
Repositioning of Quinazolinedione-Based Compounds on Soluble Epoxide Hydrolase (sEH) through 3D Structure-Based Pharmacophore Model-Driven Investigation



 , , , ,
, , , , 
Abstract
1. Introduction
2. Results and Discussion
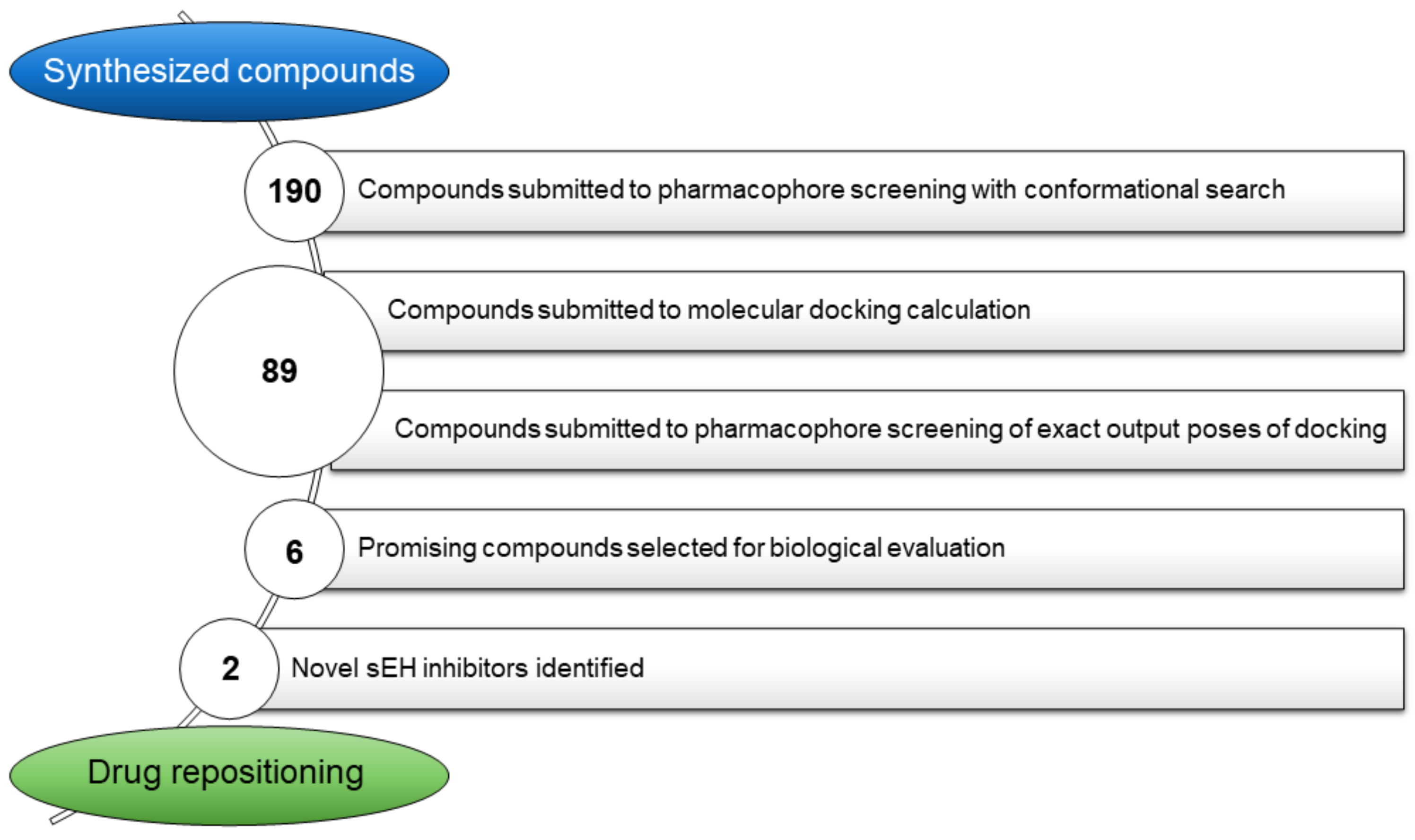
2.1. Original Building of the Library of Quinazolinedione-Based Compounds and Virtual Screening on Hsp90
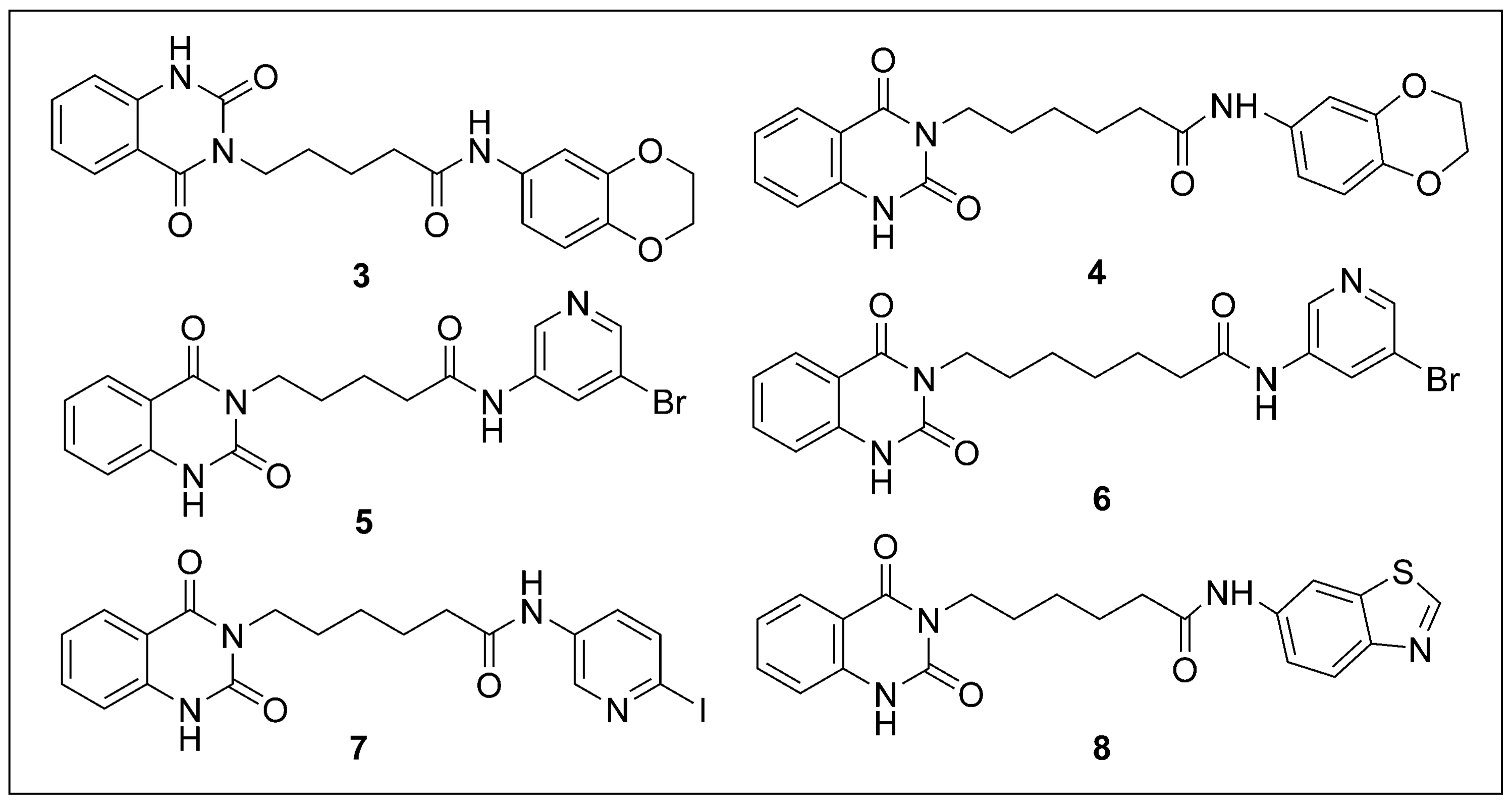
2.2. Chemical Synthesis
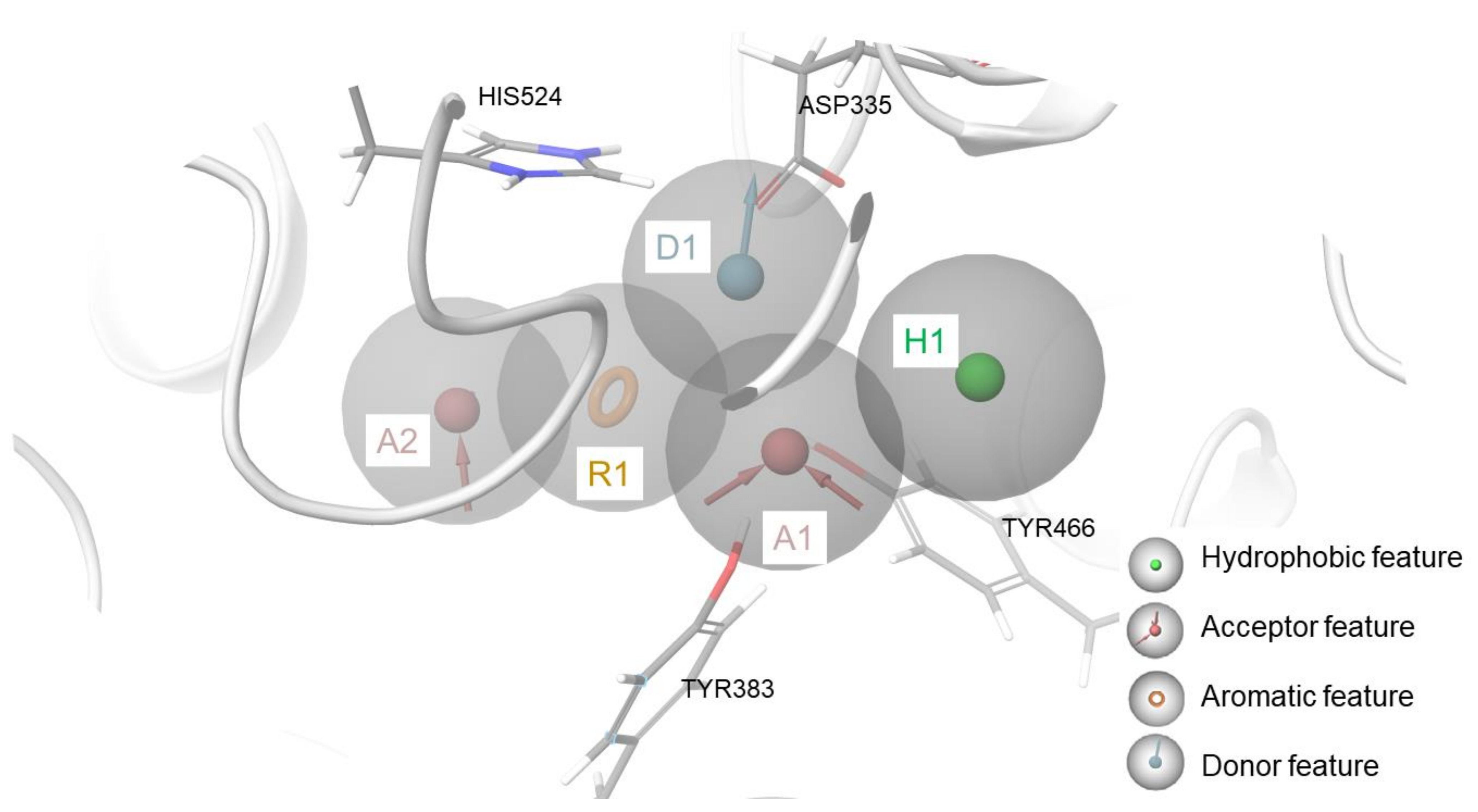
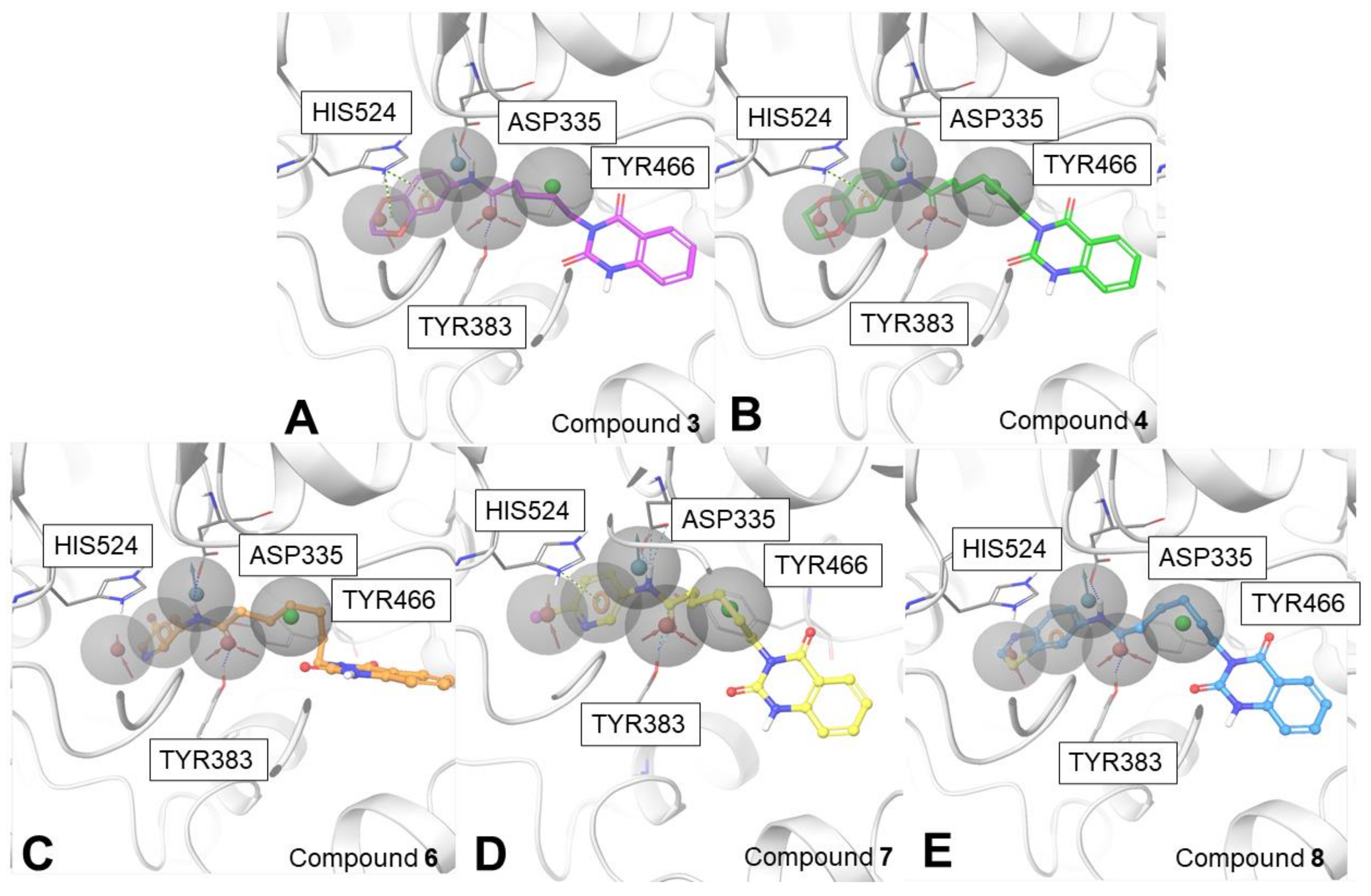
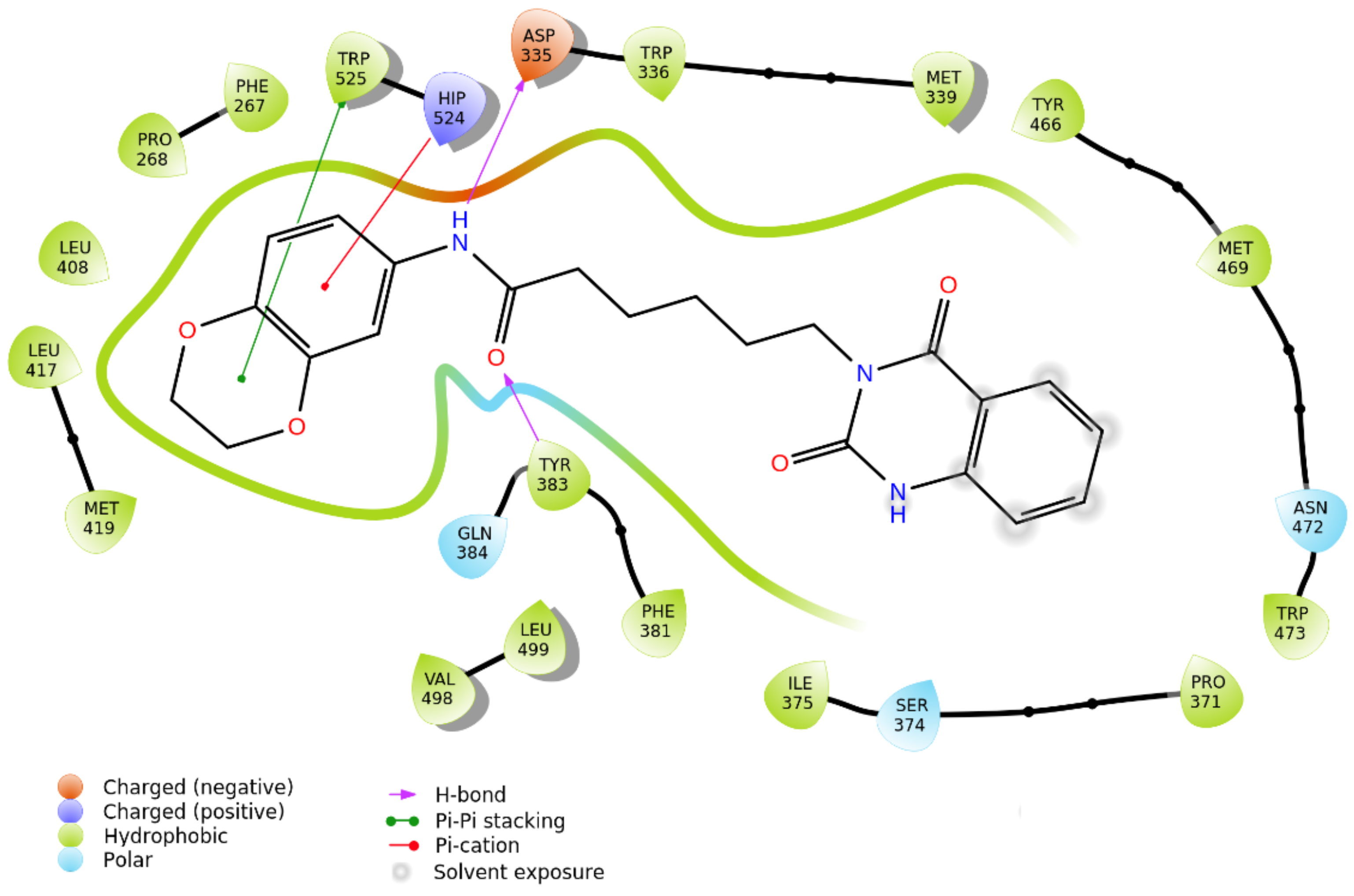
2.3. Biophysical Assays on HSP90 and Repositioning on Soluble Epoxide Hydrolase (sEH) through 3D Structure-Based Pharmacophore Model-Driven Investigation
2.4. Biological Evaluation on sEH
3. Materials and Methods
3.1. Computational Details
3.1.1. Preparation of the Library
3.1.2. Molecular Docking Experiments on sEH
3.1.3. Development of the 3D Structure-Based Pharmacophore Model for sEH
3.1.4. Pharmacophore Screening
3.2. Chemical Synthesis
3.2.1. General Procedure (A) for the Synthesis of 2a–2c
6-(2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)hexanoic acid (2b)
3.2.2. General Procedure (B) for the Synthesis of 3–8
N-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-5-(2,4-dioxo-1,2dihydroquinazolin-3(4H) yl)pentanamide 3
N-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-6-(2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)hexanamide 4
N-(5-bromopyridin-3-yl)-5-(2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)pentanamide 5
N-(5-bromopyridin-3-yl)-7-(2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)heptanamide 6
