
Three New Acrylic Acid Derivatives from Achillea mellifolium as Potential Inhibitors of Urease from Jack Bean and α-Glucosidase from Saccharomyces cerevisiae

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
2.1. Characterization of the Isolated Compounds
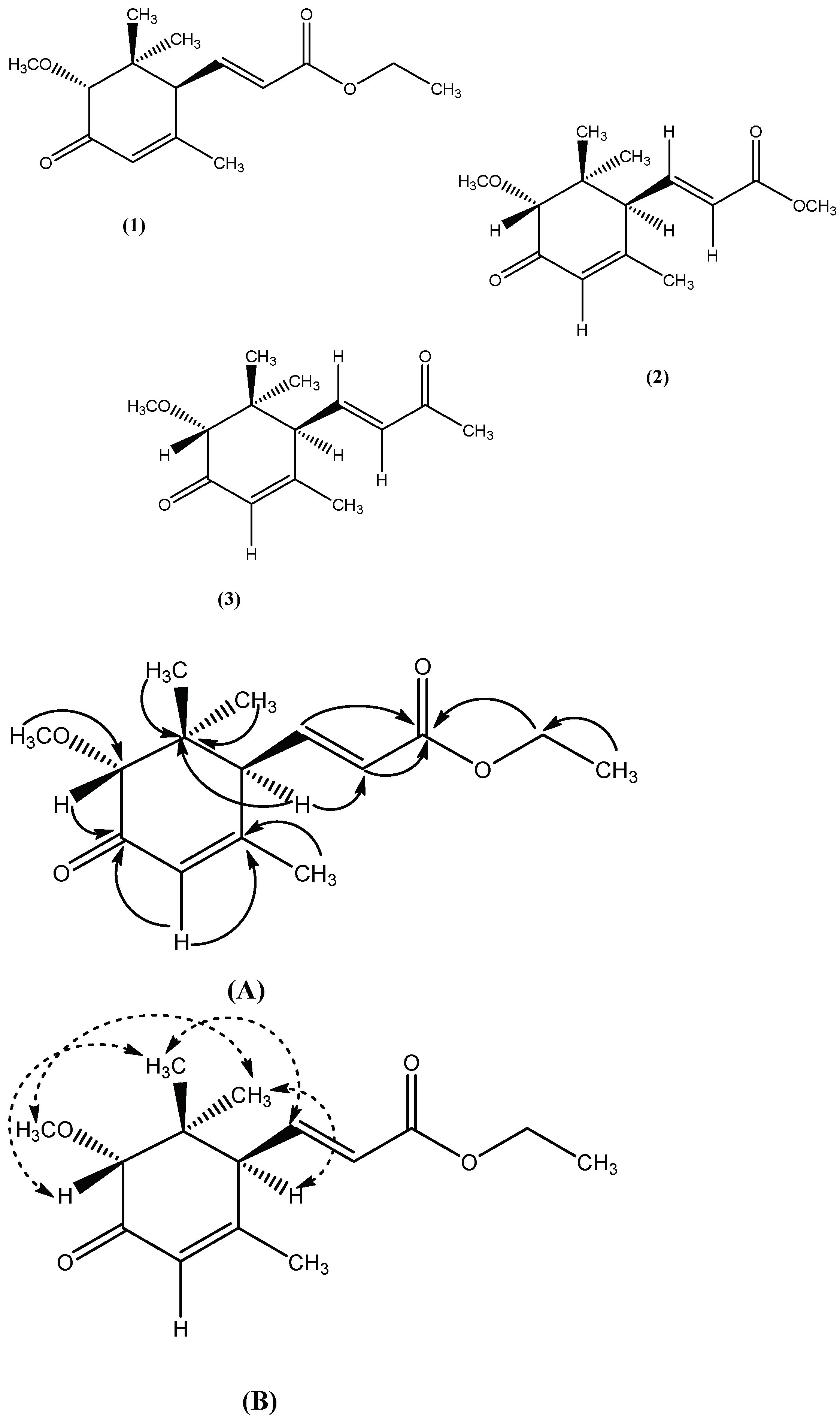
2.1.1. Characterization of Compound 1
2.1.2. Characterization of Compound 2
2.1.3. Characterization of Compound 3
 ), (B). NOE correlation (
), (B). NOE correlation ( ).
), (B). NOE correlation ().
).
), (B). NOE correlation ().
