
Synthesis of Sugar and Nucleoside Analogs and Evaluation of Their Anticancer and Analgesic Potentials

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Chemistry
2.2. Biological Study
2.2.1. Peripheral Analgesic Activity in Mice Model
2.2.2. Trypan Blue Dye Exclusion Test for Cytotoxic Activity
2.2.3. CCK-8 Cell Viability Assay for Cytotoxic Activity
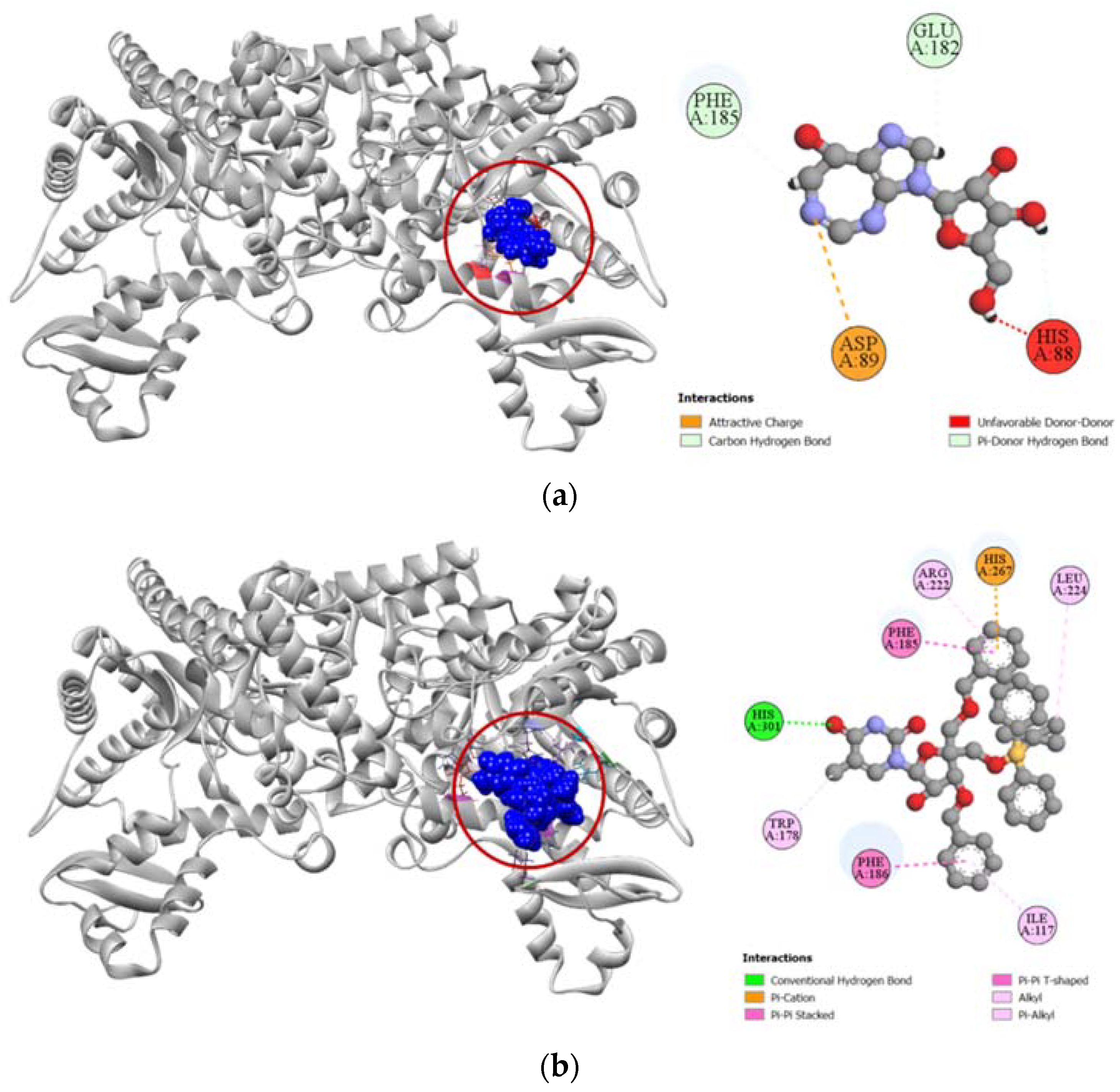
2.3. Molecular Docking Simulation
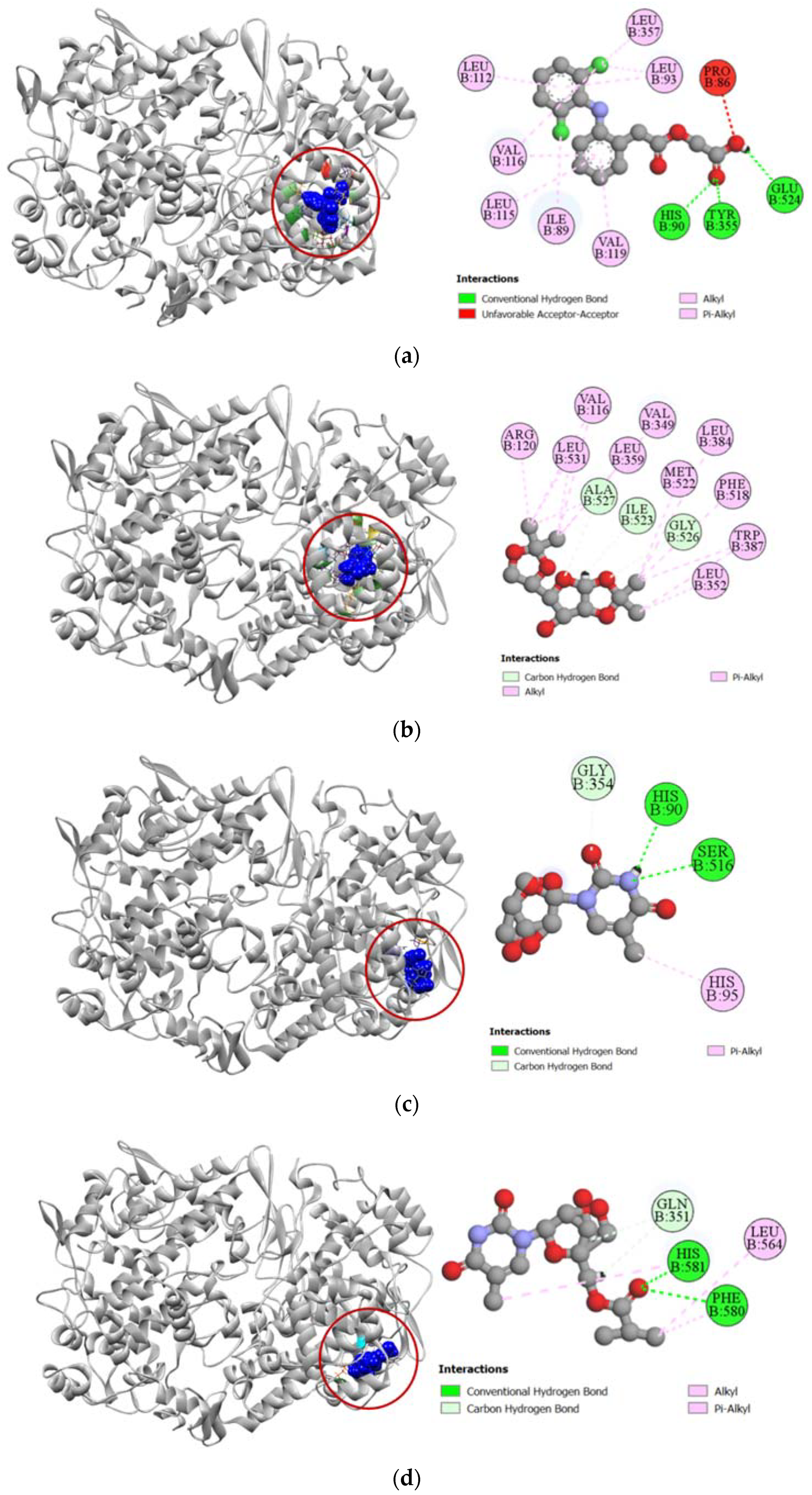
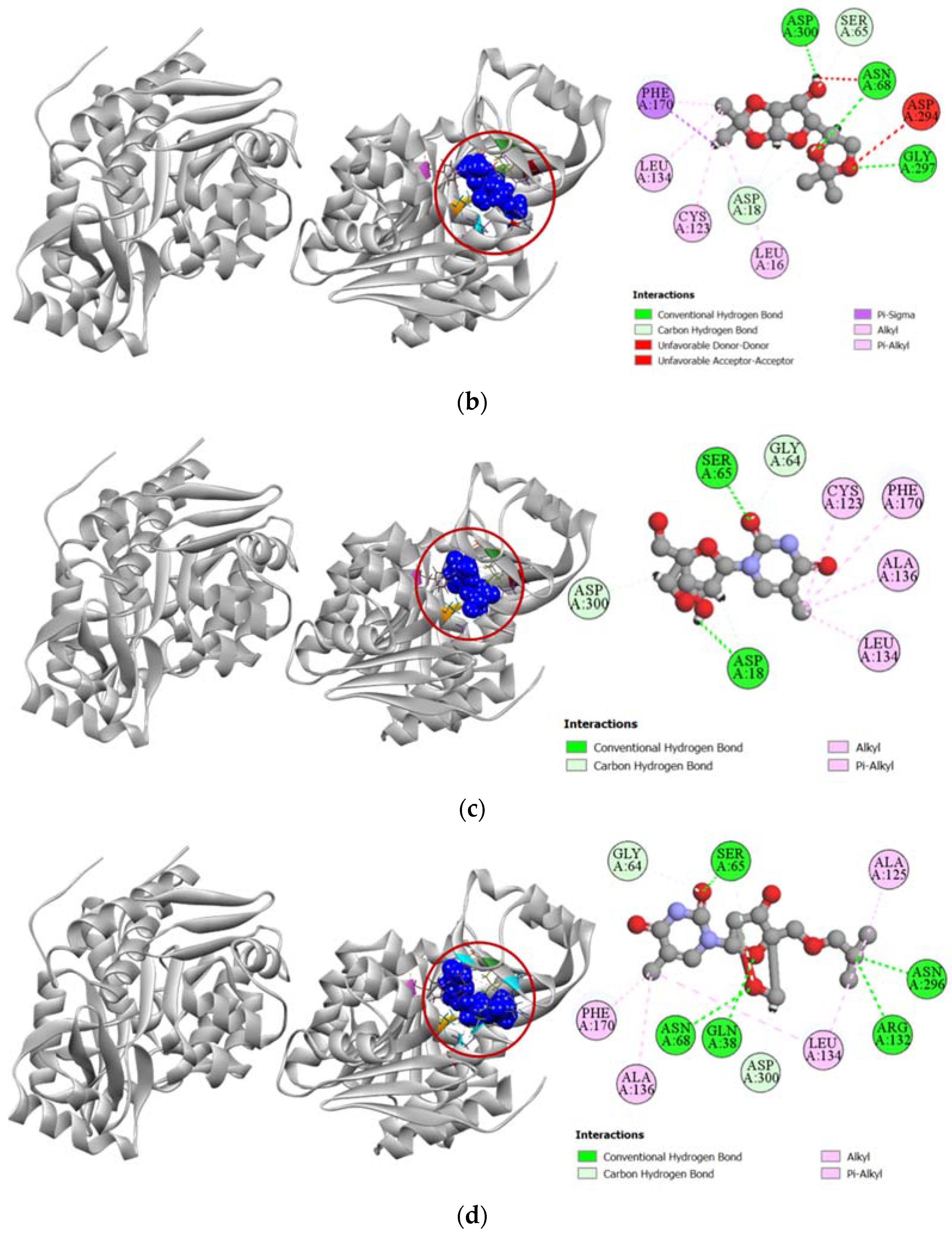
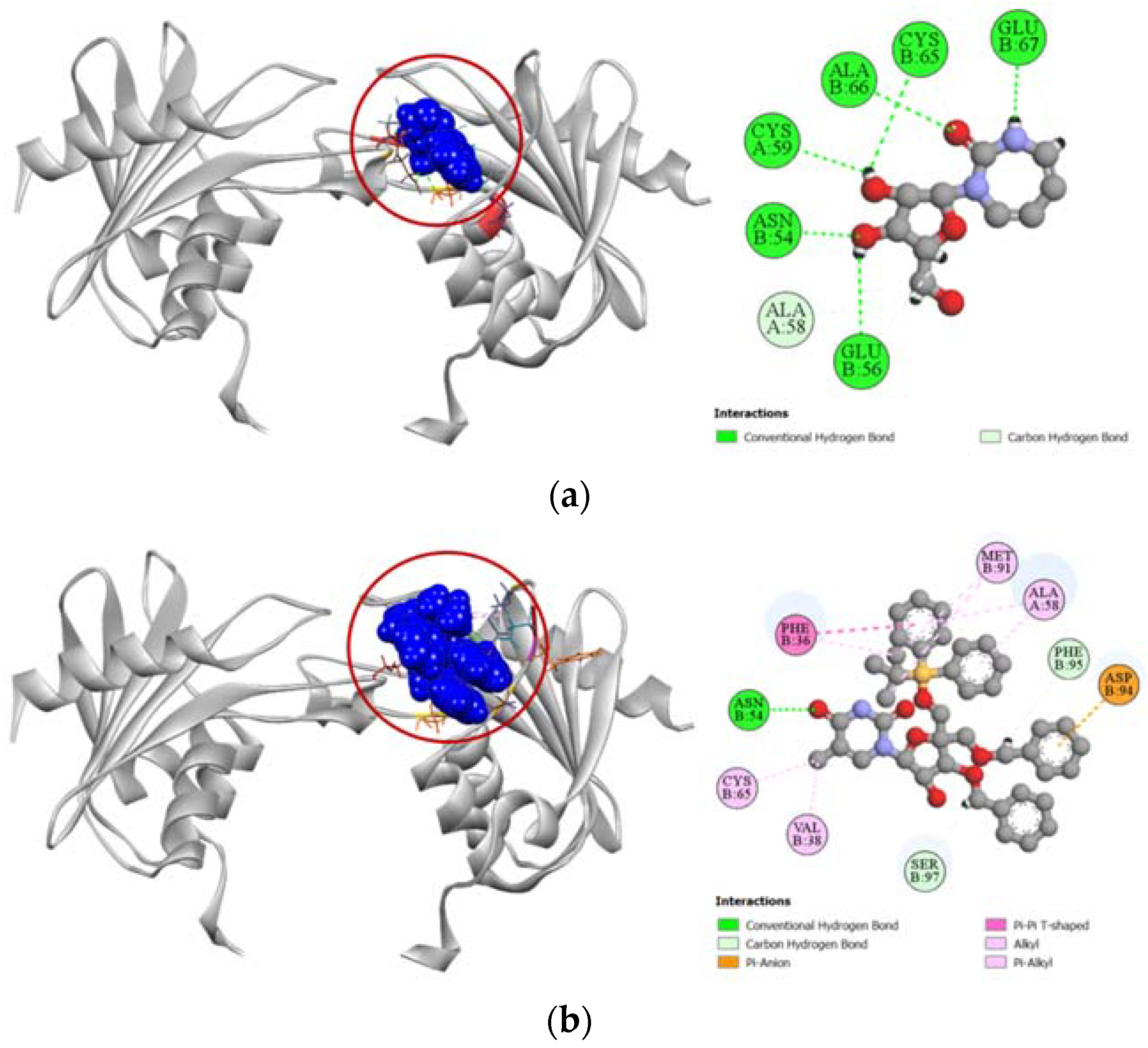
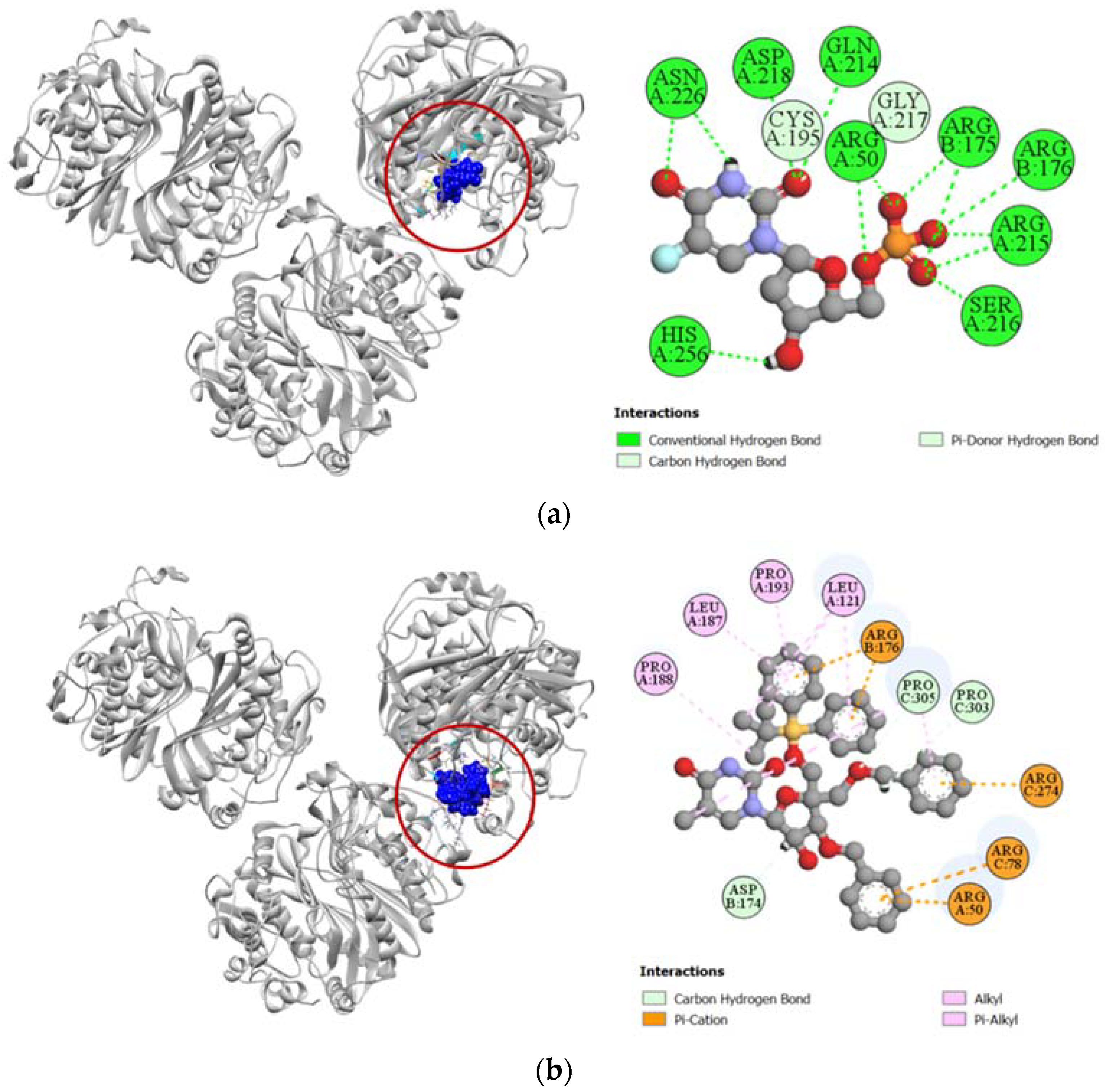
2.3.1. Docking Simulation against Target Proteins of Analgesic and Cytotoxic Activity
2.3.2. Inhibition of Cyclooxygenase-1 and Cyclooxygenase-2
2.3.3. Inhibition of Human Adenosine Kinase
2.3.4. Inhibition of Human Cytidine Deaminase
2.3.5. Inhibition of Proto-Oncogene Tyrosine-Protein Kinase Src
2.3.6. Inhibition of Human Thymidine Kinase 1
2.3.7. Inhibition of Human Thymidylate Synthase
2.3.8. Inhibition of Human Adenosine Deaminase 2
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Synthesis
4.2.1. Preparation of 1,2:5,6-Di-O-Isopropylidene-α-d-Allofuranose (2)
4.2.2. Preparation of 3-O-Benzyl-4-C-Hydroxymethyl-1,2-O-Isopropylidene-α-d-Ribofuranose (3)
4.2.3. Preparation of 1-(4-C-Acetoxymethyl-2-O-Acetyl-3,5-Di-O-Benzyl-β-d-Ribofuranosyl)Thymine (4)
4.2.4. Preparation of 1-(4-C-Hydroxymethyl-2-Hydroxyl-3,5-Di-O-Benzyl-β-d-Ribofuranosyl)Thymine (5)
4.2.5. Preparation of 3′,5′-Di-O-Benzyl-4′-C-Tert-Butyldiphenylsiloxymethyl-5-Methyluridine (6)
4.2.6. Preparation of (1S,3R,4R,7S)-7-Hydroxy-1-Hydroxymethyl-3-(Thymin-1-Yl)-2,5-Dioxabicyclo [2.2.1]Heptane (7)
4.2.7. Preparation of ((1R,3R,4R,7S)-7-Hydroxy-3-(5-Methyl-2,4-Dioxo-3,4-Dihydropyrimidin-1(2H)-yl)-2,5-Dioxabicyclo [2.2.1]Heptan-1-yl)Methyl Isobutyrate (8)
4.3. Biological Study
4.3.1. In Vivo Peripheral Analgesic Activity
4.3.2. In Vitro Cytotoxic Activity
4.3.3. Trypan Blue Dye Exclusion Method
4.3.4. In Vitro Cytotoxic Activity
4.4. In Silico Molecular Docking Simulation
4.4.1. Target Protein Selection
4.4.2. Ligand Preparation
