
Promoting Co-Crystallization in Poly(butylene succinate) and Poly(butylene fumarate) Blends via End-Group Functionalization


Abstract
1. Introduction
2. Results and Discussion
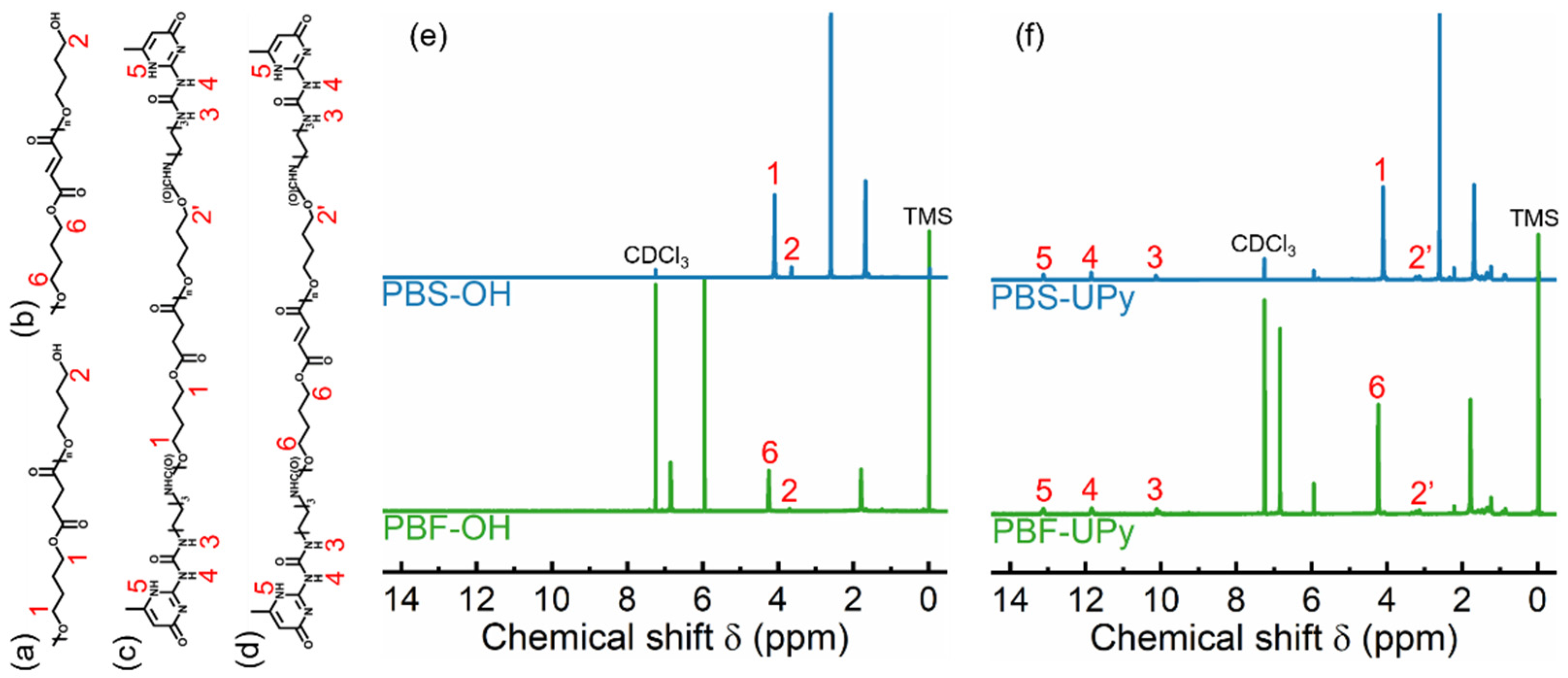
2.1. Chain Structure of Polyesters
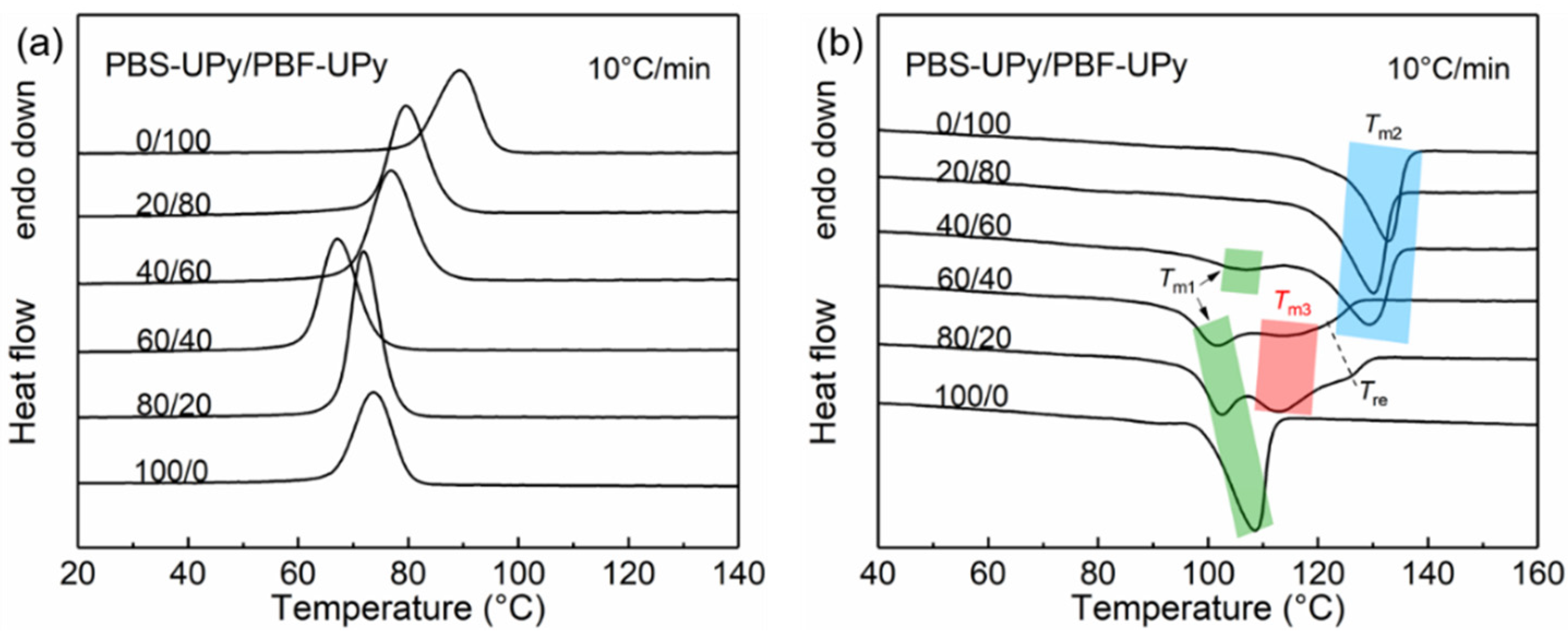
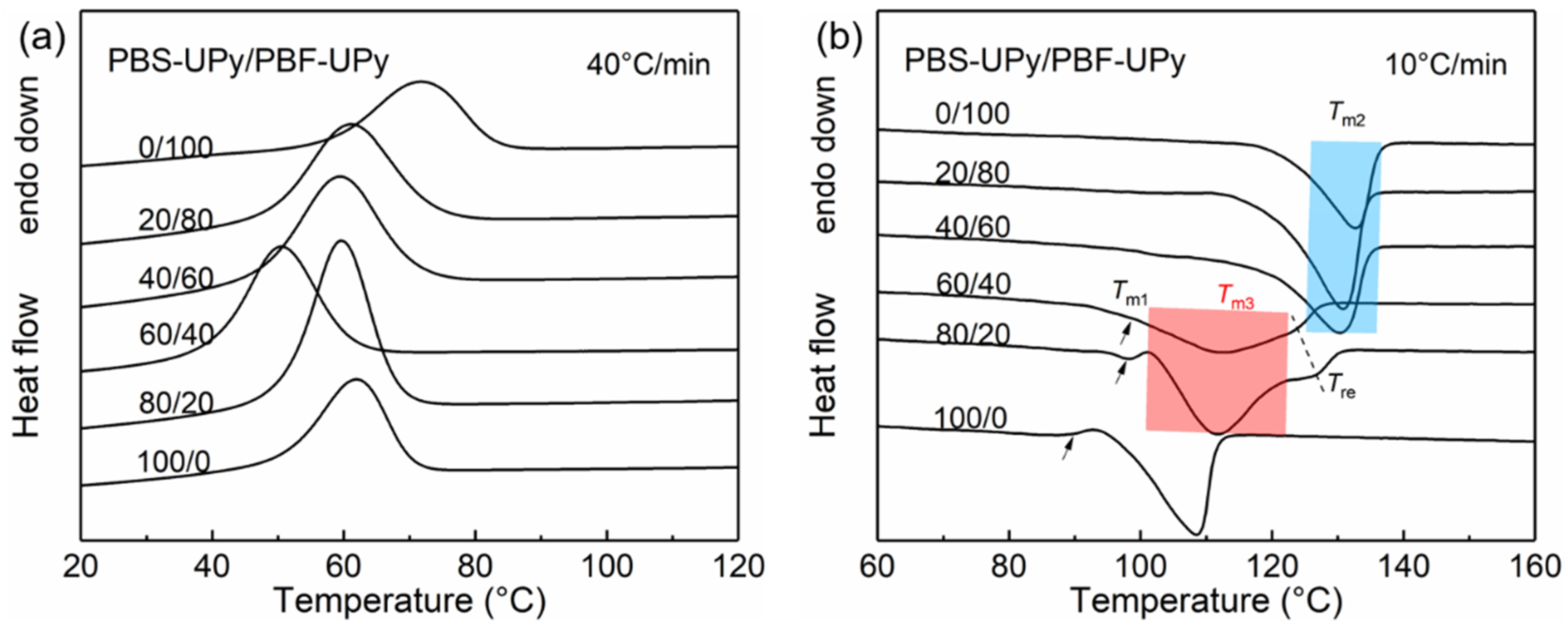
2.2. Thermal Properties of PBS-UPy/PBF-UPy Blends
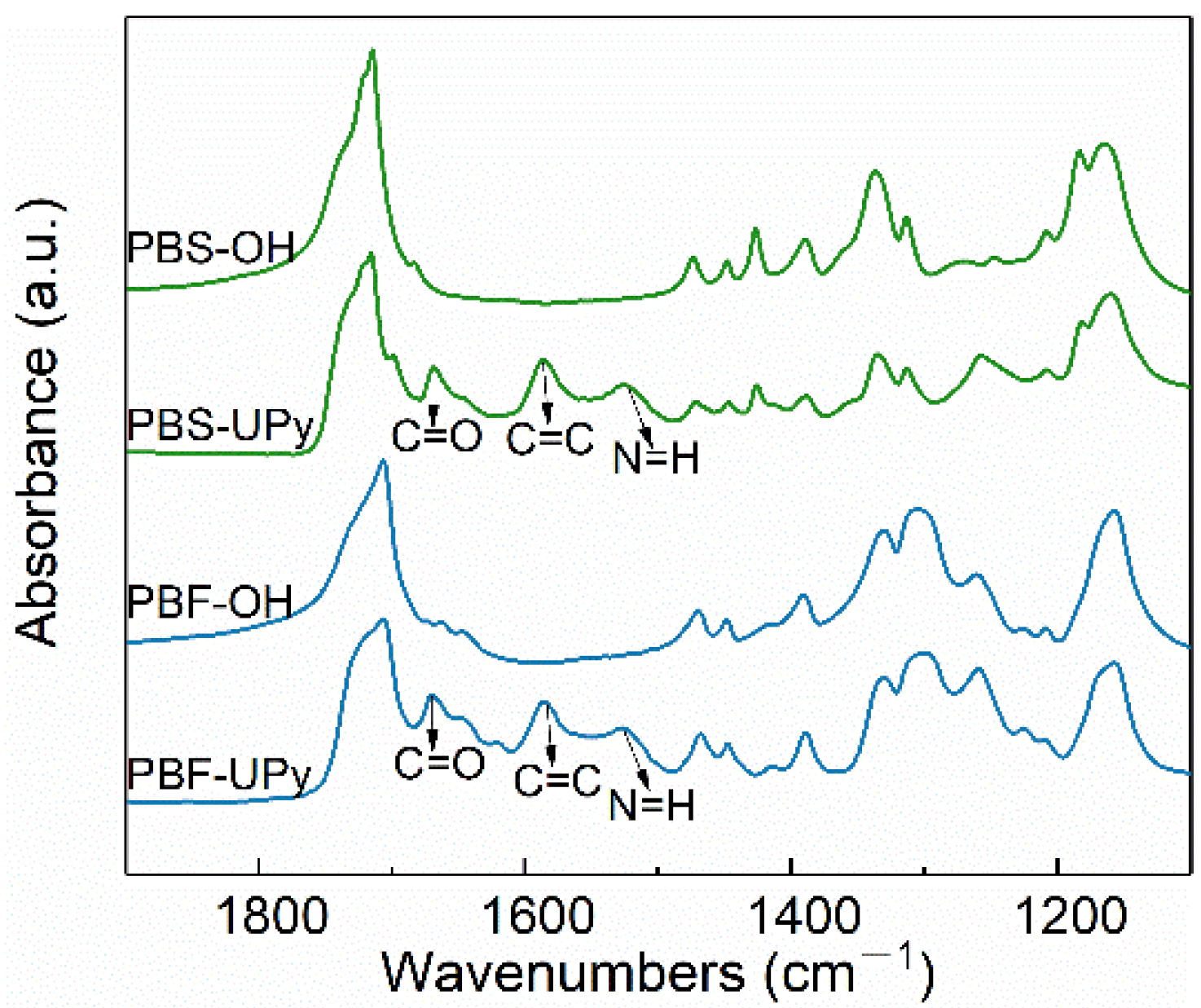
2.3. FTIR Investigation
2.4. Crystal Structure and Morphology
3. Materials and Methods
3.1. Materials
3.2. Fabrication of PBS-UPy/PBF-UPy Blends
3.3. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shokrollahi, P.; Mirzadeh, H.; Huck, W.T.S.; Scherman, O.A. Effect of self-complementary motifs on phase compatibility and material properties in blends of supramolecular polymers. Polymer 2010, 51, 6303–6312. [Google Scholar] [CrossRef]
- Kummara, S.; Tashiro, K.; Monma, T.; Horita, K. Isotope Effect on the Melt–Isothermal Crystallization of Polyoxymethylene D/H Random Copolymers and D/H Blend Samples. Macromolecules 2015, 48, 8070–8081. [Google Scholar] [CrossRef]
- Sham, C.K.; Guerra, G.; Karasz, F.E.; MacKnight, W.J. Blends of two poly(aryl ether ketones). Polymer 1988, 29, 1016–1020. [Google Scholar] [CrossRef]
- Pal, S.; Nandi, A.K. Cocrystallization Behavior of Poly(3-alkylthiophenes): Influence of Alkyl Chain Length and Head to Tail Regioregularity. Macromolecules 2003, 36, 8426–8432. [Google Scholar] [CrossRef]
- Yoshie, N.; Asaka, A.; Inoue, Y. Cocrystallization and Phase Segregation in Crystalline/Crystalline Polymer Blends of Bacterial Copolyesters. Macromolecules 2004, 37, 3770–3779. [Google Scholar] [CrossRef]