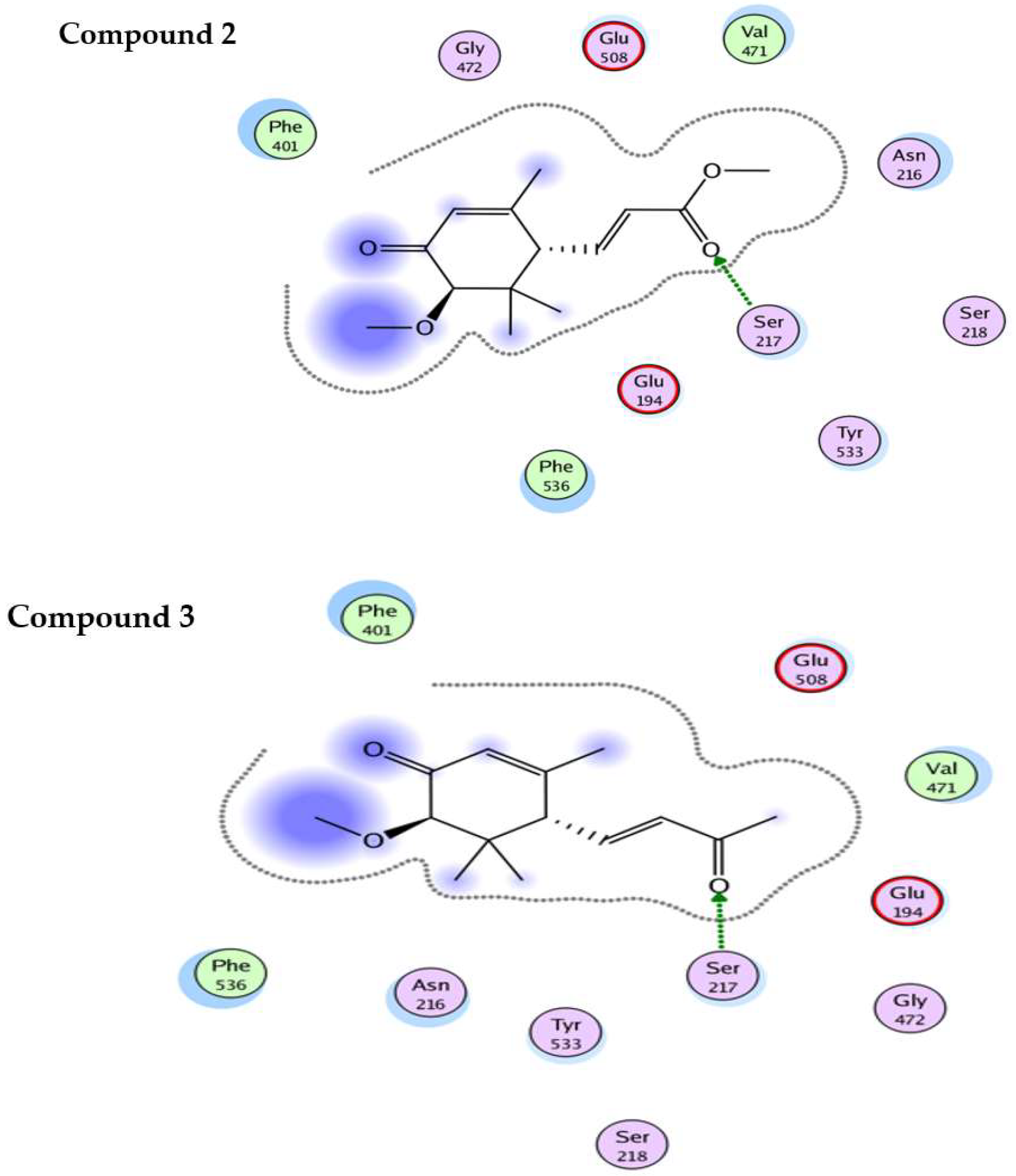
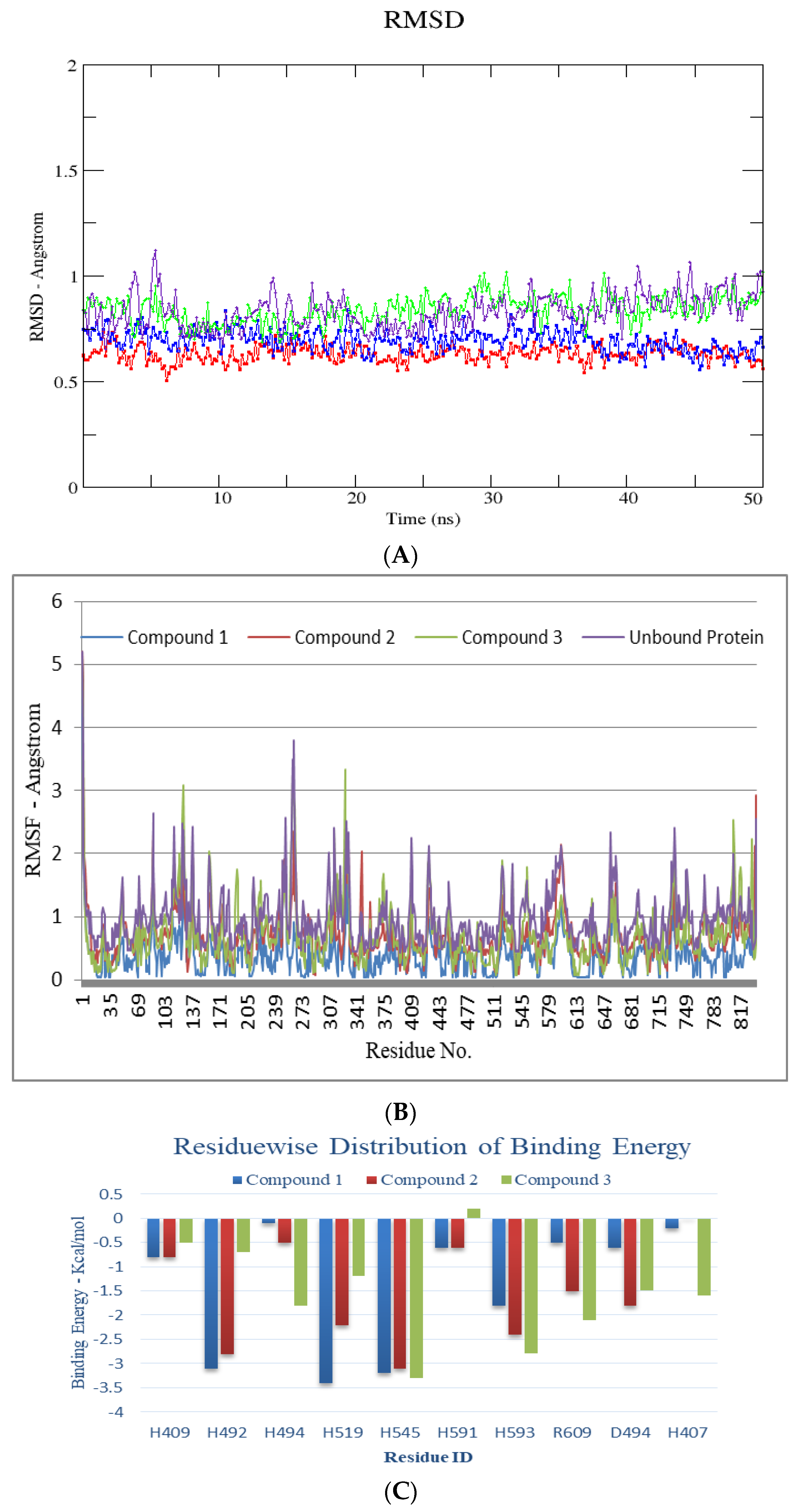
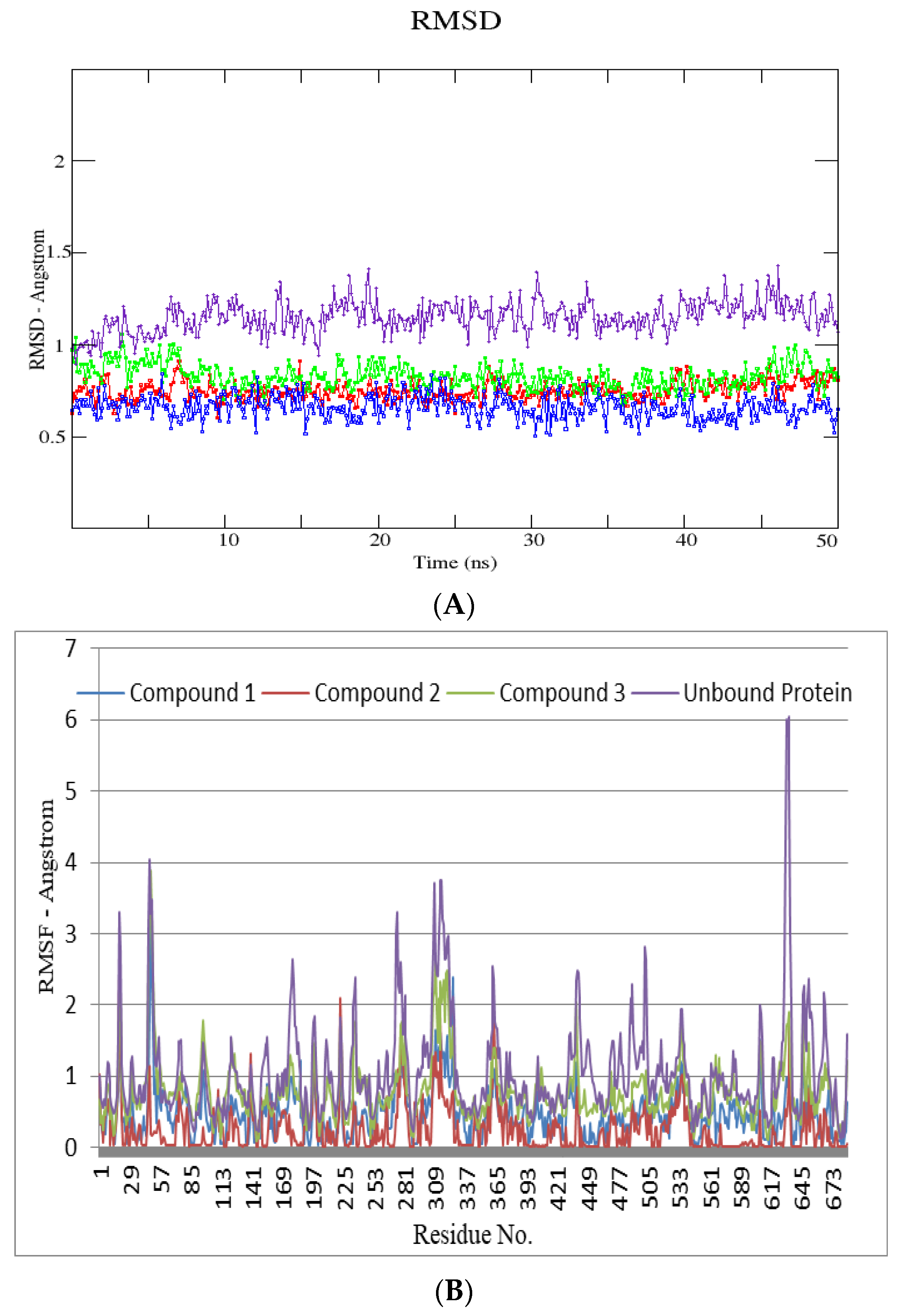
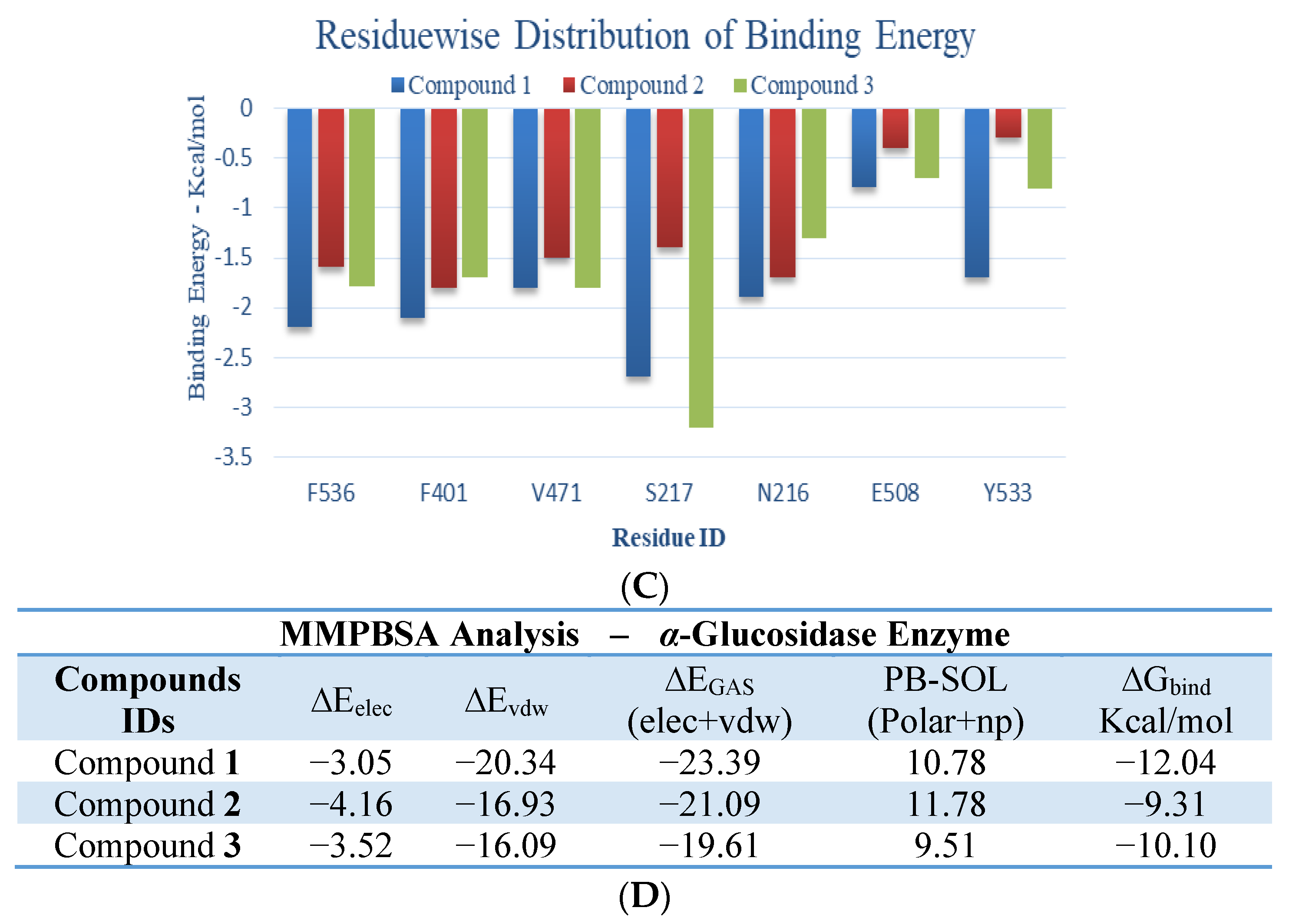
3. Discussion
4. Materials and Methods
4.1. General Experimental Techniques
4.2. Plant Material
4.3. Extraction and Isolation
4.4. Enzyme Inhibition Studies
4.4.1. Urease Inhibition Assay
4.4.2. α-Glucosidase Inhibition Assay
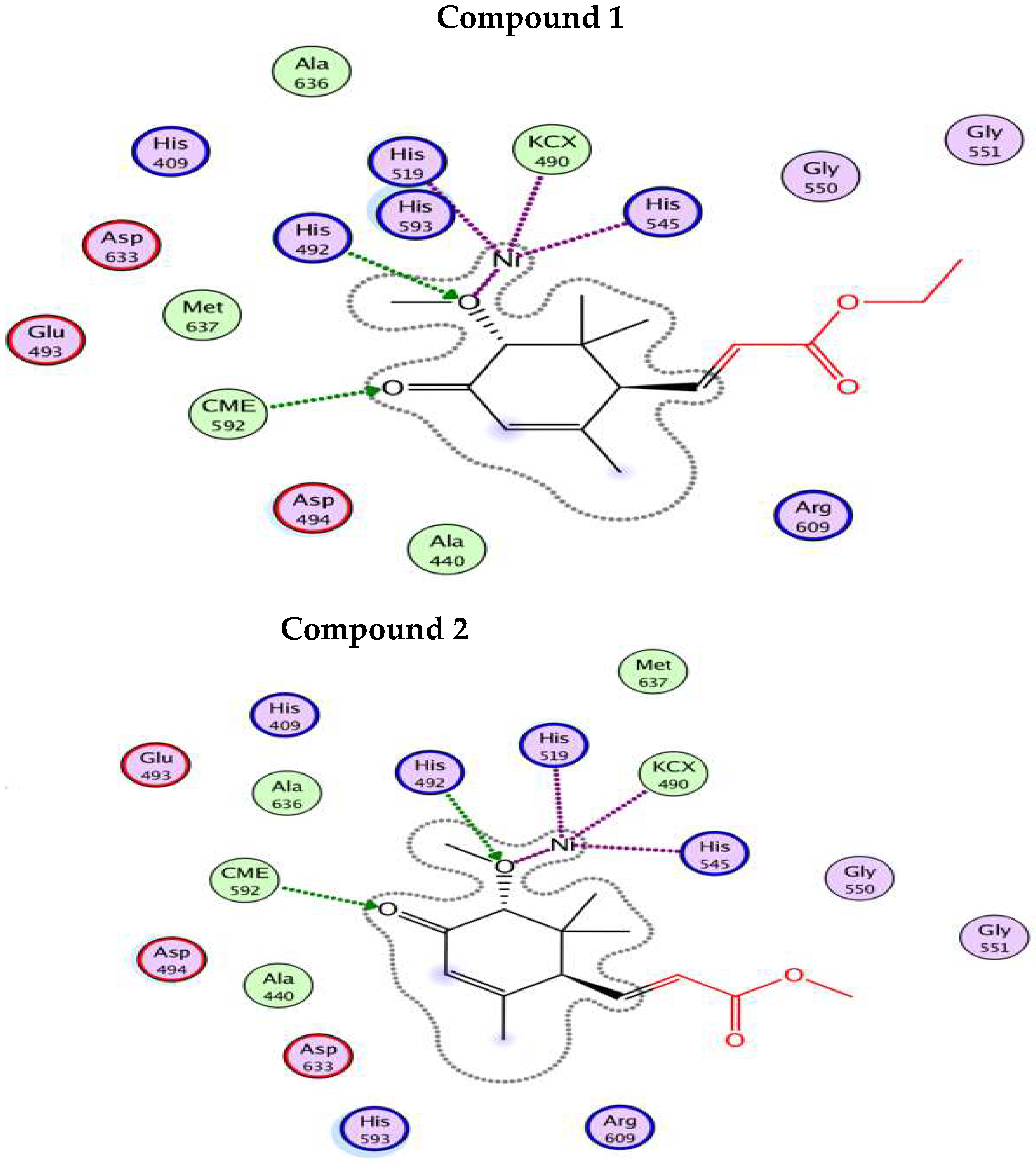
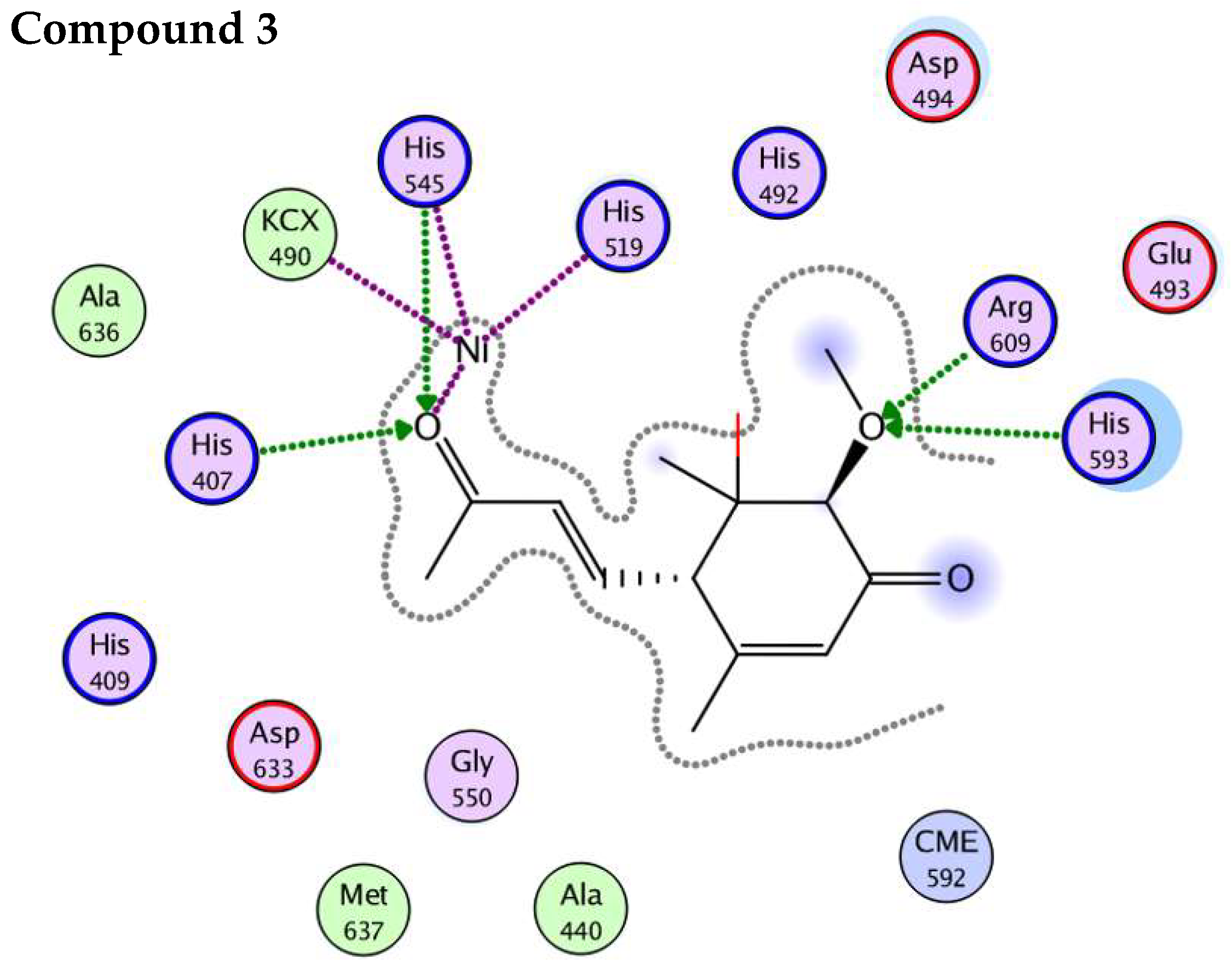
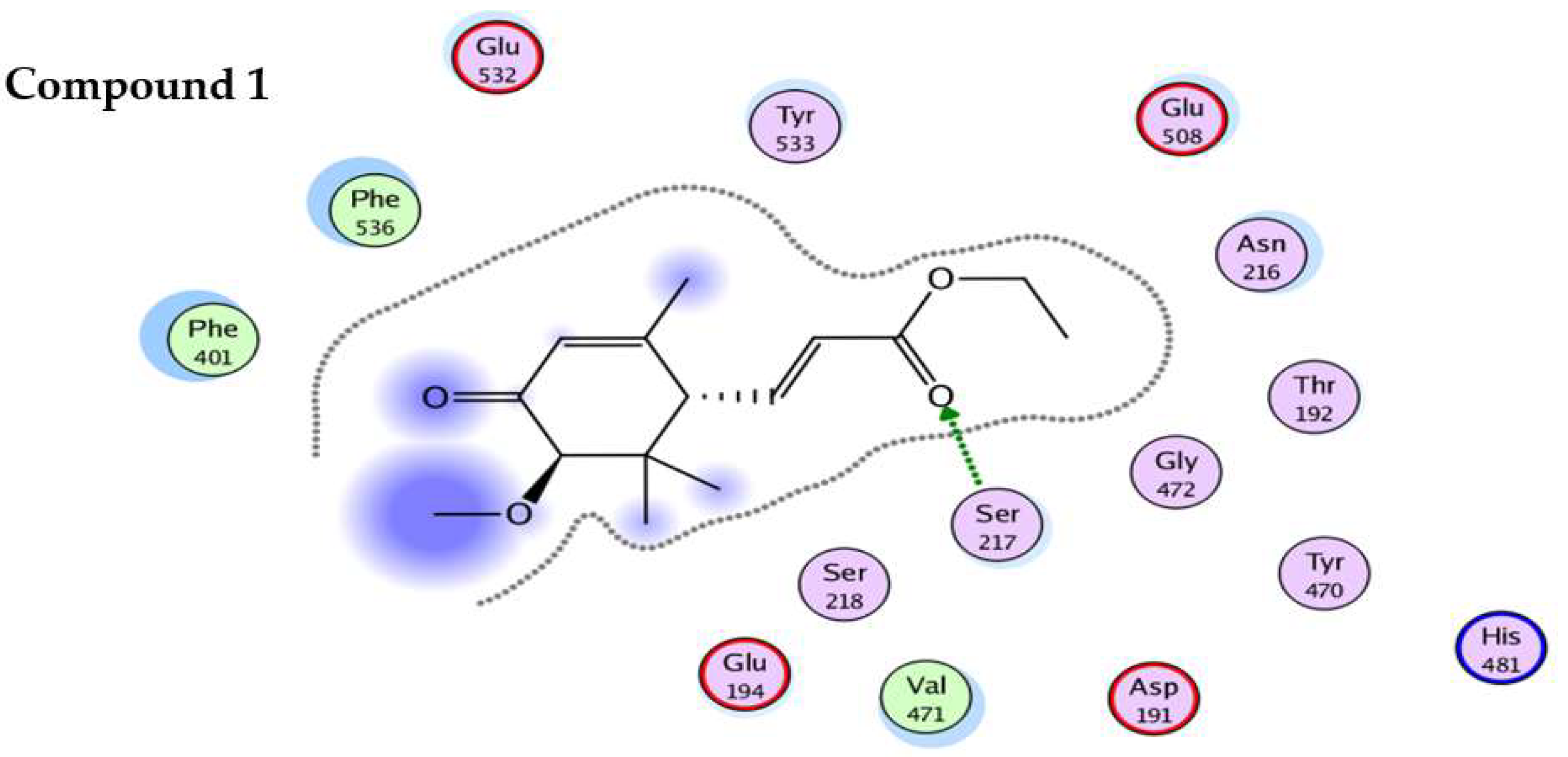
4.5. In-Silico Studies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Farooq, U.; Khan, A.; Khan, S.S.; Iqbal, S.; Sarwar, R.; Khan, S.B.; Ahmad, V.U. Isolation and structure determination of three new sesquiterpenoids from Achillea millefolium. Z. Für Nat. B 2012, 67, 421–425. [Google Scholar] [CrossRef]
- Könemann, B. The Illustrated AZ of over 10,000 Garden Plants and How to Cultivate Them; Gordon Cheers Publication: Hong Kong, China, 1999; pp. 51–53. [Google Scholar]
- Chandler, R.; Hooper, S.; Hooper, D.; Jamieson, W.; Flinn, C.; Safe, L. Herbal remedies of the Maritime Indians: Sterols and triterpenes of Achillea millefolium L.(Yarrow). J. Pharm. Sci. 1982, 71, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, M.; Goldberg, A.; Brinckmann, J. Yarrow. Herbal Medicine Expanded Commission E Monographs; Integrative Medicine Communications: Boston, MA, USA, 2000; pp. 419–423. [Google Scholar]
- Dall’Acqua, S.; Bolego, C.; Cignarella, A.; Gaion, R.M.; Innocenti, G. Vasoprotective activity of standardized Achillea millefolium extract. Phytomedicine 2011, 18, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Baytop, T. Türkiye’de bitkiler ile tedavi (Treatment with Plants in Turkey); Istanbul University Publications: Istanbul, Turkey, 1999; p. 40. [Google Scholar]
