
An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Analysis of the Anomeric Effect



Abstract
:1. Introduction
2. Background
2.1. Interacting Quantum Atoms (IQA)
2.2. Relative Energy Gradient (REG) Method
3. Computational Details and Materials
4. Results and Discussion
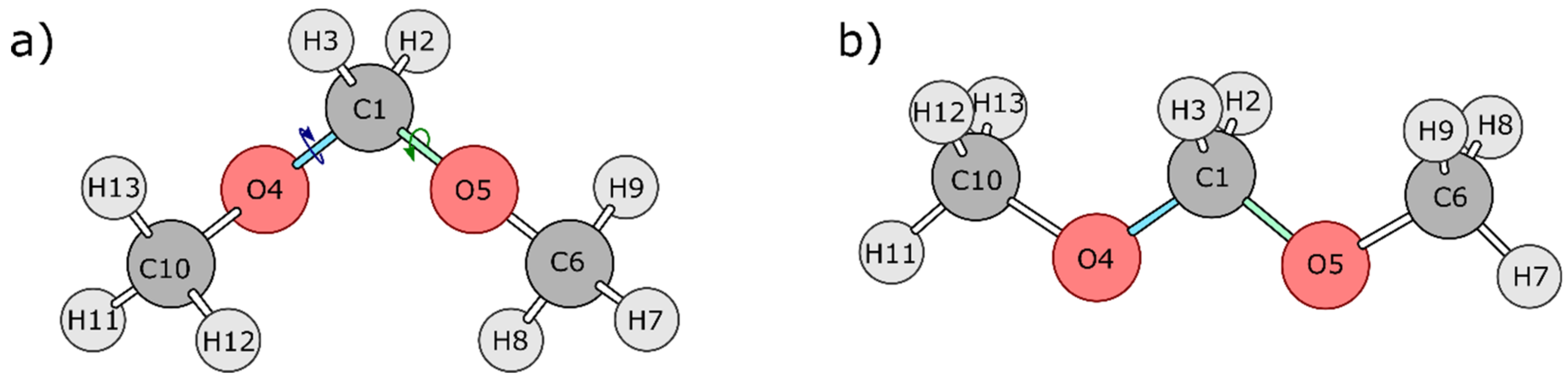
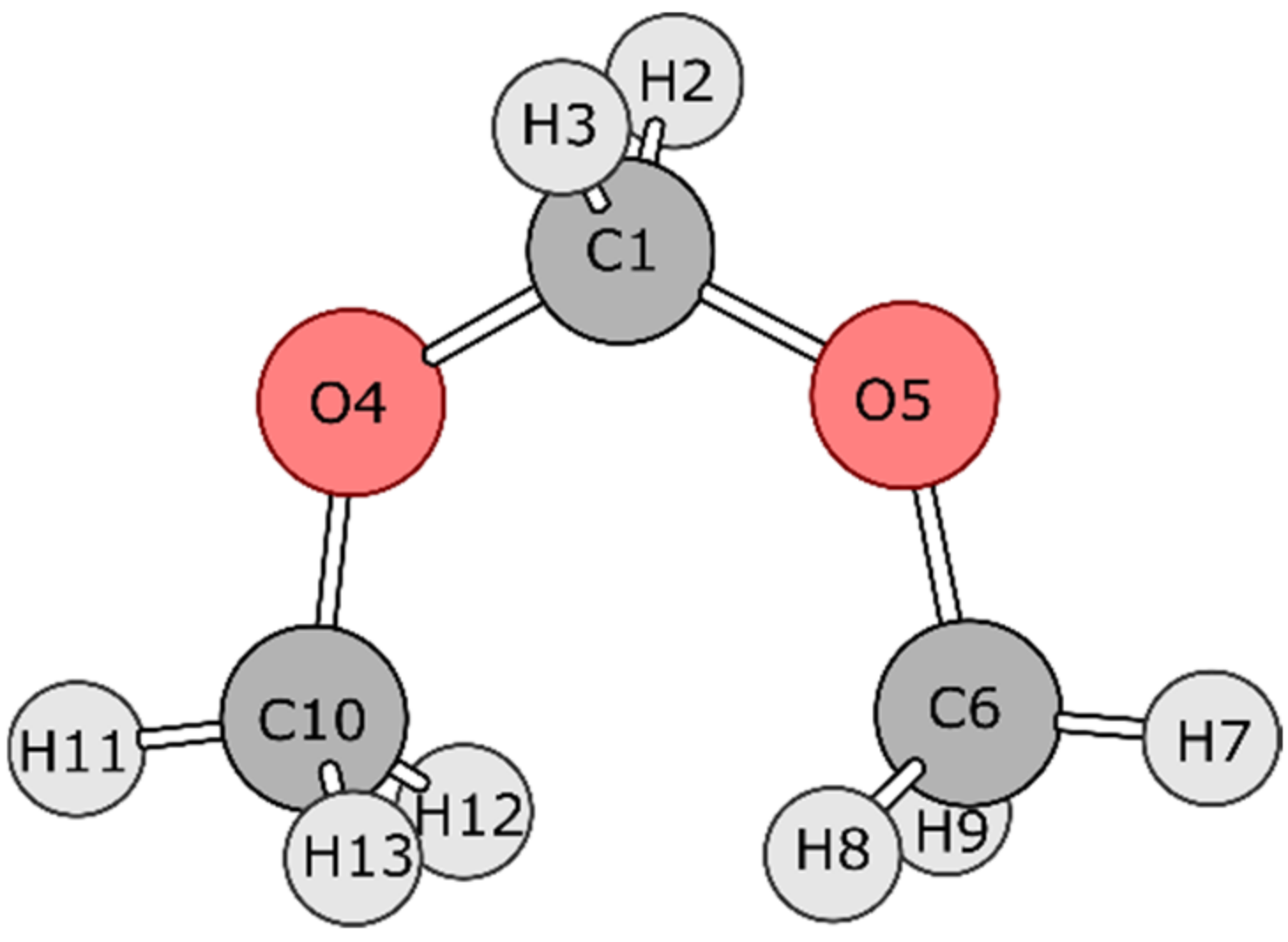
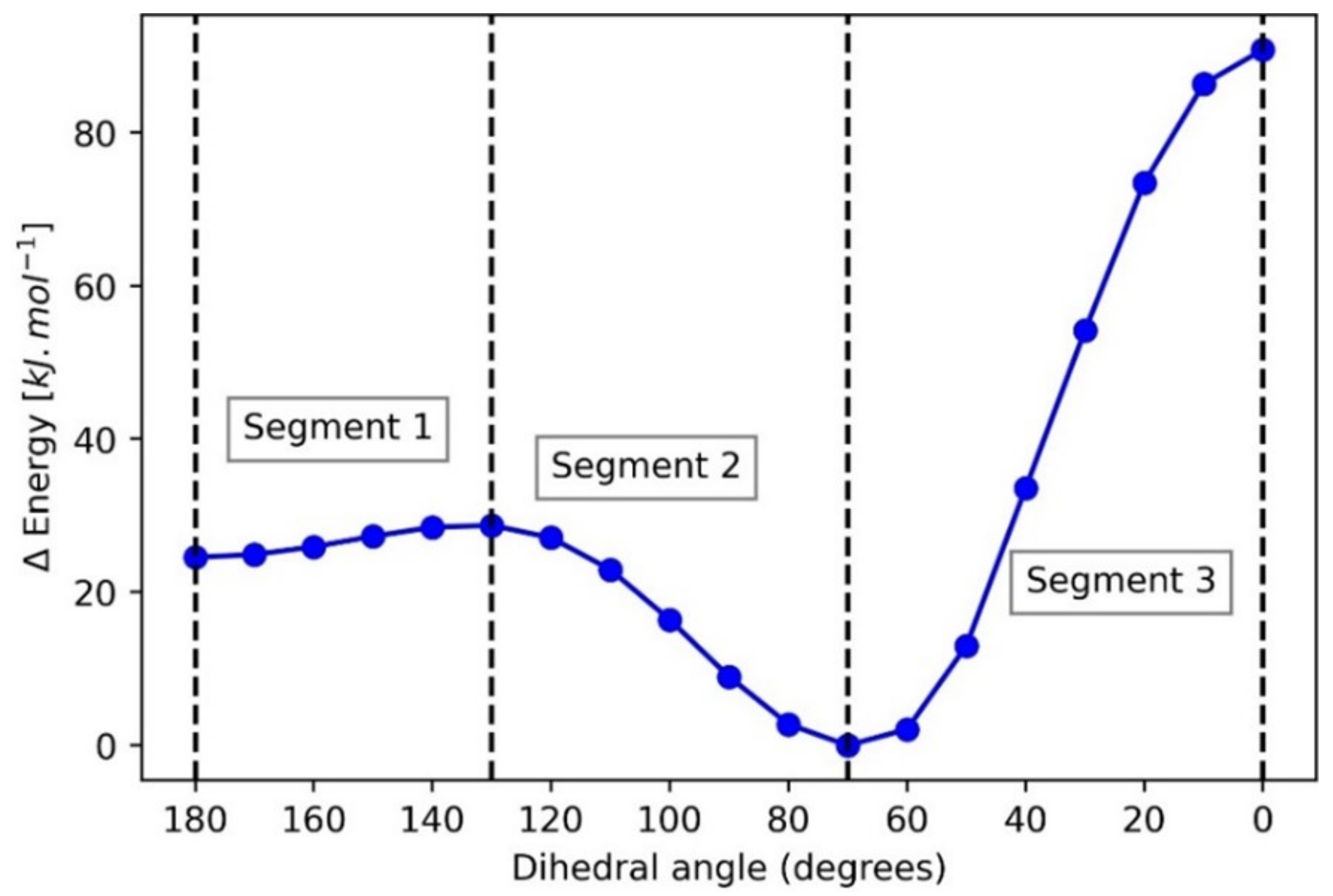
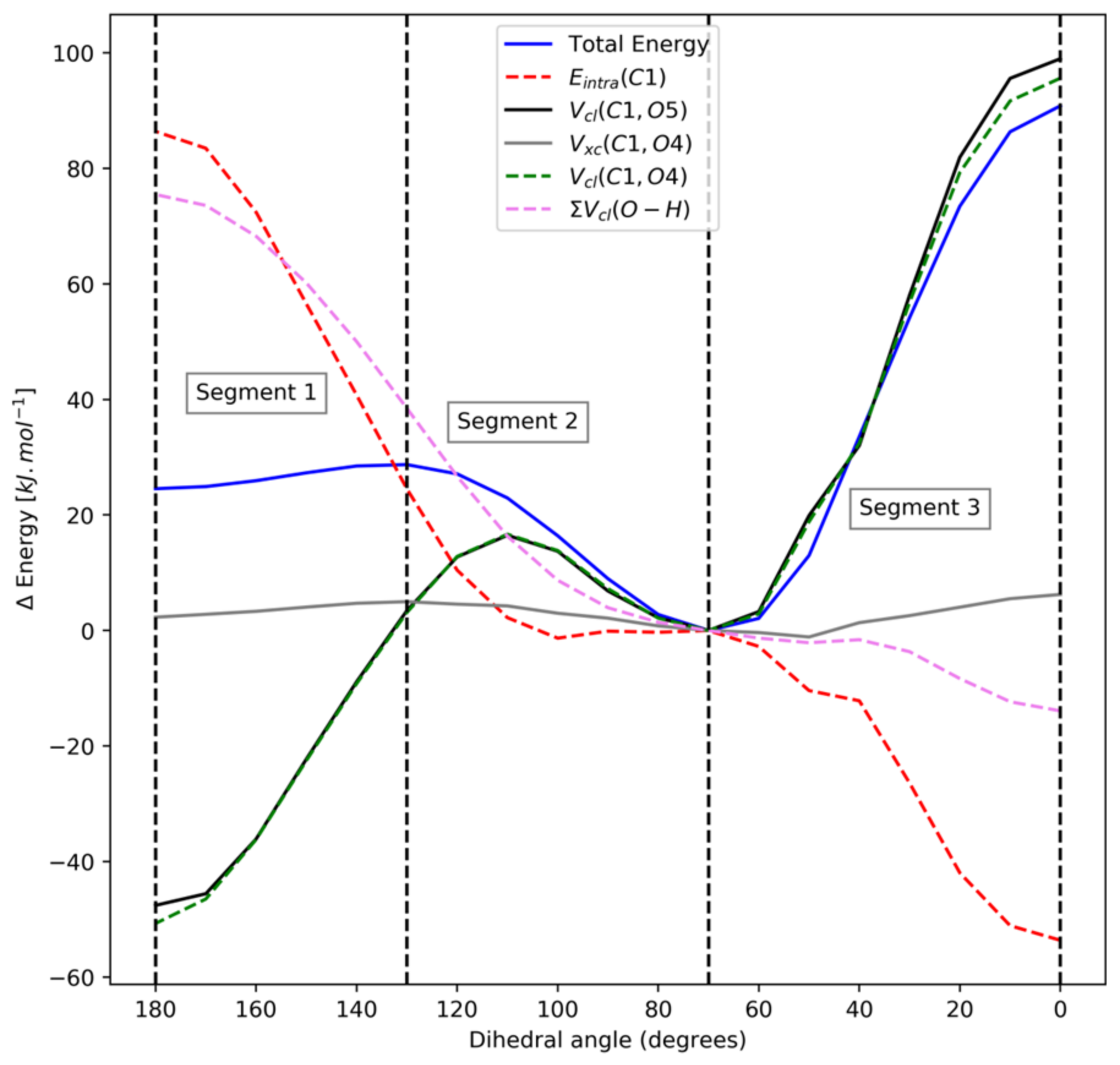
4.1. Dimethoxymethane (DMM)
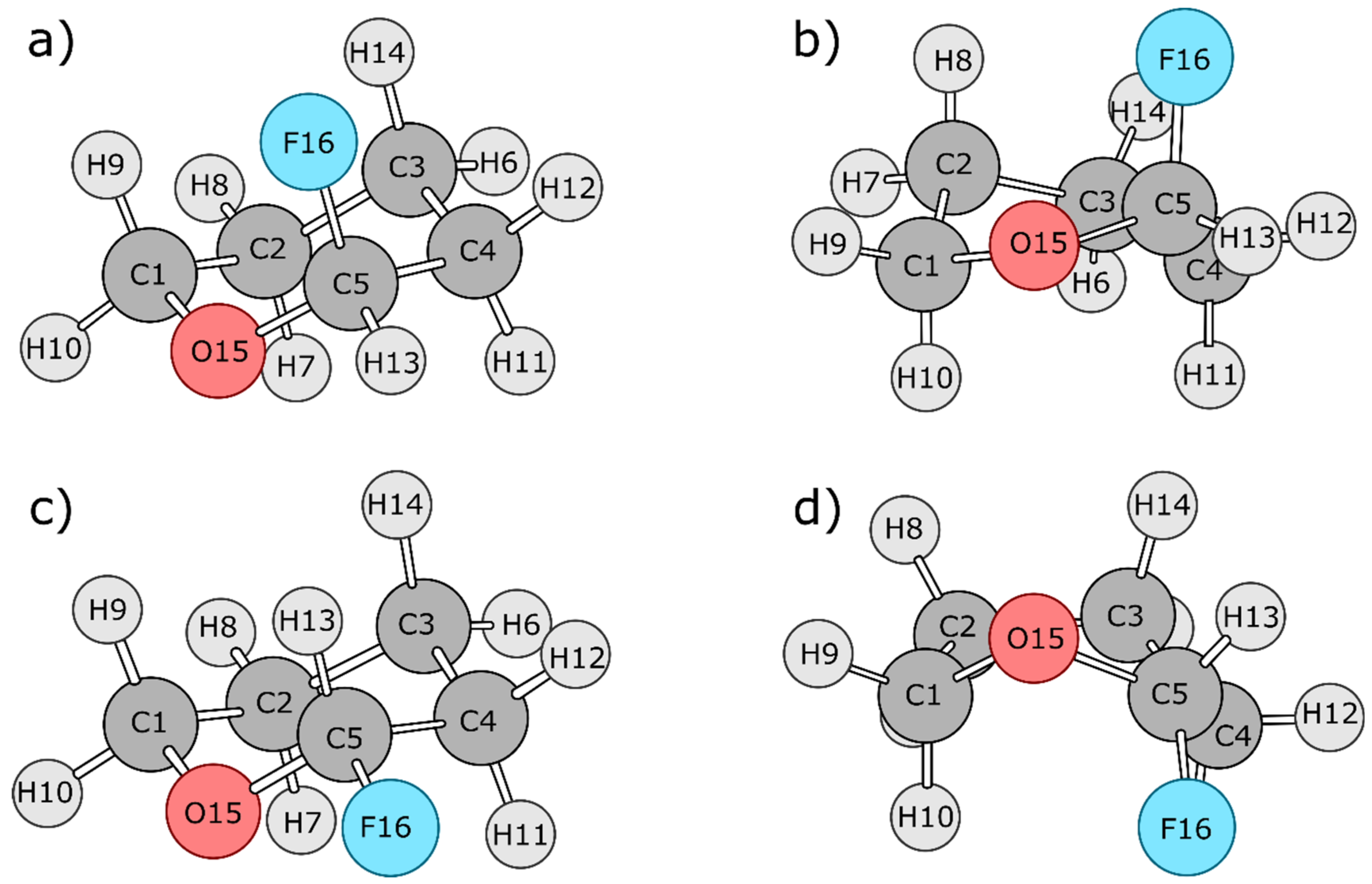
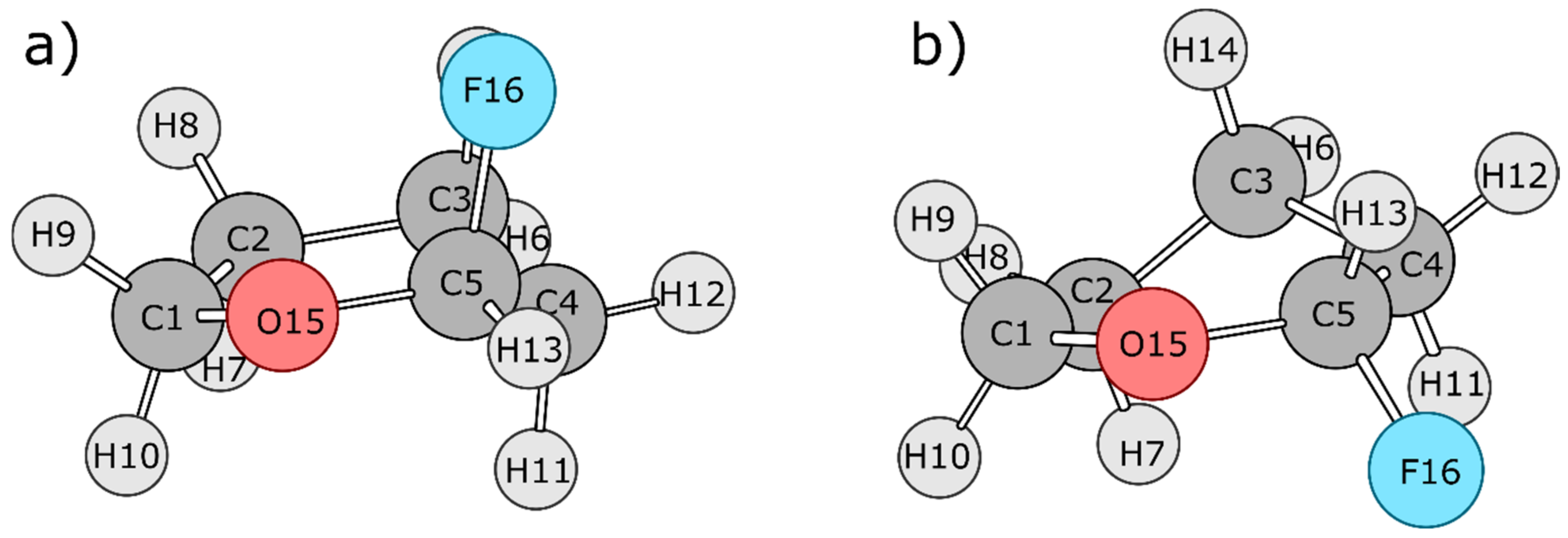
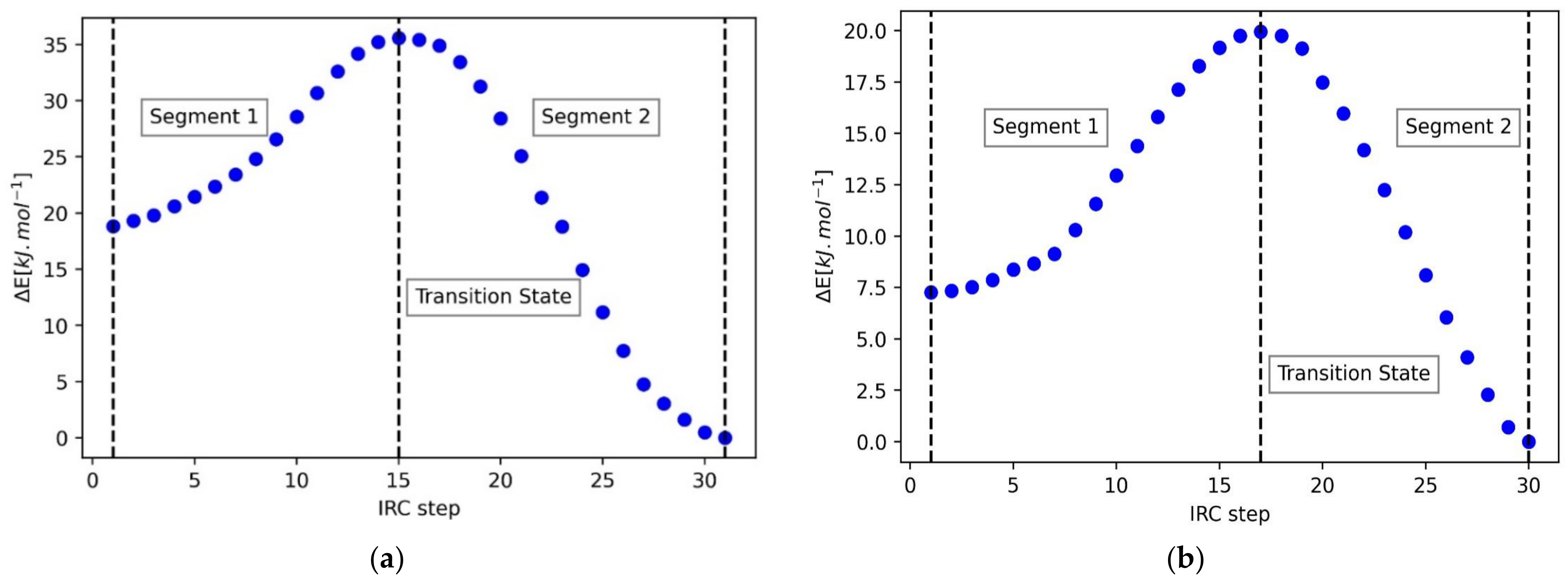
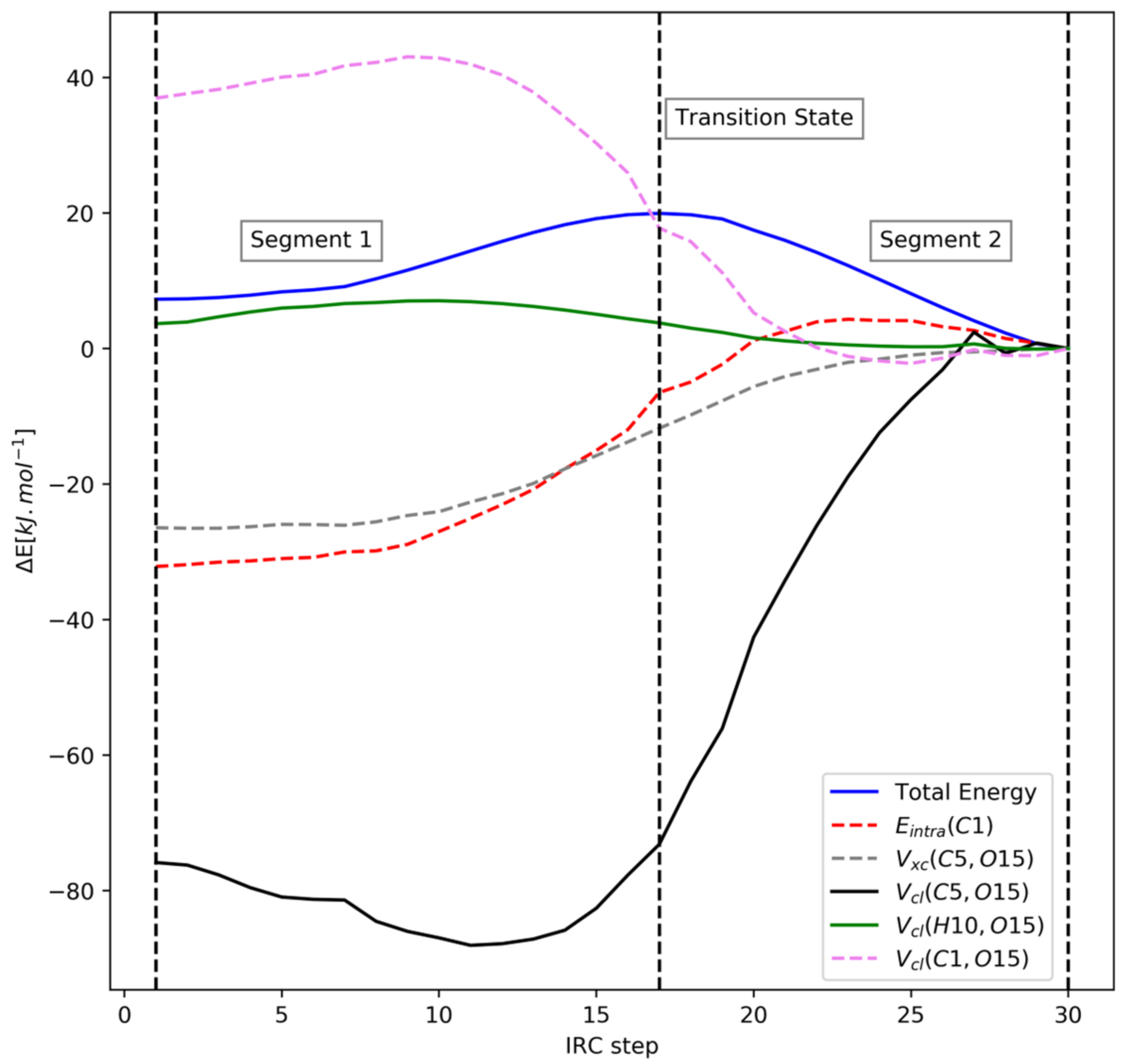
4.2. 2-Fluorotetrahydropyran (FTHP)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Edward, J.T. Stability of glycosides to acid hydrolysis. Chem. Ind. 1955, 3, 1102–1104. [Google Scholar]
- Lemieux, R.U.; Chu, P. Abstracts of Papers. In Proceedings of the 133rd National Meeting of the American Chemical Society, San Francisco, CA, USA, 13–18 April 1958. [Google Scholar]
- Lemieux, R.U. Effects of Unshared Pairs of Electrons. Pure Appl. Chem. 1971, 25, 527–548. [Google Scholar]
- Juaristi, E.; Cuevas, G. Recent studies of the anomeric effect. Tetrahedron 1992, 48, 5019–5087. [Google Scholar] [CrossRef]
