
Sesquiterpene Lactones as Promising Candidates for Cancer Therapy: Focus on Pancreatic Cancer


 and
and 
Abstract
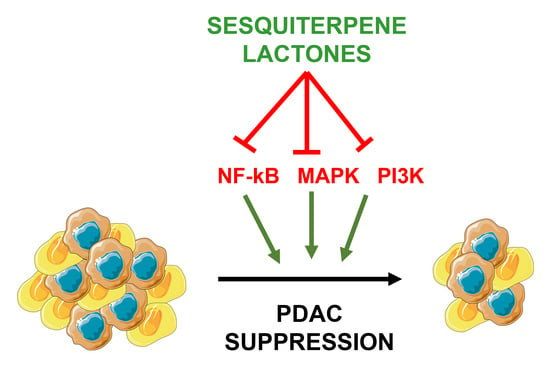
:
1. Introduction
2. Pancreatic Cancer
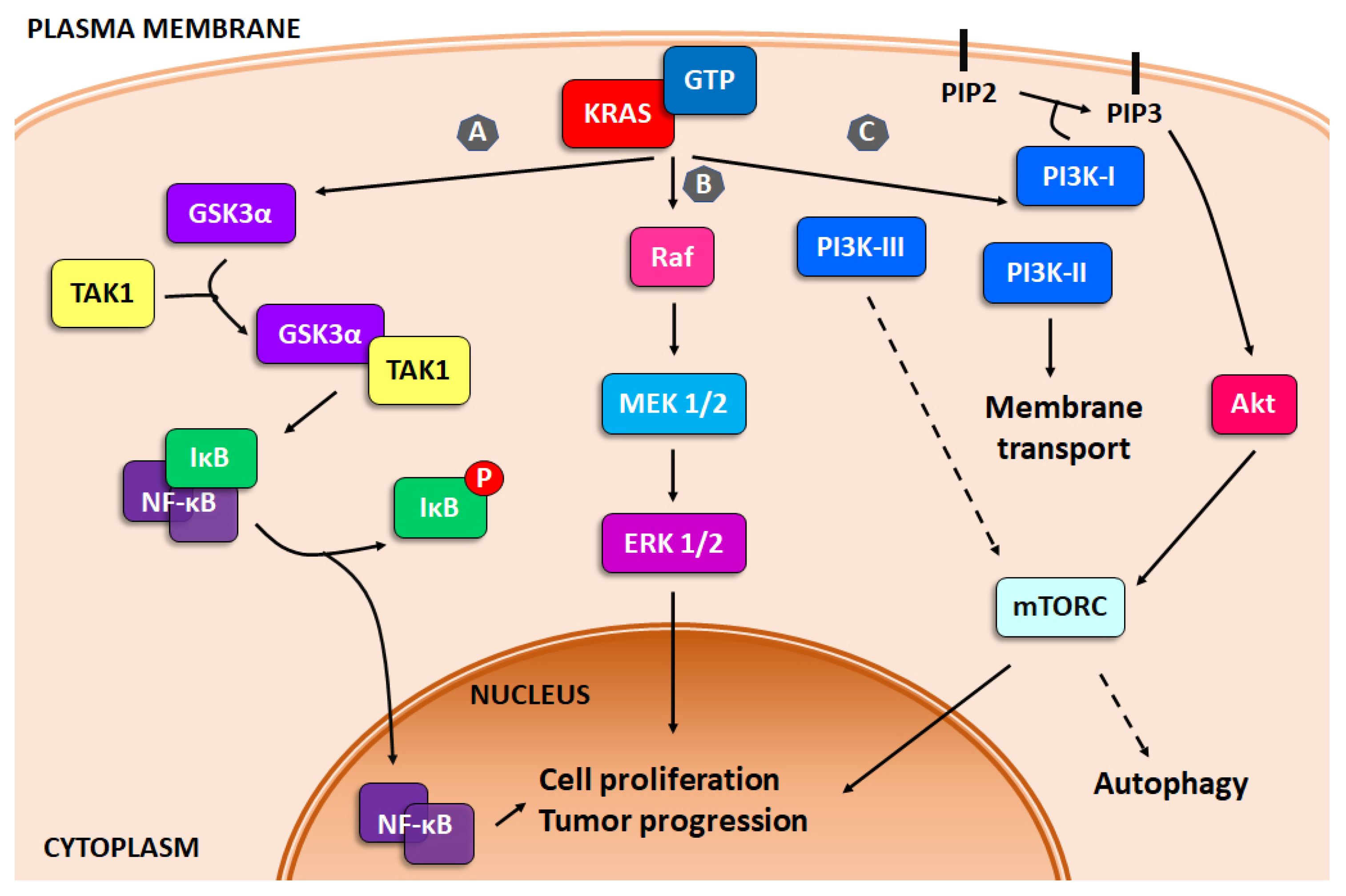
3. Activated Survival Pathways in Pancreatic Cancer: KRAS Mutation
3.1. NF-κB
3.2. MAPK
3.3. PI3K
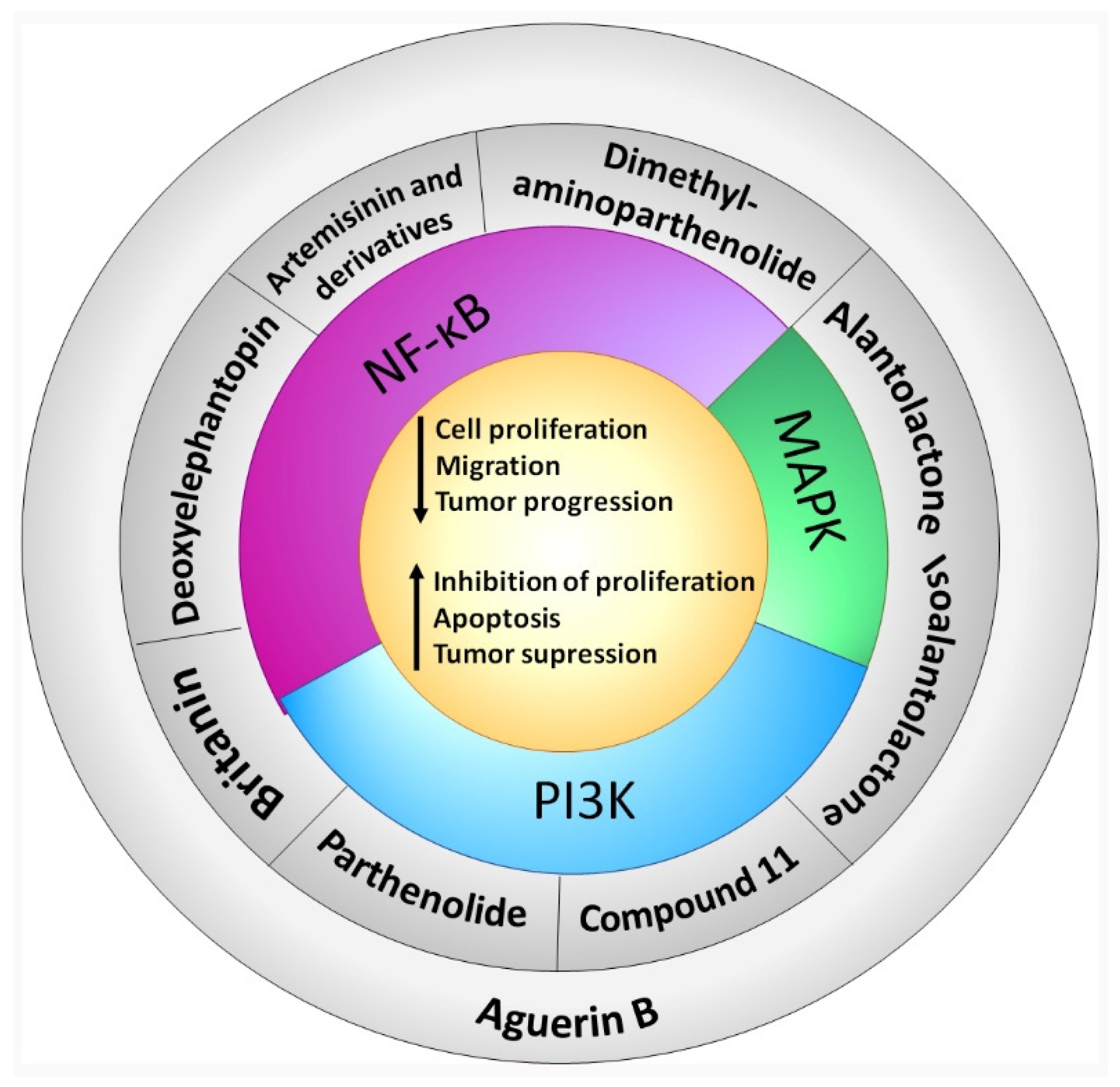
4. In Vitro and In Vivo Bioactive Sesquiterpene Lactones with Activity on KRAS Pathways
4.1. NF-κB
4.2. MAPK
4.3. PI3K
4.4. NF-κB, MAPK and PI3K Inhibition
5. Other Bioactive Sesquiterpene Lactones Tested in PDAC Models
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Sülsen, V.P.; Martino, V.S. Overview. In Sesquiterpene Lactones: Advances in their Chemistry and Biological Aspects; Sülsen, V.P., Martino, V.S., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 3–17. Available online: https://link.springer.com/chapter/10.1007/978-3-319-78274-4_1 (accessed on 21 January 2022).
- Moujir, L.; Callies, O.; Sousa, P.M.C.; Sharopov, F.; Seca, A.M.L. Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases. Appl. Sci. 2020, 10, 3001. [Google Scholar] [CrossRef]
- Babaei, G.; Aliarab, A.; Abroon, S.; Rasmi, Y.; Aziz, S.G.G. Application of Sesquiterpene Lactone: A New Promising Way for Cancer Therapy Based on Anticancer Activity. Biomed. Pharmacother. 2018, 106, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Gach, K.; Długosz, A.; Janecka, A. The Role of Oxidative Stress in Anticancer Activity of Sesquiterpene Lactones. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 477–486. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, J.; Kinghorn, A.D. Development of Anticancer Agents from Plant-Derived Sesquiterpene Lactones. Curr. Med. Chem. 2016, 23, 2397–2420. [Google Scholar] [CrossRef]
- Fabian, L.; Sulsen, V.; Frank, F.; Cazorla, S.; Malchiodi, E.; Martino, V.; Lizarraga, E.; Catalan, C.; Moglioni, A.; Muschietti, L.; et al. In Silico Study of Structural and Geometrical Requirements of Natural Sesquiterpene Lactones with Trypanocidal Activity. Mini-Rev. Med. Chem. 2013, 13, 1407–1414. [Google Scholar] [CrossRef]
- Shih, H.P.; Wang, A.; Sander, M. Pancreas Organogenesis: From Lineage Determination to Morphogenesis. Annu. Rev. Cell Dev. Biol. 2013, 29, 81–105. [Google Scholar] [CrossRef] [Green Version]
- Malinova, A.; Veghini, L.; Real, F.X.; Corbo, V. Cell Lineage Infidelity in PDAC Progression and Therapy Resistance. Front. Cell Dev. Biol. 2021, 9, 795251. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic Cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of Oncogenic KRAS in the Diagnosis, Prognosis and Treatment of Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Singh, K.; Pruski, M.; Bland, R.; Younes, M.; Guha, S.; Thosani, N.; Maitra, A.; Cash, B.D.; McAllister, F.; Logsdon, C.D.; et al. Kras Mutation Rate Precisely Orchestrates Ductal Derived Pancreatic Intraepithelial Neoplasia and Pancreatic Cancer. Lab. Investig. 2021, 101, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.A.; Gallinger, S. Pancreatic Cancer Evolution and Heterogeneity: Integrating Omics and Clinical Data. Nat. Cancer 2021, 22, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Picozzi, V.J.; Oh, S.Y.; Edwards, A.; Mandelson, M.T.; Dorer, R.; Rocha, F.G.; Alseidi, A.; Biehl, T.; Traverso, L.W.; Helton, W.S.; et al. Five-Year Actual Overall Survival in Resected Pancreatic Cancer: A Contemporary Single-Institution Experience from a Multidisciplinary Perspective. Ann. Surg. Oncol. 2017, 24, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Lemoine, N.R. Pancreatic Cancer: Molecular Pathogenesis and New Therapeutic Targets. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 412–422. [Google Scholar] [CrossRef]