4.4.3. Ligand–Protein Interaction
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cipolla, L.; La Ferla, B.; Airoldi, C.; Zona, C.; Orsato, A.; Shaikh, N.; Russo, L.; Nicotra, F. Carbohydrate mimetics and scaffolds: Sweet spots in medicinal chemistry. Future Med. Chem. 2010, 2, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. 2019 FDA drug approvals. Nat. Rev. Drug Discov. 2020, 19, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, R.; Aliota, M.; Bonnac, L. Broad-Spectrum Antiviral Strategies and Nucleoside Analogues. Viruses 2021, 13, 667. [Google Scholar] [CrossRef]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Guinan, M.; Benckendorff, C.; Smith, M.; Miller, G.J. Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues. Molecules 2020, 25, 2050. [Google Scholar] [CrossRef] [PubMed]
- Pruijssers, A.J.; Denison, M. Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 2019, 35, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Borbone, N.; Piccialli, G.; Roviello, G.N.; Oliviero, G. Nucleoside Analogs and Nucleoside Precursors as Drugs in the Fight against SARS-CoV-2 and Other Coronaviruses. Molecules 2021, 26, 986. [Google Scholar] [CrossRef]
- Rahman, S.M.A.; Seki, S.; Utsuki, K.; Obika, S.; Miyashita, K.; Imanishi, T. 2′,4′-BNANC: A Novel Bridged Nucleic Acid Analogue with Excellent Hybridizing and Nuclease Resistance Profiles. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1625–1628. [Google Scholar] [CrossRef]
- Rahman, S.M.A.; Seki, S.; Obika, S.; Yoshikawa, H.; Miyashita, K.; Imanishi, T. Design, synthesis, and properties of 2′,4′-BNANC: A bridged nucleic acid analogue. J. Am. Chem. Soc. 2008, 130, 4886–4896. [Google Scholar] [CrossRef]
- Mahmoud, S.; Hasabelnaby, S.; Hammad, S.; Sakr, T. Antiviral Nucleoside and Nucleotide Analogs: A Review. J. Adv. Pharm. Res. 2018, 2, 73–88. [Google Scholar] [CrossRef]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002, 3, 415–424. [Google Scholar] [CrossRef]
- Kimura, T.; Shimizu, T.; Funahashi, T.; Nagakura, M.; Watanabe, K.; Tachibana, K.; Kondo, S.; Ho, I.K.; Yamamoto, I. N3-phenacyluridine as a new type of antinociceptive compound in mice. Res. Commun. Mol. Pathol. Pharmacol. 2003, 113–114, 57–65. [Google Scholar]