- Salzner, U.; von Ragué Schleyer, P. Generalized Anomeric Effects and Hyperconjugation in CH2(OH)2, CH2(SH)2, CH2(SeH)2, and CH2(TeH)2. J. Am. Chem. Soc. 1993, 115, 10231–10236. [Google Scholar] [CrossRef]
- Filloux, C.M. The Problem of Origins and Origins of the Problem: Influence of Language on Studies Concerning the Anomeric Effect. Amgew. Chem. Int. Ed. 2015, 54, 8880–8894. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.; Ellencweig, A.; Tartakovsky, E.; Aped, P. Structure and conformation of heterocycles. 15. Solvent polarity and the anomeric effect. Angew. Chem. Int. Ed. 1986, 25, 287–289. [Google Scholar] [CrossRef]
- Mo, Y. Computational evidence that hyperconjugative interactions are not responsible for the anomeric effect. Nat. Chem. 2010, 2, 666–671. [Google Scholar] [CrossRef]
- Vila, A.; Mosquera, R.A. Atoms in molecules interpretation of the anomeric effect in the O-C-O unit. J. Comput. Chem. 2007, 28, 1516–1530. [Google Scholar] [CrossRef]
- Wang, C.; Ying, F.; Wu, W.; Mo, Y. How solvent influences the anomeric effect: Roles of hyperconjugative versus steric interactions on the conformational preference. J. Org. Chem. 2014, 79, 1571–1581. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Bailey, W.F.; Lambert, K.M.; Stempel, Z.D. The Anomeric Effect: It’s Complicated. J. Org. Chem. 2018, 83, 5242–5255. [Google Scholar] [CrossRef]