6-(2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)-N-(6-iodopyridin-3-yl)hexanamide 7
N-(benzo[d]thiazol-6-yl)-6-(2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)hexanamide 8
3.3. SPR Assays on Hsp90
3.4. Biological Evalutaion on sEH
3.4.1. Expression, Purification, and Activity Assay of Human Recombinant sEH
3.4.2. Cell Viability Assay on PBMC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Berdigaliyev, N.; Aljofan, M. An overview of drug discovery and development. Future Med. Chem. 2020, 12, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Dudley, J.T.; Deshpande, T.; Butte, A.J. Exploiting drug–disease relationships for computational drug repositioning. Brief. Bioinform. 2011, 12, 303–311. [Google Scholar] [CrossRef]
- Pierri, M.; Gazzillo, E.; Chini, M.G.; Ferraro, M.G.; Piccolo, M.; Maione, F.; Irace, C.; Bifulco, G.; Bruno, I.; Terracciano, S.; et al. Introducing structure-based three-dimensional pharmacophore models for accelerating the discovery of selective BRD9 binders. Bioorg. Chem. 2022, 118, 105480. [Google Scholar] [CrossRef]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef] [PubMed]
- McReynolds, C.; Hwang, S.H.; Yang, J.; Wan, D.; Wagner, K.; Morisseau, C.; Li, D.; Schmidt, W.; Hammock, B.D. Pharmaceutical effects of inhibiting the soluble epoxide hydrolase in canine osteoarthritis. Front. Pharmacol. 2019, 10, 533. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Xuan, W.; Fan, G.H. Roles of resolvins in the resolution of acute inflammation. Cell Biol. Int. 2015, 39, 3–22. [Google Scholar] [CrossRef]
- Schmelzer, K.R.; Kubala, L.; Newman, J.W.; Kim, I.-H.; Eiserich, J.P.; Hammock, B.D. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9772–9777. [Google Scholar] [CrossRef]