- Shimizu, T.; Kimura, T.; Funahashi, T.; Watanabe, K.; Ho, I.K.; Yamamoto, I. Synthesis of N3-Substituted Uridine and Related Pyrimidine Nucleosides and Their Antinociceptive Effects in Mice. Chem. Pharm. Bull. 2005, 53, 313–318. [Google Scholar] [CrossRef][Green Version]
- Spriha, S.E.; Rahman, F.I.; Rahman, S.M.A. Synthesis, in vivo and in silico analgesic and anti-inflammatory studies of α-D-ribofuranose derivatives. Saudi Pharm. J. 2021, 29, 981–991. [Google Scholar] [CrossRef]
- Rahman, F.I.; Hussain, F.; Saqueeb, N.; Rahman, S.M.A. Synthesis and evaluation of pharmacological activities of some 3-O-benzyl-4-C-(hydroxymethyl)-1,2-O-isopropylidene-α-D-ribofuranose derivatives as potential anti-inflammatory agents and analgesics. Res. Pharm. Sci. 2020, 15, 209–217. [Google Scholar] [CrossRef]
- Kaur, T.; Madgulkar, A.; Bhalekar, M.; Asgaonkar, K. Molecular Docking in Formulation and Development. Curr. Drug Discov. Technol. 2018, 16, 30–39. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva, F.P., Jr. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef]
- Obika, S.; Imanishi, T.; Kawada, Y.; Baba, T.; Fujisaka, A. Bridged Nucleic Acids: Development, Synthesis and Properties. Heterocycles 2010, 81, 1347. [Google Scholar] [CrossRef]
- Gao, J.; Luo, X.; Li, Y.; Gao, R.; Chen, H.; Ji, D. Synthesis and biological evaluation of 2-oxo-pyrazine-3-carboxamide-yl nucleoside analogues and their epimers as inhibitors of influenza a viruses. Chem. Biol. Drug Des. 2015, 85, 245–252. [Google Scholar] [CrossRef]
- Kawsar, S.M.A.; Islam, M.M.; Chowdhury, S.A.; Hasan, T.; Hossain, M.K.; Manchur, M.A.; Ozeki, Y. Design and Newly Synthesis of some 1,2-O-Isopropylidene-α-D-Glucofuranose Derivatives: Characterization and Antibacterial Screening Studies Bazı 1,2-O-izopropiliden-α-D-Glucofuranose Türevlerinin Tasarım ve Yeni Sentez Metodu: Karakterizasyonu ve Antibakteriyel Tarama Çalışmaları Research Article. Hacet. J. Biol. Chem. 2013, 41, 195–206. [Google Scholar]