- Cavalcanti, A.M.; Baggio, C.; Freitas, C.S.; Rieck, L.; de Sousa, R.S.; da Silva-Santos, J.E.; Mesia-Vela, S.; Marques, M.C.A. Safety and antiulcer efficacy studies of Achillea millefolium L. after chronic treatment in Wistar rats. J. Ethnopharmacol. 2006, 107, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Schulz, V.; Hänsel, R.; Tyler, V.E. Rational Phytotherapy: A Physician’s Guide to Herbal Medicine; Psychology Press: London, UK, 2001. [Google Scholar]
- Potrich, F.B.; Allemand, A.; da Silva, L.M.; dos Santos, A.C.; Baggio, C.H.; Freitas, C.S.; Mendes, D.A.G.B.; Andre, E.; Werner, M.F.D.P.; Marques, M.C.A. Antiulcerogenic activity of hydroalcoholic extract of Achillea millefolium L.: Involvement of the antioxidant system. J. Ethnopharmacol. 2010, 130, 85–92. [Google Scholar] [CrossRef]
- Georgieva, L.; Gadjalova, A.; Mihaylova, D.; Pavlov, A. Achillea millefolium L.-phytochemical profile and in-vitro antioxidant activity. Int. Food Res. J. 2015, 22, 1347. [Google Scholar]
- Trumbeckaite, S.; Benetis, R.; Bumblauskiene, L.; Burdulis, D.; Janulis, V.; Toleikis, A.; Viškelis, P.; Jakštas, V. Achillea millefolium L. s.l. herb extract: Antioxidant activity and effect on the rat heart mitochondrial functions. Food Chem. 2011, 127, 1540–1548. [Google Scholar] [CrossRef]
- Pires, J.M.; Mendes, F.R.; Negri, G.; Duarte-Almeida, J.M.; Carlini, E.A. Antinociceptive peripheral effect of Achillea millefolium L. and Artemisia vulgaris L.: Both plants known popularly by brand names of analgesic drugs. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2009, 23, 212–219. [Google Scholar] [CrossRef]
- Goldberg, A.S.; Mueller, E.C.; Eigen, E.; Desalva, S.J. Isolation of the anti-inflammatory principles from Achillea millefolium (Compositae). J. Pharm. Sci. 1969, 58, 938–941. [Google Scholar] [CrossRef]
- Tunon, H.; Olavsdotter, C.; Bohlin, L. Evaluation of anti-inflammatory activity of some Swedish medicinal plants. Inhibition of prostaglandin biosynthesis and PAF-induced exocytosis. J. Ethnopharmacol. 1995, 48, 61–76. [Google Scholar] [CrossRef]