- Yoshie, N.; Inoue, Y. Cocrystallization and Phase Segregation in Blends of Two Bacterial Polyesters. Macromol. Symp. 2005, 224, 59–70. [Google Scholar] [CrossRef]
- Mata, A.; Palmer, L.; Tejeda-Montes, E.; Stupp, S.I. Design of Biomolecules for Nanoengineered Biomaterials for Regenerative Medicine. In Nanotechnology in Regenerative Medicine: Methods and Protocols; Navarro, M., Planell, J.A., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 39–49. [Google Scholar] [CrossRef]
- Ruokolainen, J.; Mäkinen, R.; Torkkeli, M.; Mäkelä, T.; Serimaa, R.; Brinke, G.T.; Ikkala, O. Switching Supramolecular Polymeric Materials with Multiple Length Scales. Science 1998, 280, 557–560. [Google Scholar] [CrossRef]
- Mozhdehi, D.; Ayala, S.; Cromwell, O.R.; Guan, Z. Self-Healing Multiphase Polymers via Dynamic Metal–Ligand Interactions. J. Am. Chem. Soc. 2014, 136, 16128–16131. [Google Scholar] [CrossRef]
- Pereverzev, Y.V.; Prezhdo, O.V.; Dalton, L.R. Structural origin of the enhanced electro-optic response of dendrimeric systems. Chem. Phys. Lett. 2003, 373, 207–212. [Google Scholar] [CrossRef]
- Binder, W.H.; Bernstorff, S.; Kluger, C.; Petraru, L.; Kunz, M.J. Tunable Materials from Hydrogen-Bonded Pseudo Block Copolymers. Adv. Mater. 2005, 17, 2824–2828. [Google Scholar] [CrossRef]
- Bouteiller, L. Assembly via Hydrogen Bonds of Low Molar Mass Compounds into Supramolecular Polymers. In Hydrogen Bonded Polymers; Binder, W., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 79–112. [Google Scholar] [CrossRef]