- Testa, B. 5.06—Principles of Drug Metabolism 2: Hydrolysis and Conjugation Reactions. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 133–166. [Google Scholar]
- Newman, J.W.; Morisseau, C.; Hammock, B.D. Epoxide hydrolases: Their roles and interactions with lipid metabolism. Prog. Lipid Res. 2005, 44, 1–51. [Google Scholar] [CrossRef]
- Bettaieb, A.; Koike, S.; Chahed, S.; Zhao, Y.; Bachaalany, S.; Hashoush, N.; Graham, J.; Fatima, H.; Havel, P.J.; Gruzdev, A.; et al. Podocyte-specific soluble epoxide hydrolase deficiency in mice attenuates acute kidney injury. FEBS J. 2017, 284, 1970–1986. [Google Scholar] [CrossRef]
- Fulton, D.; Falck, J.; McGiff, J.; Carroll, M.; Quilley, J. A method for the determination of 5, 6-EET using the lactone as an intermediate in the formation of the diol. J. Lipid Res. 1998, 39, 1713–1721. [Google Scholar] [CrossRef]
- Iyer, M.R.; Kundu, B.; Wood, C.M. Soluble epoxide hydrolase inhibitors: An overview and patent review from the last decade. Expert Opin. Ther. Pat. 2022, 32, 629–647. [Google Scholar] [CrossRef]
- Pecic, S.; Deng, S.-X.; Morisseau, C.; Hammock, B.D.; Landry, D.W. Design, synthesis and evaluation of non-urea inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett. 2012, 22, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Morisseau, C.; Goodrow, M.H.; Dowdy, D.; Zheng, J.; Greene, J.F.; Sanborn, J.R.; Hammock, B.D. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc. Natl. Acad. Sci. USA 1999, 96, 8849–8854. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Yang, L.; Zhu, Y. Pharmacophore Based Drug Design Approach as a Practical Process in Drug Discovery. Curr. Comput.-Aided Drug Des. 2010, 6, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Moser, D.; Achenbach, J.; Klingler, F.-M.; Estel la, B.; Hahn, S.; Proschak, E. Evaluation of structure-derived pharmacophore of soluble epoxide hydrolase inhibitors by virtual screening. Bioorg. Med. Chem. Lett. 2012, 22, 6762–6765. [Google Scholar] [CrossRef] [PubMed]
- Bhagwati, S.; Siddiqi, M.I. Identification of potential soluble epoxide hydrolase (sEH) inhibitors by ligand-based pharmacophore model and biological evaluation. J. Biomol. Struct. Dyn. 2020, 38, 4956–4966. [Google Scholar] [CrossRef]
- Fakhar, Z.; Hejazi, L.; Tabatabai, S.A.; Munro, O.Q. Discovery of novel heterocyclic amide-based inhibitors: An integrative in-silico approach to targeting soluble epoxide hydrolase. J. Biomol. Struct. Dyn. 2021, 1–15. [Google Scholar] [CrossRef]
- Potenza, M.; Sciarretta, M.; Chini, M.G.; Saviano, A.; Maione, F.; D’Auria, M.V.; De Marino, S.; Giordano, A.; Hofstetter, R.K.; Festa, C.; et al. Structure-based screening for the discovery of 1,2,4-oxadiazoles as promising hits for the development of new anti-inflammatory agents interfering with eicosanoid biosynthesis pathways. Eur. J. Med. Chem. 2021, 224, 113693. [Google Scholar] [CrossRef] [PubMed]
- Colarusso, E.; Potenza, M.; Lauro, G.; Chini, M.G.; Sepe, V.; Zampella, A.; Fischer, K.; Hofstetter, R.K.; Werz, O.; Bifulco, G. Thiazolidin-4-one-based compounds interfere with the eicosanoid biosynthesis pathways by mPGES-1/sEH/5-LO multitarget inhibition. Eur. J. Med. Chem. Rep. 2022, 5, 100046. [Google Scholar] [CrossRef]