- Hruban, R.H.; Adsay, N.V.; Albores-Saavedra, J.; Compton, C.; Garrett, E.S.; Goodman, S.N.; Kern, S.E.; Klimstra, D.S.; Klöppel, G.; Longnecker, D.S.; et al. Pancreatic Intraepithelial Neoplasia: A New Nomenclature and Classification System for Pancreatic Duct Lesions. Am. J. Surg. Pathol. 2001, 25, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Means, A.L.; Meszoely, I.M.; Suzuki, K.; Miyamoto, Y.; Rustgi, A.K.; Coffey, R.J.; Wright, C.V.E.; Stoffers, D.A.; Leach, S.D. Pancreatic Epithelial Plasticity Mediated by Acinar Cell Transdifferentiation and Generation of Nestin-Positive Intermediates. Development 2005, 132, 3767–3776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, X.-Q.; Wang, Z.-F.; Wu, X.-L.; Zhou, C.-H.; Yan, J.-Y.; Hu, B.-Y.; et al. The Molecular Biology of Pancreatic Adenocarcinoma: Translational Challenges and Clinical Perspectives. Signal Transduct. Target. Ther. 2021, 6, 1–23. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic Cancer Genomes Reveal Aberrations in Axon Guidance Pathway Genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Camateros, P.; Cheung, W.Y. A Real-World Comparison of FOLFIRINOX, Gemcitabine Plus nab-Paclitaxel, and Gemcitabine in Advanced Pancreatic Cancers. J. Gastrointest. Cancer 2019, 50, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Sielaf, C.M.; Mousa-Shaker, A. Status and Future Directions in the Management of Pancreatic Cancer: Potential Impact of Nanotechnology. J. Cancer Res. Clin. Oncol. 2018, 144, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, A.V.; Biankin, A.V.; Parish, C.; Khachigian, L.M. Targeted Therapies in the Management of Locally Advanced and Metastatic Pancreatic Cancer: A Systematic Review. Oncotarget 2018, 9, 21613–21627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uccello, M.; Moschetta, M.; Mak, G.; Alam, T.; Murias Henriquez, C.; Arkenau, H. Towards an Optimal Treatment Algorithm for Metastatic Pancreatic Ductal Adenocarcinoma (PDA). Curr. Oncol. 2018, 25, 90–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; di Magliano, M.P. Oncogenic Kras Is Required for Both the Initiation and Maintenance of Pancreatic Cancer in Mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.; Der, C. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Raphael, B.; Al, E. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017, 32, 185–203. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, T.L.; Lertpiriyapong, K.; Cocco, L.; Martelli, A.M.; Libra, M.; Candido, S.; Montalto, G.; Cervello, M.; Steelman, L.; Abrams, S.L.; et al. Roles of EGFR and KRAS and Their Downstream Signaling Pathways in Pancreatic Cancer and Pancreatic Cancer Stem Cells. Adv. Biol. Regul. 2015, 59, 65–81. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Krappmann, D.; Vincendeau, M. Mechanisms of NF-κB Deregulation in Lymphoid Malignancies. Semin. Cancer Biol. 2016, 39, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Sun, S. The Noncanonical NF-κB Pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, A.; Bertrand, M. More to Life than NF-κB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Yang, G.; Feng, M.; Zheng, S.; Cao, Z.; Qiu, J.; You, L.; Zheng, L.; Hu, Y.; Zhang, T.; et al. NF-κB in Pancreatic Cancer: Its Key Role in Chemoresistance. Cancer Lett. 2018, 421, 127–134. [Google Scholar] [CrossRef]
- Concetti, J.; Wilson, C. NFKB1 and Cancer: Friend or Foe? Cells 2018, 7, 133. [Google Scholar] [CrossRef] [Green Version]
- Asati, V.; Mahapatra, D.; Bharti, S. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK Signaling Pathways Inhibitors as Anticancer Agents: Structural and Pharmacological Perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef]
- Qazi, A.; Hussain, A.; Hamid, A.; Qurishi, Y.; Majeed, R.; Ahmad, M.; Ahmad Najar, R.; Ahamd Bhat, J.; Kumar Singh, S.; Afzal Zargar, M.; et al. Recent Development in Targeting PI3K-Akt-mTOR Signaling for Anticancer Therapeutic Strategies. Anticancer Agents Med. Chem. 2013, 13, 1552–1564. [Google Scholar] [CrossRef]