- Takahashi, O.; Yamasaki, K.; Kohno, Y.; Ueda, K.; Suezawa, H.; Nishio, M. The origin of the generalized anomeric effect: Possibility of CH/n and CH/π hydrogen bonds. Carbohydr. Res. 2009, 344, 1225–1229. [Google Scholar] [CrossRef] [Green Version]
- Carballeira, L.; Ferez-Juste, I. Ab initio study and NBO interpretation of the anomeric effect in CH2(XH2)2 (X = N, P, As) compounds. J. Phys. Chem. A 2000, 2014, 9362–9369. [Google Scholar] [CrossRef]
- Bauerfeldt, G.F.; Cardozo, T.M.; Pereira, M.S.; Da Silva, C.O. The anomeric effect: The dominance of exchange effects in closed-shell systems. Org. Biomol. Chem. 2013, 11, 299–308. [Google Scholar] [CrossRef]
- Juaristi, E.; Cuevas, G. The Anomeric Effect; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Huang, Y.; Zhong, A.G.; Yang, Q.; Liu, S. Origin of anomeric effect: A density functional steric analysis. J. Chem. Phys. 2011, 134, 084103. [Google Scholar] [CrossRef]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theor. Comp. 2005, 1, 1096–1109. [Google Scholar] [CrossRef]