- Okada, T.; Asawa, T.; Sugiyama, Y.; Kirihara, M.; Iwai, T.; Kimura, Y. Sodium hypochlorite pentahydrate (NaOCl·5H2O) crystals as an extraâordinary oxidant for primary and secondary alcohols. Synlett 2014, 25, 596–598. [Google Scholar] [CrossRef]
- Hoffmann-Röder, A.; Johannes, M. Synthesis of a MUC1-glycopeptide–BSA conjugate vaccine bearing the 3′-deoxy-3′-fluoro-Thomsen–Friedenreich antigen. Chem. Commun. 2011, 47, 9903–9905. [Google Scholar] [CrossRef] [PubMed]
- Goud, P.M.; Venkatachalam, T.K.; Uckun, F.M. Introduction of a Carbohydrate Moiety into the Structure of Thiourea Compounds Targeting HIV-1 Reverse Transcriptase. Synth. Commun. 2003, 33, 1185–1193. [Google Scholar] [CrossRef]
- Sharma, V.K.; Kumar, M.; Olsen, C.E.; Prasad, A.K. Chemoenzymatic convergent synthesis of 2′-o,4′-c-methyleneribonucleosides. J. Org. Chem. 2014, 79, 6336–6341. [Google Scholar] [CrossRef] [PubMed]
- US7105499B2-Nucleoside Derivatives as Inhibitors of RNA-Dependent RNA Viral Polymerase-Google Patents. Available online: https://patents.google.com/patent/US7105499B2/en (accessed on 29 January 2022).
- WO1999014226A2-Bi- and Tri-Cyclic Nucleoside, Nucleotide and Oligonucleotide Analogues-Google Patents. Available online: https://patents.google.com/patent/WO1999014226A2/fi (accessed on 29 January 2022).
- Koshkin, A.A.; Singh, S.K.; Nielsen, P.; Rajwanshi, V.K.; Kumar, R.; Meldgaard, M.; Olsen, C.E.; Wengel, J. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron 1998, 54, 3607–3630. [Google Scholar] [CrossRef]
- Koshkin, A.A.; Rajwanshi, V.K.; Wengel, J. Novel convenient syntheses of LNA [2.2.1]bicyclo nucleosides. Tetrahedron Lett. 1998, 39, 4381–4384. [Google Scholar] [CrossRef]
- Yssel, A.E.J.; Vanderleyden, J.; Steenackers, H.P. Repurposing of nucleoside- and nucleobase-derivative drugs as antibiotics and biofilm inhibitors. J. Antimicrob. Chemother. 2017, 72, 2326–2333. [Google Scholar] [CrossRef]
- Mishra, S.; Upadhaya, K.; Mishra, K.B.; Shukla, A.K.; Tripathi, R.P.; Tiwari, V.K. Carbohydrate-Based Therapeutics: A Frontier in Drug Discovery and Development; Elsevier: Amsterdam, The Netherlands, 2016; Volume 49, ISBN 9780444636010. [Google Scholar]