- TOzYO, T.; Yoshimura, Y.; Sakurai, K.; Uchida, N.; Takeda, Y.; Nakai, H.; Ishii, H. Novel antitumor sesquiterpenoids in Achillea millefolium. Chem. Pharm. Bull. 1994, 42, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Baggio, C.H.; Freitas, C.S.; Nhaducue, P.F.; Rieck, L.; Marques, M.C.A. Action of crude aqueous extract of leaves of Achillea millefolium L.(Compositae) on gastrointestinal tract. Rev. Bras. De Farmacogn. 2002, 12, 31–33. [Google Scholar] [CrossRef]
- Lin, L.T.; Liu, L.T.; Chiang, L.C.; Lin, C.C. In-vitro anti-hepatoma activity of fifteen natural medicines from Canada. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2002, 16, 440–444. [Google Scholar] [CrossRef]
- Kintzios, S.; Papageorgiou, K.; Yiakoumettis, I.; Baričevič, D.; Kušar, A. Evaluation of the antioxidants activities of four Slovene medicinal plant species by traditional and novel biosensory assays. J. Pharm. Biomed. Anal. 2010, 53, 773–776. [Google Scholar] [CrossRef]
- Miller, F.; Chow, L.M. Alkaloids of Achillea millefolium LI Isolation and Characterization of Achilleine1, 2. J. Am. Chem. Soc. 1954, 76, 1353–1354. [Google Scholar] [CrossRef]
- Haggag, M.Y.; Shalaby, A.; Verzar–Petri, G. Thin layer and gas–chromatographic studies on the essential oil from Achillea millefolium. Planta Med. 1975, 27, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Kokkalou, E.; Kokkini, S.; Handidou, E. Volatile constituents of Achillea millefolium in relation to their infraspecific variation. Biochem. Syst. Ecol. 1992, 20, 664–670. [Google Scholar] [CrossRef]
- Kubelka, W.; Kastner, U.; Glasl, S.; Saukel, J.; Jurenitsch, J. Chemotaxonomic relevance of sesquiterpenes within the Achillea millefolium group. Biochem. Syst. Ecol. 1999, 27, 437–444. [Google Scholar] [CrossRef]
- Souza, T.; Rangel, V.L.B.I.; Pietro, R.C.L.R.; Santos, L.E.; Moreira, R.R.D. Phytochemical screening of Achillea Millefolium harvested at Araraquara–SP. Rev. Bras. Plant Med. Botucatu. 2006, 8, 151–154. [Google Scholar]