- Popelier, P.L.A.; Kosov, D.S. Atom-atom partitioning of intramolecular and intermolecular Coulomb energy. J. Chem. Phys. 2001, 114, 6539–6547. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules: An Introduction; Pearson Education: London, UK, 2000. [Google Scholar]
- Popelier, P.L.A. The Quantum Theory of Atoms in Molecules. In The Nature of the Chemical Bond Revisited; Frenking, G., Shaik, S., Eds.; Wiley-VCH: Hoboken, NJ, USA, 2014; pp. 271–308. [Google Scholar]
- McDonagh, J.L.; Silva, A.F.; Vincent, M.A.; Popelier, P.L.A. Quantifying electron correlation of the chemical bond. J. Phys. Chem. Letts. 2017, 8, 1937–1942. [Google Scholar] [CrossRef] [Green Version]
- Thacker, J.C.R.; Popelier, P.L.A. The ANANKE relative energy gradient (REG) method to automate IQA analysis over configurational change. Theor. Chem. Acc. 2017, 136, 86. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.D.; Jaffe, R.L.; Yoon, D.Y. Conformational Characteristics of Dimethoxymethane Based upon ab Initio Electronic Structure Calculations. J. Phys. Chem. 1994, 98, 9072–9077. [Google Scholar] [CrossRef]
- Buschbeck-Alvarado, M.E.; Hernández-Fernández, G.; Hernández-Trujillo, J.; Cortés-Guzmán, F.; Cuevas, G. Charge transfer and electron localization as the origin of the anomeric effect in the O-C-O segment of dimethoxymethane and spiroketals. J. Phys. Org. Chem. 2018, 31, 1–13. [Google Scholar] [CrossRef]