- Galan, M.C.; Benito-Alifonso, D.; Watt, G.M. Carbohydrate chemistry in drug discovery. Org. Biomol. Chem. 2011, 9, 3598–3610. [Google Scholar] [CrossRef]
- Secrist, J.A. Nucleosides as anticancer agents: From concept to the clinic. Nucleic Acids Symp. Ser. 2005, 49, 15–16. [Google Scholar] [CrossRef][Green Version]
- Jarvis, M.F.; Yu, H.; McGaraughty, S.; Wismer, C.T.; Mikusa, J.; Zhu, C.; Chu, K.; Kohlhaas, K.; Cowart, M.; Lee, C.H.; et al. Analgesic and anti-inflammatory effects of A-286501, a novel orally active adenosine kinase inhibitor. Pain 2002, 96, 107–118. [Google Scholar] [CrossRef]
- Boyer, S.H.; Ugarkar, B.G.; Solbach, J.; Kopcho, J.; Matelich, M.C.; Ollis, K.; Gomez-Galeno, J.E.; Mendonca, R.; Tsuchiya, M.; Nagahisa, A.; et al. Adenosine kinase inhibitors. 5. Synthesis, enzyme inhibition, and analgesic activity of diaryl-erythro-furanosyltubercidin analogues. J. Med. Chem. 2005, 48, 6430–6441. [Google Scholar] [CrossRef] [PubMed]
- El-Gazzar, A.B.A.; Hafez, H.N.; Nawwar, G.A.M. New acyclic nucleosides analogues as potential analgesic, anti-inflammatory, anti-oxidant and anti-microbial derived from pyrimido [4,5-b]quinolines. Eur. J. Med. Chem. 2009, 44, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Rácz, I.; Zimmer, A. Animal Models of Nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar] [CrossRef]
- Boison, D. Adenosine kinase, epilepsy and stroke: Mechanisms and therapies. Trends Pharmacol. Sci. 2006, 27, 652–658. [Google Scholar] [CrossRef]
- McCrory, C.R.; Lindahl, S.G.E. Cyclooxygenase inhibition for postoperative analgesia. Anesth. Analg. 2002, 95, 169–176. [Google Scholar] [CrossRef]
- Snášel, J.; Nauš, P.; Dostál, J.; Hnízda, A.; Fanfrlík, J.; Brynda, J.; Bourderioux, A.; Dušek, M.; Dvořáková, H.; Stolaříková, J.; et al. Structural basis for inhibition of mycobacterial and human adenosine kinase by 7-substituted 7-(Het)aryl-7-deazaadenine ribonucleosides. J. Med. Chem. 2014, 57, 8268–8279. [Google Scholar] [CrossRef]