- Safari, M.; Otaegi, I.; Aramburu, N.; Wang, Y.; Liu, G.; Dong, X.; Wang, D.; Guerrica-Echevarria, G.; Müller, A.J. Composition dependent miscibility in the crystalline state of polyamide 6/polyamide 4,10 blends: From single to double crystalline blends. Polymer 2021, 219, 123570. [Google Scholar] [CrossRef]
- Söntjens, S.H.M.; Sijbesma, R.P.; van Genderen, M.H.P.; Meijer, E.W. Stability and Lifetime of Quadruply Hydrogen Bonded 2-Ureido-4[1H]-pyrimidinone Dimers. J. Am. Chem. Soc. 2000, 122, 7487–7493. [Google Scholar] [CrossRef]
- Bao, J.; Chang, R.; Shan, G.; Bao, Y.; Pan, P. Promoted Stereocomplex Crystallization in Supramolecular Stereoblock Copolymers of Enantiomeric Poly(Lactic Acid)s. Cryst. Growth Des. 2016, 16, 1502–1511. [Google Scholar] [CrossRef]
- Bao, J.; Chang, X.; Shan, G.; Bao, Y.; Pan, P. Synthesis of end-functionalized hydrogen-bonding poly(lactic acid)s and preferential stereocomplex crystallization of their enantiomeric blends. Polym. Chem. 2016, 7, 4891–4900. [Google Scholar] [CrossRef]
- Yu, M.-M.; Yang, W.-J.; Niu, D.-Y.; Cai, X.-X.; Weng, Y.-X.; Dong, W.-F.; Chen, M.-Q.; Xu, P.-W.; Wang, Y.; Chu, H.; et al. Enhancing the Crystallization Performance of Poly(L-lactide) by Intramolecular Hybridizing with Tunable Self-assembly-type Oxalamide Segments. Chin. J. Polym. Sci. 2021, 39, 122–132. [Google Scholar] [CrossRef]
- Guo, M.; Pitet, L.M.; Wyss, H.M.; Vos, M.; Dankers, P.Y.; Meijer, E.W. Tough stimuli-responsive supramolecular hydrogels with hydrogen-bonding network junctions. J. Am. Chem. Soc. 2014, 136, 6969–6977. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, W.; Yuan, W.; Liu, K.; Zhou, J.; Shan, G.; Bao, Y.; Pan, P. Separate crystallization and melting of polymer blocks and hydrogen bonding units in double-crystalline supramolecular polymers. Polymer 2021, 222, 123670. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, X.-W.; Ye, H.-M. Dependence of Crystallization Behavior of Interacting Telechelic Poly(butylene succinate) Oligomer on Molecular Weight. Crystals 2021, 11, 1530. [Google Scholar] [CrossRef]
- Wei, X.-W.; Yang, L.-L.; Li, Y.; Meng, X.; Cai, L.-H.; Zhou, Q.; Ye, H.-M. Asymmetrical formation of isomorphism in the crystalline/crystalline blend of poly(butylene succinate) and poly(butylene fumarate). Polymer 2021, 235, 124282. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, Z.; Wu, S.; Li, C.; Zhang, D.; Xiao, Y. Novel Poly(butylene fumarate) and Poly(butylene succinate) Multiblock Copolymers Bearing Reactive Carbon–Carbon Double Bonds: Synthesis, Characterization, Cocrystallization, and Properties. Ind. Eng. Chem. Res. 2013, 52, 6147–6155. [Google Scholar] [CrossRef]
- Perrin, C.L.; Nielson, J.B. “Strong” hydrogen bonds in chemistry and biology. Annu. Rev. Phys. Chem. 1997, 48, 511–544. [Google Scholar] [CrossRef] [PubMed]
- Gorobets, N.Y.; Yermolayev, S.A.; Gurley, T.; Gurinov, A.A.; Tolstoy, P.M.; Shenderovich, I.G.; Leadbeater, N.E. Difference between 1H NMR signals of primary amide protons as a simple spectral index of the amide intramolecular hydrogen bond strength. J. Phys. Org. Chem. 2012, 25, 287–295. [Google Scholar] [CrossRef]
- Folmer, B.J.B.; Sijbesma, R.P.; Versteegen, R.M.; van der Rijt, J.A.J.; Meijer, E.W. Supramolecular Polymer Materials: Chain Extension of Telechelic Polymers Using a Reactive Hydrogen-Bonding Synthon. Adv. Mater. 2000, 12, 874–878. [Google Scholar] [CrossRef]