- Guédon, D.; Abbe, P.; Lamaison, J.L. Leaf and flower head flavonoids of Achillea millefolium L. subspecies. Biochem. Syst. Ecol. 1993, 21, 607–611. [Google Scholar] [CrossRef]
- Valant-Vetschera, K.; Wollenweber, E. Leaf flavonoids of the Achillea millefolium group part II: Distribution patterns of free aglycones in leaf exudates. Biochem. Syst. Ecol. 1988, 16, 605–614. [Google Scholar] [CrossRef]
- Orav, A.; Arak, E.; Raal, A. Phytochemical analysis of the essential oil of Achillea millefolium L. from various European Countries. Nat. Prod. Res. 2006, 20, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Kosikowska, P.; Berlicki, Ł. Urease inhibitors as potential drugs for gastric and urinary tract infections: A patent review. Expert Opin. Ther. Pat. 2011, 21, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Golbabaei, S.; Bazl, R.; Golestanian, S.; Nabati, F.; Omrany, Z.B.; Yousefi, B.; Hajiaghaee, R.; Rezazadeh, S.; Amanlou, M. Urease inhibitory activities of β-boswellic acid derivatives. DARU J. Pharm. Sci. 2013, 21, 1–6. [Google Scholar] [CrossRef]
- Kafarski, P.; Talma, M. Recent advances in design of new urease inhibitors: A review. J. Adv. Res. 2018, 13, 101–112. [Google Scholar] [CrossRef]
- Krajewska, B.; Zaborska, W.; Leszko, M.; Brzózka, Z. Inhibition of jack bean urease by a mixture of boric acid and phosphate buffer pH 6.96. Pol. J. Chem. 1999, 73, 359–366. [Google Scholar]
- Chaidam, S.; Saehlim, N.; Athipornchai, A.; Sirion, U.; Saeeng, R. Synthesis and biological evaluation of 1,6-bis-triazole-2,3,4-tri-O-benzyl-α-d-glucopyranosides as a novel α-glucosidase inhibitor in the treatment of Type 2 diabetes. Bioorganic Med. Chem. Lett. 2021, 50, 128331. [Google Scholar] [CrossRef]
- Aminu, K.S.; Uzairu, A.; Umar, A.B.; Ibrahim, M.T. Salicylic acid derivatives as potential α-glucosidase inhibitors: Drug design, molecular docking and pharmacokinetic studies. Bull. Natl. Res. Centre 2022, 46, 162. [Google Scholar] [CrossRef]