- Burke, M.P.; Borland, K.M.; Litosh, V. Base-Modified Nucleosides as Chemotherapeutic Agents: Past and Future. Curr. Top. Med. Chem. 2016, 16, 1231–1241. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Ellery, S.P.; Rivas, F.; Saye, K.; Rogers, E.; Workinger, T.J.; Schallenberger, M.; Tawatao, R.; Montero, A.; Hessell, A.; et al. Synthesis and biological evaluation of 2′,4′- and 3′,4′-bridged nucleoside analogues. Bioorg. Med. Chem. 2011, 19, 5648–5669. [Google Scholar] [CrossRef][Green Version]
- Marzabadi, C.H.; Franck, R.W.; Schinazi, R.F. The synthesis and biological evaluation of novel bridging nucleoside analogues. Bioorg. Med. Chem. 2002, 10, 273–281. [Google Scholar] [CrossRef]
- Mirza, A.Z. Advancement in the development of heterocyclic nucleosides for the treatment of cancer-A review. Nucleosides Nucleotides Nucleic Acids 2019, 38, 836–857. [Google Scholar] [CrossRef] [PubMed]
- Frances, A.; Cordelier, P. The Emerging Role of Cytidine Deaminase in Human Diseases: A New Opportunity for Therapy? Mol. Ther. 2020, 28, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.J.; Fromme, J.C.; Verdine, G.L. Structure of human cytidine deaminase bound to a potent inhibitor. J. Med. Chem. 2005, 48, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Summy, J.M. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef]
- Montero, J.C.; Seoane, S.; Ocaña, A.; Pandiella, A. Inhibition of Src Family Kinases and Receptor Tyrosine Kinases by Dasatinib: Possible Combinations in Solid Tumors. Clin. Cancer Res. 2011, 17, 5546–5552. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Zhao, L.; He, H.; Zhao, P.; Peng, Z.; Liu, F.; Chen, J.; Wu, W.; Wang, G.; et al. Knockdown of Thymidine Kinase 1 Suppresses Cell Proliferation, Invasion, Migration, and Epithelial–Mesenchymal Transition in Thyroid Carcinoma Cells. Front. Oncol. 2020, 9, 1475. [Google Scholar] [CrossRef]
- Aufderklamm, S.; Todenhöfer, T.; Gakis, G.; Kruck, S.; Hennenlotter, J.; Stenzl, A.; Schwentner, C. Thymidine kinase and cancer monitoring. Cancer Lett. 2012, 316, 6–10. [Google Scholar] [CrossRef]
- Welin, M.; Kosinska, U.; Mikkelsen, N.E.; Carnrot, C.; Zhu, C.; Wang, L.; Eriksson, S.; Munch-Petersen, B.; Eklund, H. Structures of thymidine kinase 1 of human and mycoplasmic origin. Proc. Natl. Acad. Sci. USA. 2004, 101, 17970–17975. [Google Scholar] [CrossRef]