- Liu, B.; Tang, Z.; Wang, Z.; Zhang, L.; Guo, B. Integrating transient and sacrificial bonds into biobased elastomers toward mechanical property enhancement and macroscopically responsive property. Polymer 2019, 184, 121914. [Google Scholar] [CrossRef]
- Zou, S.; Schonherr, H.; Vancso, G.J. Stretching and rupturing individual supramolecular polymer chains by AFM. Angew. Chem. Int. Ed. Engl. 2005, 44, 956–959. [Google Scholar] [CrossRef]
- Zhang, C.; Pérez-Camargo, R.A.; Zheng, L.; Zhao, Y.; Liu, G.; Wang, L.; Wang, D. Crystallization of Poly(hexamethylene carbonate)-co-poly(hexamethylene urethane) Segmental Block Copolymers: From Single to Double Crystalline Phases. Polymer 2021, 222, 123675. [Google Scholar] [CrossRef]
- Kim, J.Y.; Seo, E.S.; Kim, S.H.; Kikutani, T. Effects of annealing on structure and properties of TLCP/PEN/PET ternary blend fibers. Macromol. Res. 2003, 11, 62–68. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, J.; Li, L. Multiple melting behavior of poly(butylene succinate). Eur. Polym. J. 2007, 43, 3163–3170. [Google Scholar] [CrossRef]
- Yao, S.F.; Chen, X.T.; Ye, H.M. Investigation of Structure and Crystallization Behavior of Poly(butylene succinate) by Fourier Transform Infrared Spectroscopy. J. Phys. Chem. B 2017, 121, 9476–9485. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Li, Y.; Yao, S.-F.; Wei, X.-W.; Ye, H.-M. Unusual Spherulitic Morphology of Poly(propylene fumarate). Chin. J. Polym. Sci. 2021, 39, 493–500. [Google Scholar] [CrossRef]
- Wei, X.-W.; Zhao, X.-X.; Li, Y.; Meng, X.-Y.; Zhou, Q.; Ye, H.-M. Distinctive Polymorphism-like Isodimorphism in Poly(propylene succinate-ran-propylene fumarate). Chin. J. Polym. Sci. 2022, 40, 602–610. [Google Scholar] [CrossRef]