- Hilger, R.; Scheulen, M.; Strumberg, D. The Ras-Raf-MEK-ERK Pathway in the Treatment of Cancer. Oncol. Res. Treat. 2002, 25, 511–518. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK Mitogen-Activated Protein Kinase Cascade for the Treatment of Cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Smalley, K. A Pivotal Role for ERK in the Oncogenic Behaviour of Malignant Melanoma? Int. J. Cancer 2003, 104, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.; Er, E.; Blenis, J. The Ras-ERK and PI3K-mTOR Pathways: Cross-Talk and Compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodish, H. Molecular Cell Biology; NCBI Bookshelf W.H.Freeman & Co. Ltd.: New York, NY, USA, 2000. [Google Scholar]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF Proteins Take Centre Stage. Nat. Rev. Mol. Cell. Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Rajalingam, K.; Schreck, R.; Rapp, U.; Albert, S. Ras Oncogenes and Their Downstream Targets. Biochim. Biophys. Acta 2007, 1773, 1177–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, N.; Lyons, J. Recent Progress in Targeting the Raf/MEK/ERK Pathway with Inhibitors in Cancer Drug Discovery. Curr. Opin. Pharmacol. 2005, 5, 350–356. [Google Scholar] [CrossRef]
- Burgering, B.; Coffer, P.J. Protein Kinase B (c-Akt) in Phosphatidylinositol-3-OH Kinase Signal Transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the Phosphoinositide 3-Kinase Pathway in Cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [Green Version]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.-K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millán, J.; Cutillas, P.R.; et al. Angiogenesis Selectively Requires the p110α Isoform of PI3K to Control Endothelial Cell Migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef]
- Chi, H. Regulation and Function of mTOR Signalling in T Cell Fate Decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.-B.; Bousquet-Dubouch, M.-P.; et al. Pharmacological Targeting of the Protein Synthesis mTOR/4E-BP1 Pathway in Cancer-Associated Fibroblasts Abrogates Pancreatic Tumour Chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kγ Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN Mutations: The PI3K Pathway as an Integrator of Multiple Inputs during Tumorigenesis. Nat. Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular Function of Phosphoinositide 3-Kinases: Implications for Development, Immunity, Homeostasis, and Cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef] [PubMed]
- Suire, S.; Coadwell, J.; Ferguson, G.J.; Davidson, K.; Hawkins, P.; Stephens, L. p84, A New Gbetagamma-Activated Regulatory Subunit of the Type IB Phosphoinositide 3-kinase p110gamma. Curr. Biol. 2005, 15, 566–570. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.D.; Sukhova, G.K.; Libby, P.; Schvartz, E.; Lichtenstein, A.H.; Field, S.J.; Kennedy, C.; Madhavaraou, S.; Luo, J.; Wu, D.; et al. Deletion of the Phosphoinositide 3-kinase p110gamma Gene Attenuates Murine Atherosclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 8077–8082. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.R.; Auger, K.R. Phylogenomics of Phosphoinositide Lipid Kinases: Perspectives on the Evolution of Second Messenger Signaling and Drug Discovery. BMC Evol. Biol. 2011, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Falasca, M.; Maffucci, T. Regulation and Cellular Functions of Class II Phosphoinositide 3-kinases. Biochem. J. 2012, 443, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Long, Y.C.; Shen, H.-M. Differential Regulatory Functions of Three Classes of Phosphatidylinositol and Phosphoinositide 3-Kinases in Autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef] [Green Version]