- Weldon, A.J.; Vickrey, T.L.; Tschumper, G.S. Intrinsic conformational preferences of substituted cyclohexanes and tetrahydropyrans evaluated at the CCSD(T) complete basis set limit: Implications for the anomeric effect. J. Phys. Chem. A 2005, 109, 11073–11079. [Google Scholar] [CrossRef]
- Maxwell, P.; Martín Pendás, A.; Popelier, P.L.A. Extension of the interacting quantum atoms (IQA) approach to B3LYP level density functional theory. PhysChemChemPhys 2016, 18, 20986–21000. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.L.; Popelier, P.L.A. Exponential relationships capturing atomistic short-range repulsion from the Interacting Quantum Atoms (IQA) method. J. Phys. Chem. A 2016, 120, 9647–9659. [Google Scholar] [CrossRef]
- Symons, B.C.B.; Williamson, D.J.; Brooks, C.M.; Wilson, A.L.; Popelier, P.L.A. Does the Intra-Atomic Deformation Energy of Interacting Quantum Atoms Represent Steric Energy? Chem. Open 2019, 8, 560–570. [Google Scholar]
- Thacker, J.C.R.; Popelier, P.L.A. Fluorine Gauche Effect explained by Electrostatic Polarization instead of Hyperconjugation: An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study. J. Phys. Chem. A 2018, 122, 1439–1450. [Google Scholar] [CrossRef] [Green Version]
- Thacker, J.C.R.; Vincent, M.A.; Popelier, P.L.A. Using the Relative Energy Gradient Method with Interacting Quantum Atoms to Determine the Reaction Mechanism and Catalytic Effects in the Peptide Hydrolysis in HIV-1 Protease. Chem. Eur. J. 2018, 14, 11200–11210. [Google Scholar] [CrossRef] [Green Version]
- Popelier, P.L.A.; Maxwell, P.I.; Thacker, J.C.R.; Alkorta, I. A Relative Energy Gradient (REG) study of the planar and perpendicular torsional energy barriers in biphenyl. Theor. Chem. Acc. 2019, 138, 12. [Google Scholar] [CrossRef] [Green Version]
- Alkorta, I.; Silva, A.F.; Popelier, P.L.A. An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study of the Halogen Bond with Explicit Analysis of Electron Correlation. Molecules 2020, 25, 2674. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Bernhard Schlegel, H. Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Fukui, K. The Path of Chemical Reactions-The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian; Version 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- AIMAll; Keith, T.A. Gristmill Software. 2021. Available online: aim.tkgristmill.com (accessed on 3 August 2022).
- Duarte, L.J. Available online: github.com/ljduarte/REG.py (accessed on 3 August 2022).