- Li, Y.-J.; Ho, K.-S.; Lo, W.-T.; Yang, S.-S.; Chao, L.; Chang, Y.-C.; Han, Y.-K.; Hsieh, T.-H.; Huang, Y.-J. Synthesis and characterization of poly(n-undecyl isocyanate)/polyaniline polyblend. J. Appl. Polym. Sci. 2010, 117, 1–7. [Google Scholar] [CrossRef]
- Si, P.; Luo, F. Hydrogen bonding interaction and crystallization behavior of poly (butylene succinate-co-butylene adipate)/thiodiphenol complexes. Polym. Adv. Technol. 2016, 27, 1413–1421. [Google Scholar] [CrossRef]
- Susi, H.; Byler, D.M. Resolution-enhanced fourier transform infrared spectroscopy of enzymes. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1986; Volume 130, pp. 290–311. [Google Scholar]
- Ni, Y.-P.; Li, Q.-T.; Chen, L.; Wu, W.-S.; Qin, Z.-H.; Zhang, Y.; Chen, L.; Wang, X.-L.; Wang, Y.-Z. Semi-aromatic copolyesters with high strength and fire safety via hydrogen bonds and π-π stacking. Chem. Eng. J. 2019, 374, 694–705. [Google Scholar] [CrossRef]
- Qiu, Z.; Yan, C.; Lu, J.; Yang, W. Miscible Crystalline/Crystalline Polymer Blends of Poly(vinylidene fluoride) and Poly(butylene succinate-co-butylene adipate): Spherulitic Morphologies and Crystallization Kinetics. Macromolecules 2007, 40, 5047–5053. [Google Scholar] [CrossRef]
- Ye, H.-M.; Wang, R.-D.; Liu, J.; Xu, J.; Guo, B.-H. Isomorphism in Poly(butylene succinate-co-butylene fumarate) and Its Application as Polymeric Nucleating Agent for Poly(butylene succinate). Macromolecules 2012, 45, 5667–5675. [Google Scholar] [CrossRef]