- Rose, M.G.; Farrell, M.P.; Schmitz, J.C. Thymidylate Synthase: A Critical Target for Cancer Chemotherapy. Clin. Color. Cancer 2002, 1, 220–229. [Google Scholar] [CrossRef]
- Bagheri, S.; Saboury, A.A.; Haertlé, T. Adenosine deaminase inhibition. Int. J. Biol. Macromol. 2019, 141, 1246–1257. [Google Scholar] [CrossRef]
- Reayi, A.; Hosmane, R.S. Inhibitors of Adenosine Deaminase: Continued Studies of Structure-Activity Relationships in Analogues of Coformycin. Nucleosides Nucleotides Nucleic Acids 2004, 23, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Emele, J.F.; Shanaman, J. Bradykinin writhing: A method for measuring analgesiA. Proc. Soc. Exp. Biol. Med. 1963, 114, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Qin, X.; Xu, Z.; Song, Y.; Jiang, H.; Wu, Y.; Ruan, H.; Chen, J. Comparison of Cytotoxicity Evaluation of Anticancer Drugs between Real-Time Cell Analysis and CCK-8 Method. ACS Omega 2019, 4, 12036–12042. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Rahman, S.M.A.; Asaduzzaman, M.; Islam, A.B.M.M.K.; Chowdhury, A.K.A. Synthesis, in vitro bioassays, and computational study of heteroaryl nitazoxanide analogs. Pharmacol. Res. Perspect. 2021, 9, e00800. [Google Scholar] [CrossRef] [PubMed]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, H.; Ishiyama, M.; Ohseto, F.; Sasamoto, K.; Hamamoto, T.; Suzuki, K.; Watanabe, M. A water-soluble tetrazolium salt useful for colorimetric cell viability assay. Anal. Commun. 1999, 36, 47–50. [Google Scholar] [CrossRef]
- Hehre, W.J.; Stewart, R.F.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. I. Use of Gaussian Expansions of Slater-Type Atomic Orbitals. J. Chem. Phys. 1969, 51, 2657–2664. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Concentration (µg/mL) | Live Cell Count (Approx.) | Cell Viability % |
|---|---|---|---|
| Control | - | 20,000 | 100% |
| DMSO (2.5%) | - | 19,000 | >95% |
| 2 | 500 | 19,000 | >95% |
| 3 | 500 | 19,000 | >95% |
| 4 | 500 | 14,000–16,000 | 70–80% |
| 5 | 500 | 8000–10,000 | 40–50% |