- Gallegos, M.; Costales, A.; Martín Pendás, Á. Energetic Descriptors of Steric Hindrance in Real Space: An Improved IQA Picture. ChemPhysChem 2021, 22, 775–787. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment 1 | Segment 2 | Segment 3 | ||||||
|---|---|---|---|---|---|---|---|---|
| Contribution | REG | R | Contribution | REG | R | Contribution | REG | R |
| Vcl(C1,O4) | 11.77 | 0.99 | ∑Vcl(O,H) | 1.14 | 0.92 ± 0.01 | Vcl(C1,O5) | 1.08 | 1.00 |
| Vcl(C1,O5) | 11.34 | 0.98 | Vcl(O4,C6) | 0.79 | 0.99 | Vcl(C1,O4) | 1.05 | 1.00 |
| Vcl(C1,H2) | 3.22 | 0.96 | Vcl(O5,C10) | 0.79 | 0.99 | Eintra(C10) | 0.50 | 0.97 |
| Vcl(C1,H3) | 3.22 | 0.96 | Eintra(C10) | 0.71 | 0.99 | Eintra(C6) | 0.50 | 0.97 |
| Vcl(O5,C6) | 2.92 | 0.99 | Eintra(C6) | 0.70 | 0.99 | Vxc(O4,O5) | 0.22 | 0.99 |
| Vcl(O4,C10) | 2.91 | 0.99 | Eintra(C1) | 0.58 | 0.72 | Vcl(C6,C10) | 0.20 | 0.95 |
| Vcl(H2,O4) | −1.93 | −0.96 | Vcl(C1,O5) | 0.33 | 0.60 | Vcl(C1,C6) | 0.20 | −0.96 |
| Vcl(H3,O5) | −1.93 | −0.96 | Vcl(O5,H8) | 0.33 | 1.00 | ∑Vcl(O,H) | −0.13 | −0.93 ± 0.02 |
| Vcl(H3,O4) | −1.97 | −0.96 | Vcl(C6,C10) | −0.39 | −0.97 | Eintra(O4) | −0.30 | −0.91 |
| Vcl(H2,O5) | −1.97 | −0.96 | Vcl(C1,H2) | −0.45 | −0.92 | Eintra(O5) | −0.33 | −0.93 |
| Eintra(O5) | −4.48 | −0.99 | Vcl(C1,H3) | −0.45 | −0.92 | Vcl(O4,O5) | −0.36 | −0.99 |
| Eintra(O4) | −4.70 | −0.99 | Vcl(O4,O5) | −0.55 | −1.00 | Vcl(O5,C6) | −0.56 | −0.89 |
| ∑Vcl(O,H) | −7.80 | −0.96 ± 0.00 | Vcl(O5,C6) | −0.66 | −0.98 | Vcl(O4,C10) | −0.57 | −0.89 |
| Eintra(C1) | −13.52 | −0.98 | Vcl(O4,C10) | −0.67 | −0.98 | Eintra(C1) | −0.57 | −0.99 |
| Contribution | ∆E(gg − tt) (kJ∙mol−1) |
|---|---|
| Eintra(C1) | −87.9 |
| ∑Vcl(H,O) | −75.6 |
| Vcl(O4,C6) | −29.6 |
| Vcl(O5,C10) | −29.3 |
| Vcl(O5,C6) | 34.3 |
| Vcl(O4,C10) | 34.8 |
| Vcl(C1,O5) | 50.1 |
| Vcl(C1,O4) | 50.1 |
| Atoms | Qtt | Qgg | ∆Qgg − tt |
|---|---|---|---|
| C1 | 1.0312 | 0.9927 | −0.0385 |
| H2 | −0.0086 | 0.0225 | 0.0311 |
| H3 | −0.0086 | 0.0225 | 0.0311 |
| O4 | −1.0358 | −1.0454 | −0.0095 |
| O5 | −1.0353 | −1.0456 | −0.0102 |
| C6 | 0.4962 | 0.4813 | −0.0149 |
| H7 | 0.0351 | 0.0262 | −0.0089 |
| H8 | −0.0015 | 0.0196 | 0.0211 |
| H9 | −0.0015 | −0.0004 | 0.0011 |
| C10 | 0.4960 | 0.4811 | −0.0148 |
| H11 | 0.0352 | 0.0262 | −0.0090 |
| H12 | −0.0015 | 0.0195 | 0.0209 |
| H13 | −0.0015 | −0.0004 | 0.0010 |
| Segment 1 | Segment 2 | ||||
|---|---|---|---|---|---|
| Contribution | REG | R | Contribution | REG | R |
| Vcl(C1,O15) | 1.33 | 0.82 | Eintra(C5) | 0.62 | 0.99 |
| Eintra(C5) | 1.29 | 0.99 | Eintra(O15) | 0.57 | 0.97 |
| Eintra(O15) | 1.18 | 0.97 | Vcl(C1,O15) | 0.37 | 0.76 |
| Vcl(C5,F16) | 0.46 | 0.61 | Vcl(C1,F16) | 0.33 | 0.97 |
| Vcl(C1,H10) | 0.37 | 0.91 | Vcl(H10,O15) | 0.26 | 0.91 |
| Vcl(O15,F16) | 0.37 | 0.99 | Vcl(C5,F16) | 0.20 | 0.65 |
| Vxc(C5,F16) | 0.36 | 0.92 | Vxc(C5,O15) | 0.01 | 0.16 |
| Vxc(C5,O15) | −0.19 | −0.63 | Vcl(H14,O15) | −0.11 | −0.96 |
| Eintra(F16) | −0.26 | −0.58 | Eintra(C1) | −0.12 | −0.54 |
| Vxc(C1,H10) | −0.39 | −0.99 | Eintra(F16) | −0.20 | −0.80 |
| Vcl(H10,O15) | −0.39 | −0.88 | Vcl(C1,H10) | −0.25 | −0.94 |
| Eintra(C1) | −1.00 | −0.85 | Vcl(C1,C5) | −0.31 | −0.88 |
| Vcl(C5,O15) | −3.69 | −0.96 | Vcl(C5,O15) | −1.47 | −0.99 |
| Segment 1 | Segment 2 | ||||
|---|---|---|---|---|---|
| Contribution | REG | R | Contribution | REG | R |
| Eintra(C1) | 1.56 | 0.95 | Vcl(C5,F16) | 3.73 | 0.99 |
| Eintra(C5) | 1.45 | 0.99 | Eintra(O15) | 1.06 | 0.93 |