- Gaidarov, I.; Smith, M.E.; Domin, J.; Keen, J.H. The Class II Phosphoinositide 3-Kinase C2α Is Activated by Clathrin and Regulates Clathrin-Mediated Membrane Trafficking. Mol. Cell 2001, 7, 443–449. [Google Scholar] [CrossRef]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schöneberg, J.; Ullrich, A.; Lampe, A.; Müller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal Control of Endocytosis by Phosphatidylinositol-3,4-Bisphosphate. Nature 2013, 499, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Krag, C.; Malmberg, E.K.; Salcini, A.E. PI3KC2α, a class II PI3K, is rEquired for Dynamin-Independent Internalization Pathways. J. Cell Sci. 2010, 123, 4240–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, S.; Kiger, A.A. Coordination between RAB GTPase and Phosphoinositide Regulation and Functions. Nat. Rev. Mol. Cell Biol. 2012, 13, 463–470. [Google Scholar] [CrossRef]
- Lu, N.; Shen, Q.; Mahoney, T.R.; Neukomm, L.J.; Wang, Y.; Zhou, Z. Two PI 3-Kinases and One PI 3-Phosphatase Together Establish the Cyclic Waves of Phagosomal PtdIns(3)P Critical for the Degradation of Apoptotic Cells. PLOS Biol. 2012, 10, e1001245. [Google Scholar] [CrossRef] [Green Version]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network Organization of the Human Autophagy System. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Devereaux, K.; Dall’Armi, C.; Alcazar-Roman, A.; Ogasawara, Y.; Zhou, X.; Wang, F.; Yamamoto, A.; De Camilli, P.; Di Paolo, G. Regulation of Mammalian Autophagy by Class II and III PI 3-Kinases through PI3P Synthesis. PLoS ONE 2013, 8, e76405. [Google Scholar] [CrossRef] [Green Version]
- Backer, J.M. The Regulation and Function of Class III PI3Ks: Novel Roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Jean, S.; Kiger, A.A. Classes of Phosphoinositide 3-kinases at a Glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef] [Green Version]
- Simonsen, A.; Tooze, S.A. Coordination of Membrane Events during Autophagy by Multiple Class III PI3-Kinase Complexes. J. Cell Biol. 2009, 186, 773–782. [Google Scholar] [CrossRef]
- Raiborg, C.; Schink, K.O.; Stenmark, H. Class III Phosphatidylinositol 3-kinase and Its Catalytic Product PtdIns3P in Regulation of Endocytic Membrane Traffic. FEBS J. 2013, 280, 2730–2742. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Wu, H.; Njoku, V.; Ralstin, M.; Holcomb, B.; Crooks, P.A.; Neelakantan, S.; Sweeney, C.J.; Schmidt, C.M. Effect of Celecoxib and the Novel Anti-Cancer Agent, Dimethylamino-Parthenolide, in a Developmental Model of Pancreatic Cancer. Pancreas 2008, 37, e45–e53. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Benson, A.B.; Cardin, D.B.; Chiorean, E.G.; Chung, V.; Czito, B.; Del Chiaro, M.; et al. Pancreatic Adenocarcinoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Papademetrio, D.L.; Cavaliere, V.; Simunovich, T.; Costantino, S.; Campos, M.D.; Lombardo, T.; Kaiser, C.M.F.; Álvarez, É. Interplay between Autophagy and Apoptosis in Pancreatic Tumors in Response to Gemcitabine. Target. Oncol. 2013, 9, 123–134. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Wu, H.; Hruban, R.H.; Lowy, A.M.; Crooks, P.A.; Schmidt, C.M. Efficacy of Dimethylaminoparthenolide and Sulindac in Combination With Gemcitabine in a Genetically Engineered Mouse Model of Pancreatic Cancer. Pancreas 2013, 42, 160–167. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Wu, H.; Stantz, K.; Agaram, N.; Crooks, P.A.; Schmidt, C.M. Dimethylaminoparthenolide and Gemcitabine: A Survival Study Using a Genetically Engineered Mouse Model of Pancreatic Cancer. BMC Cancer 2013, 13, 194. [Google Scholar] [CrossRef] [Green Version]
- Lamture, G.; Crooks, P.A.; Borrelli, M.J. Actinomycin-D and Dimethylamino-parthenolide Synergism in Treating Human Pancreatic Cancer Cells. Drug Dev. Res. 2018, 79, 287–294. [Google Scholar] [CrossRef]
- Aung, W.; Sogawa, C.; Furukawa, T.; Saga, T. Anticancer Effect of Dihydroartemisinin (DHA) in a Pancreatic Tumor Model Evaluated by Conventional Methods and Optical Imaging. Anticancer Res. 2011, 31, 1549–1558. [Google Scholar]