- Chini, M.G.; Giordano, A.; Potenza, M.; Terracciano, S.; Fischer, K.; Vaccaro, M.C.; Colarusso, E.; Bruno, I.; Riccio, R.; Koeberle, A.; et al. Targeting mPGES-1 by a Combinatorial Approach: Identification of the Aminobenzothiazole Scaffold to Suppress PGE2Levels. ACS Med. Chem. Lett. 2020, 11, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, S.; Terracciano, S.; Ruggiero, D.; Potenza, M.; Vaccaro, M.C.; Fischer, K.; Werz, O.; Bruno, I.; Bifulco, G. Identification of 2-(thiophen-2-yl)acetic Acid-Based Lead Compound for mPGES-1 Inhibition. Front. Chem. 2021, 9, 676631. [Google Scholar] [CrossRef] [PubMed]
- Saviano, A.; De Vita, S.; Chini, M.G.; Marigliano, N.; Lauro, G.; Casillo, G.M.; Raucci, F.; Iorizzi, M.; Hofstetter, R.K.; Fischer, K.; et al. In Silico, In Vitro, and In Vivo Analysis of Tanshinone IIA and Cryptotanshinone from Salvia miltiorrhiza as Modulators of Cyclooxygenase-2/mPGES-1/Endothelial Prostaglandin EP3 Pathway. Biomolecules 2022, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, A.; Werz, O. Multi-target approach for natural products in inflammation. Drug Discov. Today 2014, 19, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Nakamoto, H.; Neckers, L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013, 19, 347–365. [Google Scholar] [CrossRef]
- Terracciano, S.; Russo, A.; Chini, M.G.; Vaccaro, M.C.; Potenza, M.; Vassallo, A.; Riccio, R.; Bifulco, G.; Bruno, I. Discovery of new molecular entities able to strongly interfere with Hsp90 C-terminal domain. Sci. Rep. 2018, 8, 1709. [Google Scholar] [CrossRef] [PubMed]
- CombiGlide; Schrödinger Release: New York, NY, USA, 2017.
