)-phyltetralin, and (±)-kusunokinin. J. Chem. Soc. Perkin Trans. 1 1981, 1681–1684. [Google Scholar] [CrossRef]
- Sriwiriyajan, S.; Ninpesh, T.; Sukpondma, Y.; Nasomyon, T.; Graidist, P. Cytotoxicity screening of plants of genus Piper in breast cancer cell lines. Trop. J. Pharm. Res. 2014, 13, 921–928. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | IC50 (µM) | ||
|---|---|---|---|
| (±)-Kusunokinin | (±)-TTPG-A | (±)-TTPG-B | |
| Breast cancer MCF-7 | 4.23 ± 0.21 | 3.47 ± 0.02 * | 5.94 ± 0.04 |
| MDA-MB-468 | 5.19 ± 0.02 | 6.38 ± 0.04 | 0.43 ± 0.01 * |
| MDA-MB-231 | 9.23 ± 0.12 | 6.50 ± 0.01 * | 1.83 ± 0.04 * |
| Cholangiocarcinoma KKU-M213 | 4.47 ± 0.04 | 0.07 ± 0.01 * | 0.01 ± 0.001 * |
| KKU-K100 | 4.46 ± 0.29 | 0.76 ± 0.05 * | 1.53 ± 0.01 * |
| Colon cancer HT-29 | 5.34 ± 0.03 | Not inhibited | Not inhibited |
| Ovarian cancer A2780 | 4.52 ± 0.03 | 6.13 ± 0.04 | 0.05 ± 0.01 * |
| Normal cells MMNK-1 Vero L929 | 6.79 ± 0.04 Not inhibited 9.75 ± 0.39 | 6.30 ± 0.01 14.53 ± 0.02 Not inhibited | 1.53 ± 0.01 15.15 ± 0.16 Not inhibited |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rattanaburee, T.; Sermmai, P.; Tangthana-umrung, K.; Thongpanchang, T.; Graidist, P. Anticancer Activity of (±)-Kusunokinin Derivatives towards Cholangiocarcinoma Cells. Molecules 2022, 27, 8291. https://doi.org/10.3390/molecules27238291
Rattanaburee T, Sermmai P, Tangthana-umrung K, Thongpanchang T, Graidist P. Anticancer Activity of (±)-Kusunokinin Derivatives towards Cholangiocarcinoma Cells. Molecules. 2022; 27(23):8291. https://doi.org/10.3390/molecules27238291
Chicago/Turabian StyleRattanaburee, Thidarath, Patpanat Sermmai, Kornthip Tangthana-umrung, Tienthong Thongpanchang, and Potchanapond Graidist. 2022. "Anticancer Activity of (±)-Kusunokinin Derivatives towards Cholangiocarcinoma Cells" Molecules 27, no. 23: 8291. https://doi.org/10.3390/molecules27238291
APA StyleRattanaburee, T., Sermmai, P., Tangthana-umrung, K., Thongpanchang, T., & Graidist, P. (2022). Anticancer Activity of (±)-Kusunokinin Derivatives towards Cholangiocarcinoma Cells. Molecules, 27(23), 8291. https://doi.org/10.3390/molecules27238291