- Wang, S.J.; Sun, B.; Cheng, Z.X.; Zhou, H.X.; Gao, Y.; Kong, R.; Chen, H.; Jiang, H.C.; Pan, S.H.; Xue, D.B.; et al. Dihydroartemisinin Inhibits Angiogenesis in Pancreatic Cancer by Targeting the NF-κB Pathway. Cancer Chemother. Pharmacol. 2011, 68, 1421–1430. [Google Scholar] [CrossRef]
- Chen, H.; Sun, B.; Pan, S.; Jiang, H.; Sun, X. Dihydroartemisinin Inhibits Growth of Pancreatic Cancer Cells In Vitro and In Vivo. Anti-Cancer Drugs 2009, 20, 131–140. [Google Scholar] [CrossRef]
- Chen, H.; Sun, B.; Pan, S.; Li, J.; Xue, D.; Meng, Q.; Jiang, H. Study on Anticancer Effect of Dihydroartemisinin on Pancreatic Cancer. Zhonghua Wai Ke Za Zhi 2009, 47, 1002–1005. [Google Scholar] [PubMed]
- Chen, H.; Sun, B.; Wang, S.; Pan, S.; Gao, Y.; Bai, X.; Xue, D. Growth Inhibitory Effects of Dihydroartemisinin on Pancreatic Cancer Cells: Involvement of Cell Cycle Arrest and Inactivation of Nuclear Factor-κB. J. Cancer Res. Clin. Oncol. 2010, 136, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Gao, Y.; Chen, H.; Kong, R.; Jiang, H.C.; Pan, S.H.; Xue, D.B.; Bau, X.W.; Sun, B. Dihydroartemisinin Inactivates NF-kappaB and Potentiates the Anti-tumor Effect of Gemcitabine on Pancreatic Cancer Both in Vitro and in Vivo. Cancer Lett. 2010, 293, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, B.; Pan, S.; Chen, H.; Kong, R.; Li, J.; Xue, D.; Bai, X.; Jiang, H. Experimental Study of the Function and Mechanism Combining Dihydroartemisinin and Gemcitabine in Treating Pancreatic Cancer. Zhonghua Wai Ke Za Zhi 2010, 48, 530–534. [Google Scholar] [PubMed]
- Kong, R.; Jia, G.; Cheng, Z.; Wang, Y.; Mu, M.; Wang, S.; Pan, S.; Gao, Y.; Jiang, H.; Dong, D.; et al. Dihydroartemisinin Enhances Apo2L/TRAIL-Mediated Apoptosis in Pancreatic Cancer Cells via ROS-Mediated Up-Regulation of Death Receptor 5. PLoS ONE 2012, 7, e37222. [Google Scholar] [CrossRef]
- Zhou, Z.-H.; Chen, F.-X.; Xu, W.-R.; Qian, H.; Sun, L.-Q.; Lü, X.-T.; Chen, L.; Zhang, J.; Ji, H.-C.; Fei, S.-J. Enhancement Effect of Dihydroartemisinin on Human γδ T Cell Proliferation and Killing Pancreatic Cancer Cells. Int. Immunopharmacol. 2013, 17, 850–857. [Google Scholar] [CrossRef]
- Jia, G.; Kong, R.; Ma, Z.-B.; Han, B.; Wang, Y.-W.; Pan, S.-H.; Li, Y.-H.; Sun, B. The activation of c-Jun NH2-Terminal Kinase Is Required for Dihydroartemisinin-Induced Autophagy in Pancreatic Cancer Cells. J. Exp. Clin. Cancer Res. 2014, 33, 8. [Google Scholar] [CrossRef] [Green Version]
- Gaur, R.; Pathania, A.S.; Malik, F.A.; Bhakuni, R.S.; Verma, R.K. Synthesis of a Series of Novel Dihydroartemisinin Monomers and Dimers Containing Chalcone as a Linker and Their Anticancer Activity. Eur. J. Med. Chem. 2016, 122, 232–246. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Kong, R.; Xue, D.; Pan, S.; Chen, H.; Sun, B. Dihydroartemisinin Suppresses Pancreatic Cancer Cells via a microRNA-mRNA Regulatory Network. Oncotarget 2016, 7, 62460–62473. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-W.; Zhang, W.; Xu, N.; Li, Y.; Zhang, H.; Lv, S.-J.; Zhu, W.-H. Dihydroartemisinin Inhibits Proliferation of Pancreatic Cancer JF-305 Cells by Regulating Expression of Apoptosis Related Proteins and Production of Reactive Oxygen Species. China J. Chin. Mater. Med. 2017, 42, 3026–3030. [Google Scholar]
- Du, J.; Wang, X.; Li, Y.; Ren, X.; Zhou, Y.; Hu, W.; Zhou, C.; Jing, Q.; Yang, C.; Wang, L.; et al. DHA Exhibits Synergistic Therapeutic Efficacy with Cisplatin to Induce Ferroptosis in Pancreatic Ductal Adenocarcinoma via Modulation of Iron Metabolism. Cell Death Dis. 2021, 12, 705. [Google Scholar] [CrossRef] [PubMed]