- Teracciano, S.; Chini, M.G.; Vaccaro, M.C.; Strocchia, M.; Foglia, A.; Vassallo, A.; Saturnino, C.; Riccio, R.; Bifulco, G.; Bruno, I. Identification of the key structural elements of a dihydropyrimidinone core driving toward more potent Hsp90 C-terminal inhibitors. Chem. Commun. 2016, 52, 12857–12860. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, S.; Foglia, A.; Chini, M.G.; Vaccaro, M.C.; Russo, A.; Piaz, F.D.; Saturnino, C.; Riccio, R.; Bifulco, G.; Bruno, I. New dihydropyrimidin-2(1H)-one based Hsp90 C-terminal inhibitors. RSC Adv. 2016, 6, 82330–82340. [Google Scholar] [CrossRef]
- Bunnett, J.F.; Naff, M.B. Kinetics of reactions of amines with isatoic anhydride. J. Am. Chem. Soc. 1966, 88, 4001–4008. [Google Scholar] [CrossRef]
- Staiger, R.P.; Wagner, E.C. Isatoic anhydride. III. Reactions with primary and secondary amines. J. Org. Chem. 1953, 18, 1427–1439. [Google Scholar] [CrossRef]
- Williams, A.; Ibrahim, I.T. Carbodiimide chemistry: Recent advances. Chem. Rev. 1981, 81, 589–636. [Google Scholar] [CrossRef]
- Dal Piaz, F.; Vassallo, A.; Temraz, A.; Cotugno, R.; Belisario, M.A.; Bifulco, G.; Chini, M.G.; Pisano, C.; De Tommasi, N.; Braca, A. A chemical-biological study reveals C9-type iridoids as novel heat shock protein 90 (Hsp90) inhibitors. J. Med. Chem. 2013, 56, 1583–1595. [Google Scholar] [CrossRef]
- Terracciano, S.; Chini, M.G.; Dal Piaz, F.; Vassallo, A.; Riccio, R.; Bruno, I.; Bifulco, G. Dimeric and trimeric triazole based molecules as a new class of Hsp90 molecular chaperone inhibitors. Eur. J. Med. Chem. 2013, 65, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Strocchia, M.; Terracciano, S.; Chini, M.G.; Vassallo, A.; Vaccaro, M.C.; Dal Piaz, F.; Leone, A.; Riccio, R.; Bruno, I.; Bifulco, G. Targeting the Hsp90 C-terminal domain by the chemically accessible dihydropyrimidinone scaffold. Chem. Commun. 2015, 51, 3850–3853. [Google Scholar] [CrossRef]
- Chini, M.G.; Malafronte, N.; Vaccaro, M.C.; Gualtieri, M.J.; Vassallo, A.; Vasaturo, M.; Castellano, S.; Milite, C.; Leone, A.; Bifulco, G.; et al. Identification of Limonol Derivatives as Heat Shock Protein 90 (Hsp90) Inhibitors through a Multidisciplinary Approach. Chem. Eur. J. 2016, 22, 13236–13250. [Google Scholar] [CrossRef] [PubMed]
- Dal Piaz, F.; Vassallo, A.; Chini, M.G.; Cordero, F.M.; Cardona, F.; Pisano, C.; Bifulco, G.; De Tommasi, N.; Brandi, A. Natural Iminosugar (+)-Lentiginosine Inhibits ATPase and Chaperone Activity of Hsp90. PLoS ONE 2012, 7, e43316. [Google Scholar] [CrossRef]
- D’Ambola, M.; Fiengo, L.; Chini, M.G.; Cotugno, R.; Bader, A.; Bifulco, G.; Braca, A.; De Tommasi, N.; Dal Piaz, F. Fusicoccane Diterpenes from Hypoestes forsskaolii as Heat Shock Protein 90 (Hsp90) Modulators. J. Nat. Prod. 2019, 82, 539–549. [Google Scholar] [CrossRef]
- Öster, L.; Tapani, S.; Xue, Y.; Käck, H. Successful generation of structural information for fragment-based drug discovery. Drug Discov. Today 2015, 20, 1104–1111. [Google Scholar] [CrossRef]
- Gomez, G.A.; Morisseau, C.; Hammock, B.D.; Christianson, D.W. Structure of Human Epoxide Hydrolase Reveals Mechanistic Inferences on Bifunctional Catalysis in Epoxide and Phosphate Ester Hydrolysis. Biochemistry 2004, 43, 4716–4723. [Google Scholar] [CrossRef]
- Amano, Y.; Yamaguchi, T.; Tanabe, E. Structural insights into binding of inhibitors to soluble epoxide hydrolase gained by fragment screening and X-ray crystallography. Biorg. Med. Chem. 2014, 22, 2427–2434. [Google Scholar] [CrossRef]
- Pecic, S.; Pakhomova, S.; Newcomer, M.E.; Morisseau, C.; Hammock, B.D.; Zhu, Z.; Rinderspacher, A.; Deng, S.-X. Synthesis and structure–activity relationship of piperidine-derived non-urea soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 417–421. [Google Scholar] [CrossRef]
- Lee, K.S.S.; Liu, J.-Y.; Wagner, K.M.; Pakhomova, S.; Dong, H.; Morisseau, C.; Fu, S.H.; Yang, J.; Wang, P.; Ulu, A.; et al. Optimized Inhibitors of Soluble Epoxide Hydrolase Improve in Vitro Target Residence Time and in Vivo Efficacy. J. Med. Chem. 2014, 57, 7016–7030. [Google Scholar] [CrossRef]
- Argiriadi, M.A.; Morisseau, C.; Goodrow, M.H.; Dowdy, D.L.; Hammock, B.D.; Christianson, D.W. Binding of Alkylurea Inhibitors to Epoxide Hydrolase Implicates Active Site Tyrosines in Substrate Activation. J. Biol. Chem. 2000, 275, 15265–15270. [Google Scholar] [CrossRef]