- Ye, H.-M.; Tang, Y.-R.; Song, Y.-Y.; Xu, J.; Guo, B.-H.; Zhou, Q. Aliphatic copolyester with isomorphism in limited composition range. Polymer 2014, 55, 5811–5820. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Tm1 (°C) | ΔHm1 (J/g) | Tm2 (°C) | ΔHm2 (J/g) | Tm3 (°C) | ΔHm3 (J/g) |
|---|---|---|---|---|---|---|
| PBF-UPy | – | – | 132.8 | 34.2 | – | – |
| PBS-UPy/PBF-UPy-20/80 | – | – | 131.0 | 45.1 | – | – |
| PBS-UPy/PBF-UPy-40/60 | – | – | 130.4 | 36.6 | – | – |
| PBS-UPy/PBF-UPy-60/40 | 99.0 | 3.2 | – | – | 112.3 | 26.0 |
| PBS-UPy/PBF-UPy-80/20 | 98.0 | 0.9 | – | – | 111.7 | 45.5 |
| PBS-UPy | 108.4/89.5 | 35.8 | – | – | – | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, X.-W.; Chen, C.; Wu, T.-Y.; Cai, L.-H.; Ye, H.-M. Promoting Co-Crystallization in Poly(butylene succinate) and Poly(butylene fumarate) Blends via End-Group Functionalization. Molecules 2022, 27, 7086. https://doi.org/10.3390/molecules27207086
Wei X-W, Chen C, Wu T-Y, Cai L-H, Ye H-M. Promoting Co-Crystallization in Poly(butylene succinate) and Poly(butylene fumarate) Blends via End-Group Functionalization. Molecules. 2022; 27(20):7086. https://doi.org/10.3390/molecules27207086
Chicago/Turabian StyleWei, Xue-Wei, Cong Chen, Tian-Yu Wu, Li-Hai Cai, and Hai-Mu Ye. 2022. "Promoting Co-Crystallization in Poly(butylene succinate) and Poly(butylene fumarate) Blends via End-Group Functionalization" Molecules 27, no. 20: 7086. https://doi.org/10.3390/molecules27207086
APA StyleWei, X.-W., Chen, C., Wu, T.-Y., Cai, L.-H., & Ye, H.-M. (2022). Promoting Co-Crystallization in Poly(butylene succinate) and Poly(butylene fumarate) Blends via End-Group Functionalization. Molecules, 27(20), 7086. https://doi.org/10.3390/molecules27207086