- Du, J.-H.; Zhang, H.-D.; Ma, Z.-J.; Ji, K.-M. Artesunate Induces Oncosis-like Cell Death In Vitro and Has Antitumor Activity against Pancreatic Cancer Xenografts In Vivo. Cancer Chemother. Pharmacol. 2009, 65, 895–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cui, Y. Synergism of Cytotoxicity Effects of Triptolide and Artesunate Combination Treatment in Pancreatic Cancer Cell Lines. Asian Pac. J. Cancer Prev. 2013, 14, 5243–5248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Zhang, Z.; Wang, M.; Cao, X.; Qi, J.; Wang, D.; Gong, A.; Zhu, H. Role of GRP78 Inhibiting Artesunate-Induced Ferroptosis in KRAS Mutant Pancreatic Cancer Cells. Drug Des. Dev. Ther. 2019, 13, 2135–2144. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Zhou, Y.; Chen, Y.; Zhou, L.; Liang, J. A Novel Natural Product, Britanin, Inhibits Tumor Growth of Pancreatic Cancer by Suppressing Nuclear Factor-κB Activation. Cancer Chemother. Pharmacol. 2020, 85, 699–709. [Google Scholar] [CrossRef]
- Ji, D.; Zhong, X.; Huang, P.; Kang, P.; Leng, K.; Zheng, W.; Wang, Z.; Xu, Y.; Cui, Y. Deoxyelephantopin Induces Apoptosis via Oxidative Stress and Enhances Gemcitabine Sensitivity In Vitro and In Vivo through Targeting the NF-κB Signaling Pathway in Pancreatic Cancer. Aging 2020, 12, 11116–11138. [Google Scholar] [CrossRef]
- Rasul, A.; Khan, M.; Ali, M.; Li, J.; Li, X. Targeting Apoptosis Pathways in Cancer with Alantolactone and Isoalantolactone. Sci. World J. 2013, 2013, 248532. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Ding, C.; Rasul, A.; Yi, F.; Li, T.; Gao, H.; Gao, R.; Zhong, L.; Zhang, K.; Fang, X.; et al. Isoalantolactone Induces Reactive Oxygen Species Mediated Apoptosis in Pancreatic Carcinoma PANC-1 Cells. Int. J. Biol. Sci. 2012, 8, 533–547. [Google Scholar] [CrossRef] [Green Version]
- Wood, C.D.; Thornton, T.M.; Sabio, G.; Davis, R.A.; Rincon, M. Nuclear Localization of p38 MAPK in Response to DNA Damage. Int. J. Biol. Sci. 2009, 5, 428–437. [Google Scholar] [CrossRef] [Green Version]
- Gruber, R.; Panayiotou, R.; Nye, E.; Spencer-Dene, B.; Stamp, G.; Behrens, A. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-regulation of JAK–STAT3 Signaling. Gastroenterology 2016, 151, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Daglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Lin, J. Seed-in-Soil: Pancreatic Cancer Influenced by Tumor Microenvironment. Cancers 2017, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Yang, L.; Kang, Y.; Chen, M.; Lin, S.; Xiang, Y.; Li, C.; Dai, X.; Huang, X.; Liang, G.; et al. Alantolactone Sensitizes Human Pancreatic Cancer Cells to EGFR Inhibitors through the Inhibition of STAT3 Signaling. Mol. Carcinog. 2018, 58, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.Y.; Zhang, Q.; Zhang, B.; Yang, B.; Lin, N.M. Active Ingredients of Inula helenium L. Exhibits Similar Anti-cancer Effects as Isoalantolactone in Pancreatic Cancer Cells. Nat. Prod. Res. 2019, 34, 2539–2544. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Nakshatri, H.; Sweeney, C.J.; Marshall, M.S.; Wiebke, E.A.; Schmidt, C.M. Parthenolide and Sulindac Cooperate to Mediate Growth Suppression and Inhibit the Nuclear Factor-kappaB Pathway in Pancreatic Carcinoma Cells. Mol. Cancer Ther. 2005, 4, 587–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-W.; Cai, M.-X.; Xin, Y.; Wu, Q.-S.; Ma, J.; Yang, P.; Xie, H.-Y.; Huang, D.-S. Parthenolide Induces Proliferation Inhibition and Apoptosis of Pancreatic Cancer Cells In Vitro. J. Exp. Clin. Cancer Res. 2010, 29, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wang, X.; Sun, J.; Yang, Y.; Li, W.; Song, J. Parthenolide Suppresses Pancreatic Cell Growth by Autophagy-Mediated Apoptosis. OncoTargets Ther. 2017, 10, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Omar, A.M.; Dibwe, D.F.; Tawila, A.M.; Sun, S.; Kim, M.J.; Awale, S. Chemical Constituents from Artemisia vulgaris and Their Antiausterity Activities against the PANC-1 Human Pancreatic Cancer Cell Line. Nat. Prod. Res. 2019, 35, 4279–4285. [Google Scholar] [CrossRef]
- Moeinifard, M.; Hassan, Z.M.; Fallahian, F.; Hamzeloo-Moghadam, M.; Taghikhani, M. Britannin Induces Apoptosis through AKT-FOXO1 Pathway in Human Pancreatic Cancer Cells. Biomed. Pharmacother. 2017, 94, 1101–1110. [Google Scholar] [CrossRef]