| Vxc(C5,O15) | 0.96 | 0.95 | Vxc(C1,O15) | 0.78 | 0.98 |
| Vcl(C1,F16) | 0.94 | 0.94 | Vcl(C1,O15) | 0.70 | 0.75 |
| Vcl(H9,O15) | 0.73 | 0.97 | Vcl(C5,H13) | 0.66 | 0.97 |
| Vxc(C1,O15) | 0.66 | 0.97 | Vxc(C5,F16) | 0.62 | 0.99 |
| Vcl(H13,O15) | 0.48 | 0.94 | Eintra(H13) | 0.35 | 0.97 |
| Vxc(H14,O15) | 0.40 | 1.00 | Vcl(C5,H9) | 0.30 | 0.91 |
| Vxc(O15,F16) | −0.47 | −0.94 | Vxc(C5,H13) | −0.28 | −1.00 |
| Vcl(C5,H9) | −0.62 | −0.98 | Vcl(H9,O15) | −0.32 | −0.88 |
| Vcl(C5,F16) | −0.71 | −0.55 | Vxc(C5,O15) | −0.47 | −0.88 |
| Vcl(C1,H9) | −0.75 | −0.99 | Vcl(H13,O15) | −0.51 | −0.97 |
| Vcl(C5,H13) | −0.78 | −0.96 | Vcl(C1,C5) | −0.59 | −0.99 |
| Vcl(C1,O15) | −0.91 | −0.65 | Eintra(C5) | −1.27 | −0.97 |
| Vcl(C1,C5) | −1.03 | −0.88 | Eintra(F16) | −1.39 | −0.95 |
| Eintra(O15) | −1.11 | −0.99 | Vcl(C5,O15) | −3.34 | −0.93 |
| R Isomer | S Isomer | ||
|---|---|---|---|
| Contribution | ∆E (Chair Minus Twist-Boat) | Contribution | ∆E (Chair Minus Twist-Boat) |
| Vcl(H10,O15) | −18.53 | Vcl(C5,F16) | −96.47 |
| Eintra(C1) | −14.20 | Eintra(O15) | −39.15 |
| Vcl(C5,O15) | −9.03 | Vcl(C1,O15) | −36.96 |
| Vcl(C1,F16) | −7.80 | Vcl(C5,H13) | −27.14 |
| Vcl(H10,F16) | −7.39 | Vcl(C1,H9) | −18.34 |
| Vxc(C1,C4) | 7.18 | Eintra(C1) | 32.17 |
| Vcl(C1,O15) | 11.61 | Eintra(F16) | 36.07 |
| Vcl(C5,H10) | 14.23 | Eintra(C5) | 48.22 |
| Vcl(C1,H10) | 17.24 | Vcl(C5,O15) | 75.86 |
| R Isomer | S Isomer | |||||
|---|---|---|---|---|---|---|
| Atoms | Twist-Boat | Chair | Chair-Twist-Boat | Twist-Boat | Chair | Chair-Twist-Boat |
| C1 | 0.4649 | 0.4557 | −0.0092 | 0.4540 | 0.4778 | 0.0238 |
| C2 | 0.0431 | 0.0461 | 0.0030 | 0.0393 | 0.0455 | 0.0062 |
| C3 | 0.0488 | 0.0506 | 0.0019 | 0.0592 | 0.0472 | −0.0120 |
| C4 | 0.0485 | 0.0518 | 0.0033 | 0.0406 | 0.0444 | 0.0038 |
| C5 | 0.9782 | 0.9811 | 0.0029 | 0.9865 | 1.0053 | 0.0187 |
| H6 | −0.0125 | −0.0094 | 0.0031 | −0.0065 | −0.0037 | 0.0028 |
| H7 | −0.0057 | −0.0125 | −0.0068 | −0.0117 | −0.0021 | 0.0096 |
| H8 | 0.0103 | −0.0048 | −0.0151 | −0.0071 | −0.0062 | 0.0009 |
| H9 | 0.0274 | 0.0177 | −0.0096 | 0.0235 | −0.0083 | −0.0319 |
| H10 | −0.0053 | 0.0242 | 0.0295 | 0.0219 | 0.0289 | 0.0070 |
| H11 | −0.0033 | −0.0010 | 0.0023 | 0.0201 | 0.0164 | −0.0038 |
| H12 | 0.0141 | 0.0098 | −0.0043 | −0.0002 | 0.0043 | 0.0045 |
| H13 | 0.0466 | 0.0510 | 0.0044 | 0.0504 | 0.0226 | −0.0278 |
| H14 | 0.0069 | 0.0052 | −0.0016 | −0.0056 | −0.0197 | −0.0141 |
| O15 | −1.0438 | −1.0462 | −0.0025 | −1.0454 | −1.0344 | 0.0110 |
| F16 | −0.6184 | −0.6191 | −0.0008 | −0.6192 | −0.6171 | 0.0022 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, D.; Duarte, L.J.; Popelier, P.L.A. An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Analysis of the Anomeric Effect. Molecules 2022, 27, 5003. https://doi.org/10.3390/molecules27155003
Khan D, Duarte LJ, Popelier PLA. An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Analysis of the Anomeric Effect. Molecules. 2022; 27(15):5003. https://doi.org/10.3390/molecules27155003
Chicago/Turabian StyleKhan, Danish, Leonardo J. Duarte, and Paul L. A. Popelier. 2022. "An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Analysis of the Anomeric Effect" Molecules 27, no. 15: 5003. https://doi.org/10.3390/molecules27155003
APA StyleKhan, D., Duarte, L. J., & Popelier, P. L. A. (2022). An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Analysis of the Anomeric Effect. Molecules, 27(15), 5003. https://doi.org/10.3390/molecules27155003