- Kodani, S.D.; Bhakta, S.; Hwang, S.H.; Pakhomova, S.; Newcomer, M.E.; Morisseau, C.; Hammock, B.D. Identification and optimization of soluble epoxide hydrolase inhibitors with dual potency towards fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2018, 28, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Lukin, A.; Kramer, J.; Hartmann, M.; Weizel, L.; Hernandez-Olmos, V.; Falahati, K.; Burghardt, I.; Kalinchenkova, N.; Bagnyukova, D.; Zhurilo, N.; et al. Discovery of polar spirocyclic orally bioavailable urea inhibitors of soluble epoxide hydrolase. Bioorg. Chem. 2018, 80, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Hiesinger, K.; Kramer, J.S.; Achenbach, J.; Moser, D.; Weber, J.; Wittmann, S.K.; Morisseau, C.; Angioni, C.; Geisslinger, G.; Kahnt, A.S.; et al. Computer-Aided Selective Optimization of Side Activities of Talinolol. ACS Med. Chem. Lett. 2019, 10, 899–903. [Google Scholar] [CrossRef]
- Hiesinger, K.; Kramer, J.S.; Beyer, S.; Eckes, T.; Brunst, S.; Flauaus, C.; Wittmann, S.K.; Weizel, L.; Kaiser, A.; Kretschmer, S.B.M.; et al. Design, Synthesis, and Structure–Activity Relationship Studies of Dual Inhibitors of Soluble Epoxide Hydrolase and 5-Lipoxygenase. J. Med. Chem. 2020, 63, 11498–11521. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, D.; Tsuda, Y.; Shiyama, T.; Nishimura, T.; Chiyo, N.; Tominaga, Y.; Sawada, N.; Mimoto, T.; Kusunose, N. A Practical Use of Ligand Efficiency Indices Out of the Fragment-Based Approach: Ligand Efficiency-Guided Lead Identification of Soluble Epoxide Hydrolase Inhibitors. J. Med. Chem. 2010, 54, 851–857. [Google Scholar] [CrossRef]
- Kim, I.-H.; Morisseau, C.; Watanabe, T.; Hammock, B.D. Design, Synthesis, and Biological Activity of 1,3-Disubstituted Ureas as Potent Inhibitors of the Soluble Epoxide Hydrolase of Increased Water Solubility. J. Med. Chem. 2004, 47, 2110–2122. [Google Scholar] [CrossRef]
- Blöcher, R.; Lamers, C.; Wittmann, S.K.; Diehl, O.; Hanke, T.; Merk, D.; Steinhilber, D.; Schubert-Zsilavecz, M.; Kahnt, A.S.; Proschak, E. Design and synthesis of fused soluble epoxide hydrolase/peroxisome proliferator-activated receptor modulators. MedChemComm 2016, 7, 1209–1216. [Google Scholar] [CrossRef]
- Hwang, S.H.; Tsai, H.-J.; Liu, J.-Y.; Morisseau, C.; Hammock, B.D. Orally Bioavailable Potent Soluble Epoxide Hydrolase Inhibitors. J. Med. Chem. 2007, 50, 3825–3840. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, Y.; Gong, G.; Smith, D.H.; Yan, F.; Rinderspacher, A.; Feng, Y.; Zhu, Z.; Li, X.; Deng, S.-X.; et al. Discovery of potent non-urea inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett. 2009, 19, 2354–2359. [Google Scholar] [CrossRef][Green Version]
- Morisseau, C.; Pakhomova, S.; Hwang, S.H.; Newcomer, M.E.; Hammock, B.D. Inhibition of soluble epoxide hydrolase by fulvestrant and sulfoxides. Bioorg. Med. Chem. Lett. 2013, 23, 3818–3821. [Google Scholar] [CrossRef][Green Version]
- Structure of 5AK5. Available online: http://www.bindingmoad.org/pdbrecords/index/5AK5 (accessed on 4 February 2021).
- Kotev, M.; Soliva, R.; Orozco, M. Challenges of docking in large, flexible and promiscuous binding sites. Biorg. Med. Chem. 2016, 24, 4961–4969. [Google Scholar] [CrossRef] [PubMed]
- Structure of 5ALG. Available online: http://www.bindingmoad.org/pdbrecords/index/5ALG (accessed on 4 February 2021).
- Structure of 5ALP. Available online: http://www.bindingmoad.org/pdbrecords/index/5ALP (accessed on 4 February 2021).
- Structure of 5ALU. Available online: http://www.bindingmoad.org/pdbrecords/index/5ALU (accessed on 4 February 2021).
- Anandan, S.-K.; Webb, H.K.; Chen, D.; Wang, Y.-X.; Aavula, B.R.; Cases, S.; Cheng, Y.; Do, Z.N.; Mehra, U.; Tran, V.; et al. 1-(1-Acetyl-piperidin-4-yl)-3-adamantan-1-yl-urea (AR9281) as a potent, selective, and orally available soluble epoxide hydrolase inhibitor with efficacy in rodent models of hypertension and dysglycemia. Bioorg. Med. Chem. Lett. 2011, 21, 983–988. [Google Scholar] [CrossRef]
- Shen, H.C.; Ding, F.-X.; Wang, S.; Xu, S.; Chen, H.-S.; Tong, X.; Tong, V.; Mitra, K.; Kumar, S.; Zhang, X.; et al. Discovery of spirocyclic secondary amine-derived tertiary ureas as highly potent, selective and bioavailable soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3398–3404. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput.-Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Phase; Schrödinger Release: New York, NY, USA, 2021.