| 6 | 500 | <1000 | <5% |
| 7 | 500 | 19,000 | >95% |
| 8 | 500 | 14,000–16,000 | 70–80% |
| Compound | Concentration (µg/mL) | Cell Viability % | Mean Absorbance at 490 nm | % Growth Inhibition | IC50 |
|---|---|---|---|---|---|
| DMSO (2.5%) | - | 100 | 3.9405 | 0 | - |
| 5 | 15.62 | 99.48 | 3.8395 | 0.52 | >250 µg/mL |
| 31.25 | 99.61 | 3.8445 | 0.39 | ||
| 62.5 | 99.68 | 3.8475 | 0.32 | ||
| 125 | 99.98 | 3.859 | 0.02 | ||
| 250 | 99.62 | 3.845 | 0.38 | ||
| 6 | 15.62 | 97.94 | 3.8595 | 2.06 | 54 µg/mL (76.4 µM) |
| 31.25 | 91.91 | 3.622 | 8.09 | ||
| 62.5 | 35.03 | 1.3805 | 64.97 | ||
| 125 | 30.89 | 1.2175 | 69.11 | ||
| 250 | 25.69 | 1.0125 | 74.31 |
| Compound | 1EQG | 5IKT | 4O1L |
|---|---|---|---|
| Standard | −7.7 | −8.4 | −8.9 |
| 2 | −6.9 | −7.1 | −6.5 |
| 7 | −6.1 | −7.0 | −7.9 |
| 8 | −6.8 | −6.8 | −7.4 |
| Compound | 1MQ0 | 3G5D | 1XBT | 6ZXO | 3LGG |
|---|---|---|---|---|---|
| Standard | −6.8 | −9.4 | −8.0 | −9.1 | −7.4 |
| 6 | −7.1 | −7.6 | −8.0 | −7.8 | −8.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, F.; Rahman, F.I.; Saha, P.; Mikami, A.; Osawa, T.; Obika, S.; Rahman, S.M.A. Synthesis of Sugar and Nucleoside Analogs and Evaluation of Their Anticancer and Analgesic Potentials. Molecules 2022, 27, 3499. https://doi.org/10.3390/molecules27113499
Hussain F, Rahman FI, Saha P, Mikami A, Osawa T, Obika S, Rahman SMA. Synthesis of Sugar and Nucleoside Analogs and Evaluation of Their Anticancer and Analgesic Potentials. Molecules. 2022; 27(11):3499. https://doi.org/10.3390/molecules27113499
Chicago/Turabian StyleHussain, Fahad, Fahad Imtiaz Rahman, Poushali Saha, Atsushi Mikami, Takashi Osawa, Satoshi Obika, and S. M. Abdur Rahman. 2022. "Synthesis of Sugar and Nucleoside Analogs and Evaluation of Their Anticancer and Analgesic Potentials" Molecules 27, no. 11: 3499. https://doi.org/10.3390/molecules27113499
APA StyleHussain, F., Rahman, F. I., Saha, P., Mikami, A., Osawa, T., Obika, S., & Rahman, S. M. A. (2022). Synthesis of Sugar and Nucleoside Analogs and Evaluation of Their Anticancer and Analgesic Potentials. Molecules, 27(11), 3499. https://doi.org/10.3390/molecules27113499