- Xu, W.; Yang, J.; Zhu, X.; Hu, Y.K.; Xu, S.H.; Li, Y.Y.; Zhao, Y. Ionol Derivatives from Euphorbia tirucalli. Rec. Nat. Prod. 2017, 11, 285–289. [Google Scholar]
- Hassan, S.T.; Žemlička, M. Plant-derived urease inhibitors as alternative chemotherapeutic agents. Arch. Der Pharm. 2016, 349, 507–522. [Google Scholar] [CrossRef]
- Mobley, H.; Hausinger, R. Microbial ureases: Significance, regulation, and molecular characterization. Microbiol. Rev. 1989, 53, 85–108. [Google Scholar] [CrossRef]
- Moradi-Afrapoli, F.; Asghari, B.; Saeidnia, S.; Ajani, Y.; Mirjani, M.; Malmir, M.; Dolatabadi Bazaz, R.; Hadjiakhoondi, A.; Salehi, P.; Hamburger, M.; et al. In vitro α-glucosidase inhibitory activity of phenolic constituents from aerial parts of Polygonum hyrcanicum. DARU J. Pharm. Sci. 2012, 20, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gloster, T.M.; Turkenburg, J.P.; Potts, J.R.; Henrissat, B.; Davies, G.J. Divergence of catalytic mechanism within a glycosidase family provides insight into evolution of carbohydrate metabolism by human gut flora. Chem. Biol. 2008, 15, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Zaman, Z.; Khan, S.; Nouroz, F.; Farooq, U.; Urooj, A. Targeting protein tyrosine phosphatase to unravel possible inhibitors for Streptococcus pneumoniae using molecular docking, molecular dynamics simulations coupled with free energy calculations. Life Sci. 2021, 264, 118621. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon No. | Compound 1 | Compound 2 | Compound 3 |
|---|---|---|---|
| 1H-NMR (δH ppm) | 1H-NMR (δH ppm) | 1H-NMR (δH ppm) | |
| 1 | - | - | - |
| 2 | 3.80 (1H, s, H-2) | 3.74 (1H, s, H-2) | 3.69 (1H, s, H-2) |
| 3 | - | - | - |
| 4 | 5.80 (1H, s, H-4) | 5.82 (1H, s, H-4) | 5.85 (1H, s, H-4) |
| 5 | - | - | - |