- Zhang, C.; Huang, L.; Xiong, J.; Xie, L.; Ying, S.; Jia, Y.; Yao, Y.; Song, X.; Zeng, Z.; Yuan, J. Isoalantolactone Inhibits Pancreatic Cancer Proliferation by Regulation of PI3K and Wnt Signal Pathway. PLoS ONE 2021, 16, e0247752. [Google Scholar] [CrossRef]
- Chicca, A.; Tebano, M.; Adinolfi, B.; Ertugrul, K.; Flamini, G.; Nieri, P. Anti-proliferative Activity of Aguerin B and a New Rare Nor-Guaianolide Lactone Isolated from the Aerial Parts of Centaurea Deflexa. Eur. J. Med. Chem. 2011, 46, 3066–3070. [Google Scholar] [CrossRef] [PubMed]
- Amini, E.; Nabiuni, M.; Behzad, S.B.; Seyfi, D.; Eisvand, F.; Sahebkar, A.; Shakeri, A. Anticancer Potential of Aguerin B, a Sesquiterpene Lactone Isolated from Centaurea behen in Metastatic Breast Cancer Cells. Recent Pat. Anticancer Drug Discov. 2020, 15, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Fidler, I.J. Hypoxia Increases Resistance of Human Pancreatic Cancer Cells to Apoptosis Induced by Gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef] [Green Version]
- Papademetrio, D.L.; Lompardía, S.L.; Simunovich, T.; Costantino, S.; Mihalez, C.Y.; Cavaliere, V.; Álvarez, É. Inhibition of Survival Pathways MAPK and NF-kB Triggers Apoptosis in Pancreatic Ductal Adenocarcinoma Cells via Suppression of Autophagy. Target. Oncol. 2016, 11, 183–195. [Google Scholar] [CrossRef]
- Hoffmann, R.; Von Schwarzenberg, K.; López-Antón, N.; Rudy, A.; Wanner, G.; Dirsch, V.M.; Vollmar, A.M. Helenalin Bypasses Bcl-2-mediated Cell Death Resistance by Inhibiting NF-κB and Promoting Reactive Oxygen Species Generation. Biochem. Pharmacol. 2011, 82, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Wu, Q.; Li, H.; Bae, G.-U.; Kim, A.K.; Ryu, J.-H. A Sesquiterpene Lactone from Siegesbeckia Glabrescens Suppresses Hedgehog/Gli-Mediated Transcription in Pancreatic Cancer Cells. Oncol. Lett. 2016, 12, 2912–2917. [Google Scholar] [CrossRef] [Green Version]
- Au, T.H.; Skarbek, C.; Pethe, S.; Labruere, R.; Baltaze, J.-P.; Nguyen, T.P.H.; Vu, T.T.H.; Vo-Thanh, G. Structural Modification and Biological Activity Studies of Tagitinin C and Its Derivatives. Tetrahedron 2021, 92, 132248. [Google Scholar] [CrossRef]
- De Ford, C.; Ulloa, J.L.; Catalán, C.A.N.; Grau, A.; Martino, V.S.; Muschietti, L.V.; Merfort, I. The Sesquiterpene Lactone Polymatin B from Smallanthus Sonchifolius Induces Different Cell Death Mechanisms in Three Cancer Cell Lines. Phytochemistry 2015, 117, 332–339. [Google Scholar] [CrossRef]
- Zeng, Y.-T.; Jiang, J.-M.; Lao, H.-Y.; Guo, J.-W.; Lun, Y.-N.; Yang, M. Antitumor and Apoptotic Activities of the Chemical Constituents from the Ethyl Acetate Extract of Artemisia Indica. Mol. Med. Rep. 2014, 11, 2234–2240. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, Y.; Zhang, W.-L.; Dong, M.-S.; Zhang, J.-H. Sclareolide Enhances Gemcitabine-Induced Cell Death through Mediating the NICD and Gli1 Pathways in Gemcitabine-Resistant Human Pancreatic Cancer. Mol. Med. Rep. 2017, 15, 1461–1470. [Google Scholar] [CrossRef]
- Zhou, N.; Li, J.-J.; Wu, Y.; Wang, Z.-Y.; Wang, X.; Yin, Z.-P.; Liu, Y.-R.; Shang, X.-Y. New Polymerized Sesquiterpene Lactones from Ainsliaea Yunnanensis and Their Activity Evaluation. Nat. Prod. Res. 2021, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Goswami, A.; Shah, B.A.; Batra, N.; Kumar, A.; Guru, S.K.; Bhushan, S.; Malik, F.A.; Joshi, A.; Singh, J. Multiple Pharmacological Properties of a Novel Parthenin Analog P16 as Evident by its Cytostatic and Antiangiogenic Potential Against Pancreatic Adenocarcinoma PANC -1 Cells. Anti-Cancer Agents Med. Chem. 2016, 15, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Güçlü, E.; Çınar Ayan, İ.; Gül Dursun, H.; Vural, H. Tomentosin Induces Apoptosis in Pancreatic Cancer Cells through Increasing Reactive Oxygen Species and Decreasing Mitochondrial Membrane Potential. 2022. Available online: https://www.researchsquare.com/article/rs-1508545/latest.pdf (accessed on 3 May 2022).