- Laoui, A.; Polyakov, V.R. Web services as applications’ integration tool: QikProp case study. J. Comput. Chem. 2011, 32, 1944–1951. [Google Scholar] [CrossRef] [PubMed]
- Qikprop; Schrödinger Release: New York, NY, USA, 2017.
- Protein Preparation Wizard; Epik; Schrödinger Release: New York, NY, USA, 2017.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Glide; Schrödinger Release: New York, NY, USA, 2021.
- Vaisburg, A.; Bernstein, N.; Frechette, S.; Allan, M.; Abou-Khalil, E.; Leit, S.; Moradei, O.; Bouchain, G.; Wang, J.; Woo, S.H.; et al. (2-Amino-phenyl)-amides of ω-substituted alkanoic acids as new histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Carpino, L.A.; El-Faham, A. The diisopropylcarbodiimide/1-hydroxy-7-azabenzotriazole system: Segment coupling and stepwise peptide assembly. Tetrahedron 1999, 55, 6813–6830. [Google Scholar] [CrossRef]
- Wixtrom, R.N.; Silva, M.H.; Hammock, B.D. Affinity purification of cytosolic epoxide hydrolase using derivatized epoxy-activated Sepharose gels. Anal. Biochem. 1988, 169, 71–80. [Google Scholar] [CrossRef]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90–nucleotide–p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef]
- Lee, C.-C.; Lin, T.-W.; Ko, T.-P.; Wang, A.H.J. The Hexameric Structures of Human Heat Shock Protein 90. PLoS ONE 2011, 6, e19961. [Google Scholar] [CrossRef]
- Terracciano, S.; Chini, M.G.; Vaccaro, M.C.; Strocchia, M.; Foglia, A.; Vassallo, A.; Saturnino, C.; Riccio, R.; Bifulco, G.; Bruno, I. Correction: Identification of the key structural elements of a dihydropyrimidinone core driving toward more potent Hsp90 C-terminal inhibitors. Chem. Commun. 2016, 52, 13515. [Google Scholar] [CrossRef]
- Vassallo, A.; Vaccaro, M.C.; De Tommasi, N.; Dal Piaz, F.; Leone, A. Identification of the Plant Compound Geraniin as a Novel Hsp90 Inhibitor. PLoS ONE 2013, 8, e74266. [Google Scholar] [CrossRef]
- Glide; Schrödinger Release: New York, NY, USA, 2017.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Num Sites Matched | PhaseScreen Score | Docking Score |
|---|---|---|---|
| 3 | 5/5 | 0.82 | −9.9 |
| 4 | 5/5 | 0.84 | −10.1 |
| 5 | 4/5 | 0.97 | −8.5 |
| 6 | 5/5 | 0.64 | −7.9 |
| 7 | 5/5 | 0.63 | −6.7 |
| 8 | 5/5 | 0.74 | −9.8 |
| Compound | sEH Residual Activity (%) |
|---|---|
| 3 | 42.4 ± 1.3 |
| 4 | 33.8 ± 1.9 |
| 5 | 80.4 ± 3.8 |
| 6 | 48.6 ± 3.8 |
| 7 | 67.2 ± 4.4 |
| 8 | 51.7 ± 3.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gazzillo, E.; Terracciano, S.; Ruggiero, D.; Potenza, M.; Chini, M.G.; Lauro, G.; Fischer, K.; Hofstetter, R.K.; Giordano, A.; Werz, O.; et al. Repositioning of Quinazolinedione-Based Compounds on Soluble Epoxide Hydrolase (sEH) through 3D Structure-Based Pharmacophore Model-Driven Investigation. Molecules 2022, 27, 3866. https://doi.org/10.3390/molecules27123866
Gazzillo E, Terracciano S, Ruggiero D, Potenza M, Chini MG, Lauro G, Fischer K, Hofstetter RK, Giordano A, Werz O, et al. Repositioning of Quinazolinedione-Based Compounds on Soluble Epoxide Hydrolase (sEH) through 3D Structure-Based Pharmacophore Model-Driven Investigation. Molecules. 2022; 27(12):3866. https://doi.org/10.3390/molecules27123866
Chicago/Turabian StyleGazzillo, Erica, Stefania Terracciano, Dafne Ruggiero, Marianna Potenza, Maria Giovanna Chini, Gianluigi Lauro, Katrin Fischer, Robert Klaus Hofstetter, Assunta Giordano, Oliver Werz, and et al. 2022. "Repositioning of Quinazolinedione-Based Compounds on Soluble Epoxide Hydrolase (sEH) through 3D Structure-Based Pharmacophore Model-Driven Investigation" Molecules 27, no. 12: 3866. https://doi.org/10.3390/molecules27123866
APA StyleGazzillo, E., Terracciano, S., Ruggiero, D., Potenza, M., Chini, M. G., Lauro, G., Fischer, K., Hofstetter, R. K., Giordano, A., Werz, O., Bruno, I., & Bifulco, G. (2022). Repositioning of Quinazolinedione-Based Compounds on Soluble Epoxide Hydrolase (sEH) through 3D Structure-Based Pharmacophore Model-Driven Investigation. Molecules, 27(12), 3866. https://doi.org/10.3390/molecules27123866