| 6 | 2.81 (1H, d, J = 8.4 Hz, H-6) | 2.78 (1H, d, J = 8.8 Hz, H-6) | 2.80 (1H, d, J = 8.0 Hz, H-6) |
| 7 | 6.30 (1H, dd, J = 15.8, 7.9 Hz, H-7) | 6.28 (1H, dd, J = 16.4, 8.8 Hz, H-7) | 6.27 (1H, dd, J = 16.1, 8.5 Hz, H-7) |
| 8 | 5.98 (1H, d, J = 15.8 Hz, H-8) | 5.92 (1H, d, J = 16.4 Hz, H-8) | 5.96 (1H, d, J = 15.7 Hz, H-8) |
| 9 | - | - | - |
| 10 | 3.78 (2H, q, J = 6.8 Hz, H-10) | 3.85 (3H, s, H-10) | 2.10 (3H, s, H-10) |
| 11 | 1.35 (3H, t, J = 8.2 Hz, H-11) | 0.92 (3H, s, H-11) | 0.97 (3H, s, H-11) |
| 12 | 0.98 (3H, s, H-12) | 1.08 (3H, s, H-12) | 1.02 (3H, s, H-12) |
| 13 | 1.02 (3H, s, H-13) | 3.35 (3H, s, H-13) | 3.40 (3H, s, H-13) |
| 14 | 3.42 (3H, s, H-14) | 1.90 (3H, s, H-14) | 1.88 (3H, s, H-14) |
| 15 | 1.95 (3H, s, H-15) | - | - |
| Carbon No. | Compound 1 | Compound 2 | Compound 3 |
|---|---|---|---|
| 13C-NMR (δC ppm) | 13C-NMR (δC ppm) | 13C-NMR (δC ppm) | |
| 1 | 39.6 | 38.2 | 40.2 |
| 2 | 72.1 | 70.8 | 72.3 |
| 3 | 194.8 | 192.1 | 193.6 |
| 4 | 125.1 | 126.7 | 128.7 |
| 5 | 164.4 | 167.1 | 165.6 |
| 6 | 59.2 | 58.1 | 59.1 |
| 7 | 132.4 | 134.8 | 131.8 |
| 8 | 137.6 | 141.1 | 136.5 |
| 9 | 170.1 | 172.1 | 184.4 |
| 10 | 70.6 | 56.1 | 24.1 |
| 11 | 18.1 | 19.8 | 20.8 |
| 12 | 19.6 | 23.4 | 23.6 |
| 13 | 23.6 | 57.9 | 22.2 |
| 14 | 55.7 | 22.6 | 57.2 |
| 15 | 22.6 | - | - |
| S. No. | Urease Inhibition (IC50 ± S.E.M); μΜ | Binding Score (kcal/mol) | α Glucosidase Inhibition (IC50 ± S.E.M); μΜ | Binding Score (kcal/mol) |
|---|---|---|---|---|
| Comp-1 | 16.87 ± 0.02 | −7.011 | 331.47 ± 0.04 | −3.291 |
| Comp-2 | 13.71 ± 0.07 | −7.224 | 294.18 ± 0.07 | −2.783 |
| Comp-3 | 10.46 ± 0.03 | −9.831 | 310.68 ± 0.05 | −4.103 |
| Thiourea (standard) | 21.5 ± 0.01 | −3.332 | - | - |
| Acarbose (standard) | - | - | 287.1 ± 0.03 | −8.462 |
| Name | Compound 1 | Compound 2 | Compound 3 |
|---|---|---|---|
| Mol. weight | 266.33 | 252.31 | 236.31 |
| Number of hydrogen bond acceptors | 26 | 24 | 23 |
| Number of hydrogen bond donors | 0 | 0 | 0 |