- Mandal, S.K.; Debnath, U.; Kumar, A.; Thomas, S.; Mandal, S.C.; Choudhury, M.D.; Palit, P. Natural Sesquiterpene Lactones in the Prevention and Treatment of Inflammatory Disorders and cancer: A Systematic Study of this Emerging Therapeutic Approach based on Chemical and Pharmacological Aspect. Lett. Drug Des. Discov. 2020, 17, 1102–1116. [Google Scholar] [CrossRef]
- Schmidt, T.J. Structure-Activity and Activity-Activity Relationships of Sesquiterpene Lactones. In Sesquiterpene Lactones: Advances in Their Chemistry and Biological Aspects; Sülsen, V., Martino, V., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 349–371. [Google Scholar] [CrossRef]
- Sülsen, V.; Elso, O.; Borgo, J.; Laurella, L.; Catalán, C.A. Recent Patents Involving Sesquiterpene Lactones with Therapeutic Application. In Studies in Natural Products Chemistry. Bioactive Natural Products; Atta-ur-Rahman, Ed.; Elsevier Science Publishers: Amsterdam, The Netherlands, 2021; Volume 69, Chapter 4; pp. 129–194. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Klempnauer, K.-H. Natural Products with Antitumor Potential Targeting the MYB-C/EBPβ-p300 Transcription Module. Molecules 2022, 27, 2077. [Google Scholar] [CrossRef]
- Ren, Y.; Kinghorn, A.D. Development of Potential Antitumor Agents from the Scaffolds of Plant-Derived Terpenoid Lactones. J. Med. Chem. 2020, 63, 15410–15448. [Google Scholar] [CrossRef]
- Paço, A.; Brás, T.; Santos, J.O.; Sampaio, P.; Gomes, A.C.; Duarte, M.F. Anti-Inflammatory and Immunoregulatory Action of Sesquiterpene Lactones. Molecules 2022, 27, 1142. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Compound Number | Bioactive Compound Name | Chemical Structures |
|---|---|---|
| 1 | Parthenolide |  |
| 2 | Dimethylaminoparthenolide (DMAPT) | 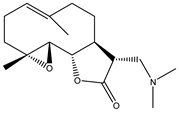 |
| 3 | Artemisinin | 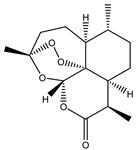 |
| 4 | Dihydroartemisinin (DHA) | 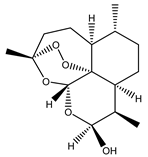 |
| 5 | Artesunate | 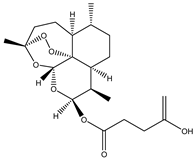 |
| 6 | Britanin |  |
| 7 | Deoxyelephantopin (DET) | 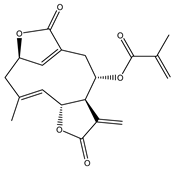 |
| 8 | Alantolactone |  |
| 9 | Isoalantolactone | 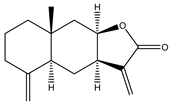 |
| 10 | Alloalantolactone |  |
| 11 | 2α-chloro-3β,9β-dihydroxy-1β,10β-epoxy-4α,6αH-guai-11(13)-en-12,5-olide | 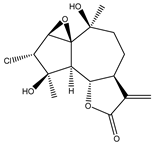 |
| 12 | Aguerin B | 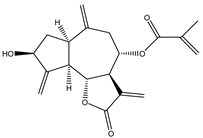 |
| 13 | 15-nor-guaianolide | 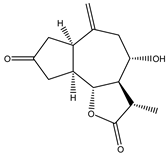 |
| Compound Number | Bioactive Compound Name | Chemical Structures |
|---|---|---|
| 14 | Helenalin | 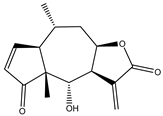 |
| 15 | 2-propenoic acid, 2-methyl-2,3,3a,4,5,8,9,10,11,11a,-decahydro-6,10-bis (hydroxymethyl)-3-methylene-2-oxocyclodeca (b) furan-4-yl ester | 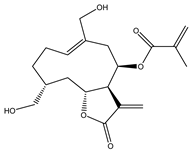 |
| 16 | Tagitinine C |  |
| 17 | 8β-angeloyloxy-9α-hydroxy-14-oxo-acanthospermolide | 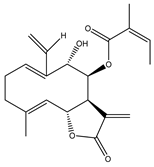 |
| 18 | Uvedalin |  |
| 19 | Enhydrin | 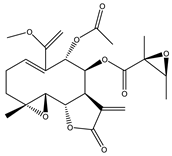 |
| 20 | Polymatin B | 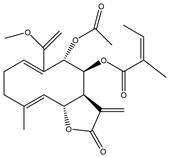 |
| 21 | Sonchifolin |  |
| 22 | Fluctuanin | 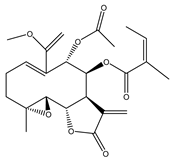 |
| 23 | Ludartin | 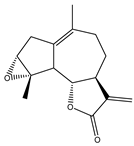 |
| 24 | Sclareolide |  |
| 25 | Gochnatiolide C | 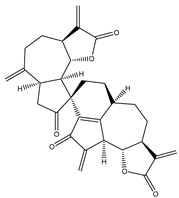 |
| 26 | Parthenin analogue | 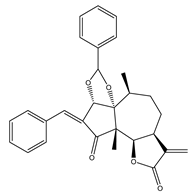 |
| 27 | Tomentosin |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laurella, L.C.; Mirakian, N.T.; Garcia, M.N.; Grasso, D.H.; Sülsen, V.P.; Papademetrio, D.L. Sesquiterpene Lactones as Promising Candidates for Cancer Therapy: Focus on Pancreatic Cancer. Molecules 2022, 27, 3492. https://doi.org/10.3390/molecules27113492
Laurella LC, Mirakian NT, Garcia MN, Grasso DH, Sülsen VP, Papademetrio DL. Sesquiterpene Lactones as Promising Candidates for Cancer Therapy: Focus on Pancreatic Cancer. Molecules. 2022; 27(11):3492. https://doi.org/10.3390/molecules27113492
Chicago/Turabian StyleLaurella, Laura Cecilia, Nadia Talin Mirakian, Maria Noé Garcia, Daniel Héctor Grasso, Valeria Patricia Sülsen, and Daniela Laura Papademetrio. 2022. "Sesquiterpene Lactones as Promising Candidates for Cancer Therapy: Focus on Pancreatic Cancer" Molecules 27, no. 11: 3492. https://doi.org/10.3390/molecules27113492
APA StyleLaurella, L. C., Mirakian, N. T., Garcia, M. N., Grasso, D. H., Sülsen, V. P., & Papademetrio, D. L. (2022). Sesquiterpene Lactones as Promising Candidates for Cancer Therapy: Focus on Pancreatic Cancer. Molecules, 27(11), 3492. https://doi.org/10.3390/molecules27113492