| Number of rotatable bonds | 5 | 4 | 3 |
| Molecular refractivity | 73.47 | 68.66 | 67.58 |
| Topological Polar Surface Area | 52.6 | 52.6 | 43.37 |
| octanol/water partition coefficient(logP) | 2.29 | 1.9 | 2.32 |
| GI Absorption | High | ||
| Predicted Toxicity Class | 4 | 4 | 5 |
| Lipinski Rule Violation | 0 | ||
| Predicted LD50 | 900 mg/kg | 900 mg/kg | 2842 mg/kg |
| Toxicity Test | Non-toxic All | Non-toxic All | Non-toxic all except Mitochondrial membrane potential |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farooq, U.; Khan, S.; Naz, S.; Wani, T.A.; Bukhari, S.M.; Aborode, A.T.; Shahzad, S.A.; Zargar, S. Three New Acrylic Acid Derivatives from Achillea mellifolium as Potential Inhibitors of Urease from Jack Bean and α-Glucosidase from Saccharomyces cerevisiae. Molecules 2022, 27, 5004. https://doi.org/10.3390/molecules27155004
Farooq U, Khan S, Naz S, Wani TA, Bukhari SM, Aborode AT, Shahzad SA, Zargar S. Three New Acrylic Acid Derivatives from Achillea mellifolium as Potential Inhibitors of Urease from Jack Bean and α-Glucosidase from Saccharomyces cerevisiae. Molecules. 2022; 27(15):5004. https://doi.org/10.3390/molecules27155004
Chicago/Turabian StyleFarooq, Umar, Sara Khan, Sadia Naz, Tanveer A. Wani, Syed Majid Bukhari, Abullahi Tunde Aborode, Sohail Anjum Shahzad, and Seema Zargar. 2022. "Three New Acrylic Acid Derivatives from Achillea mellifolium as Potential Inhibitors of Urease from Jack Bean and α-Glucosidase from Saccharomyces cerevisiae" Molecules 27, no. 15: 5004. https://doi.org/10.3390/molecules27155004
APA StyleFarooq, U., Khan, S., Naz, S., Wani, T. A., Bukhari, S. M., Aborode, A. T., Shahzad, S. A., & Zargar, S. (2022). Three New Acrylic Acid Derivatives from Achillea mellifolium as Potential Inhibitors of Urease from Jack Bean and α-Glucosidase from Saccharomyces cerevisiae. Molecules, 27(15), 5004. https://doi.org/10.3390/molecules27155004