
Influence of Genistein on Hepatic Lipid Metabolism in an In Vitro Model of Hepatic Steatosis

 ,
, 

Abstract
1. Introduction
2. Results
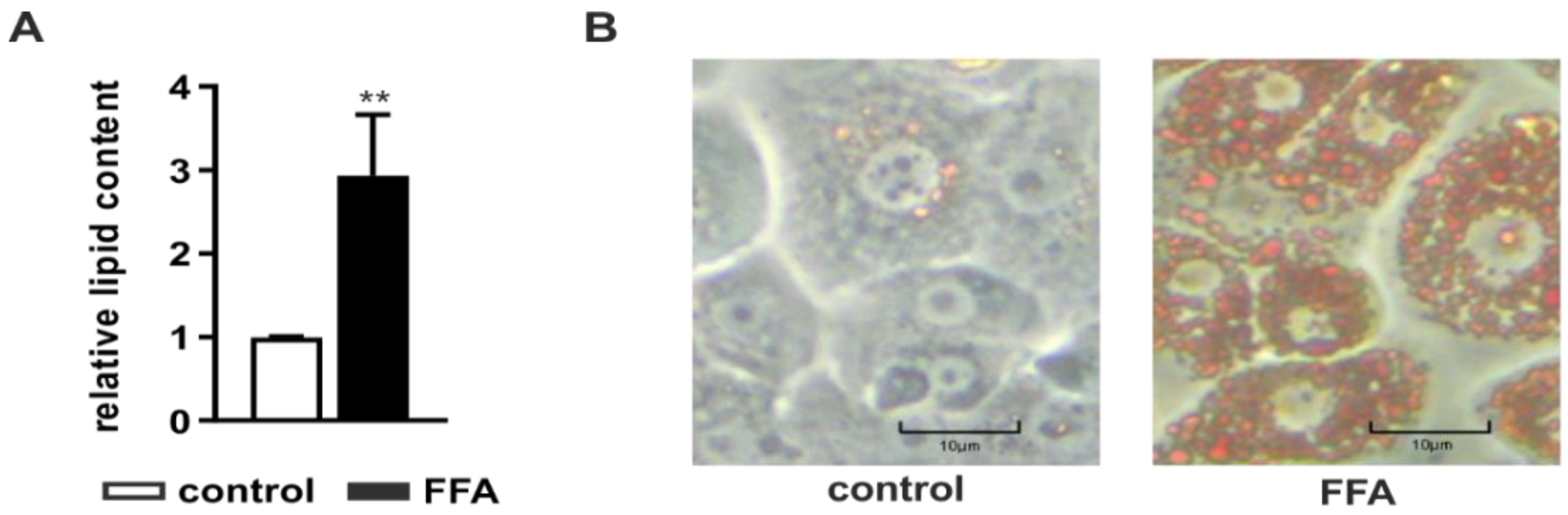
2.1. Incubation of PHHs with Free Fatty Acids Leads to Intracellular Lipid Accumulation
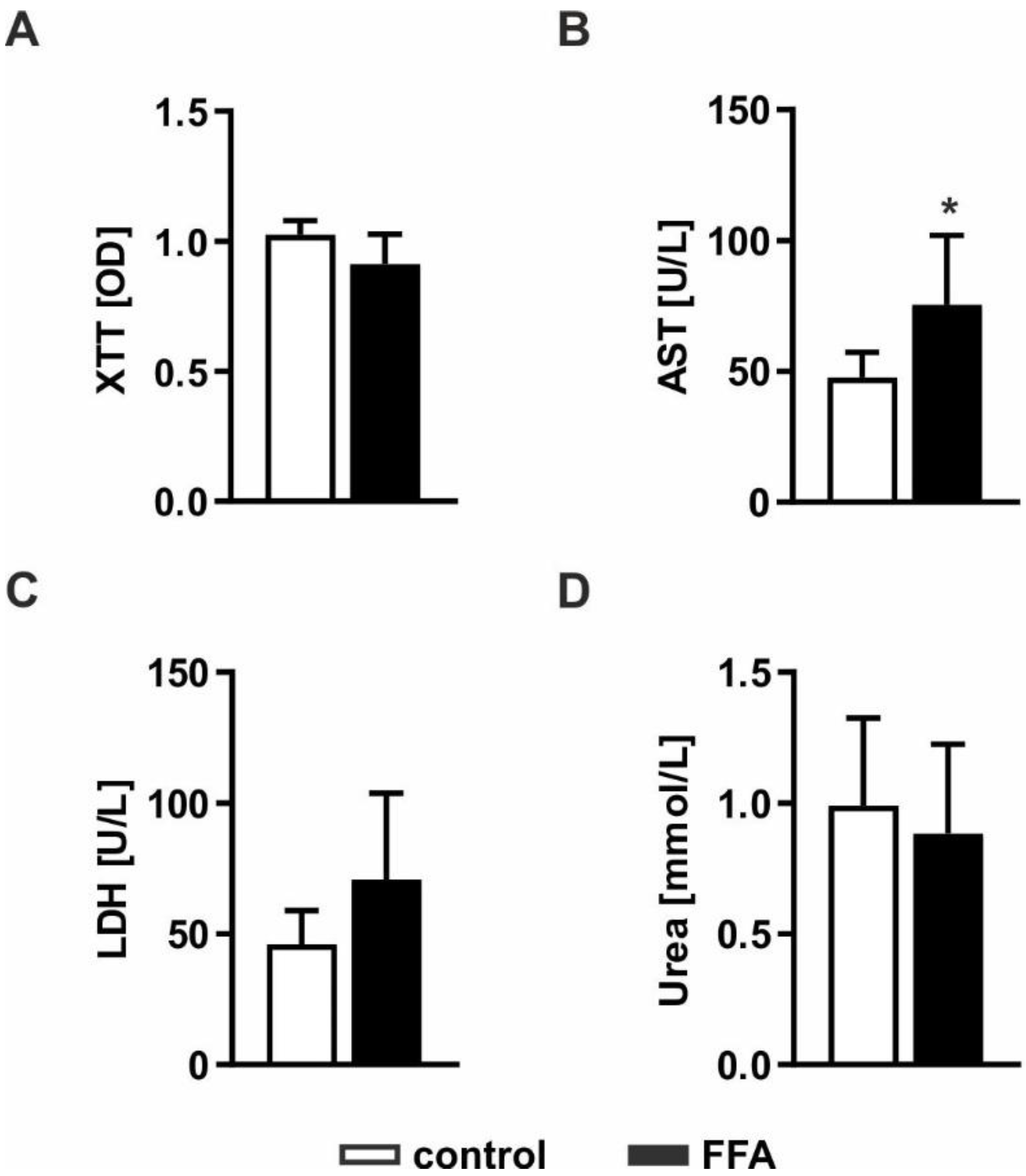
2.2. The Hepatocellular Steatosis Model Shows Only Mild Lipotoxic Effects
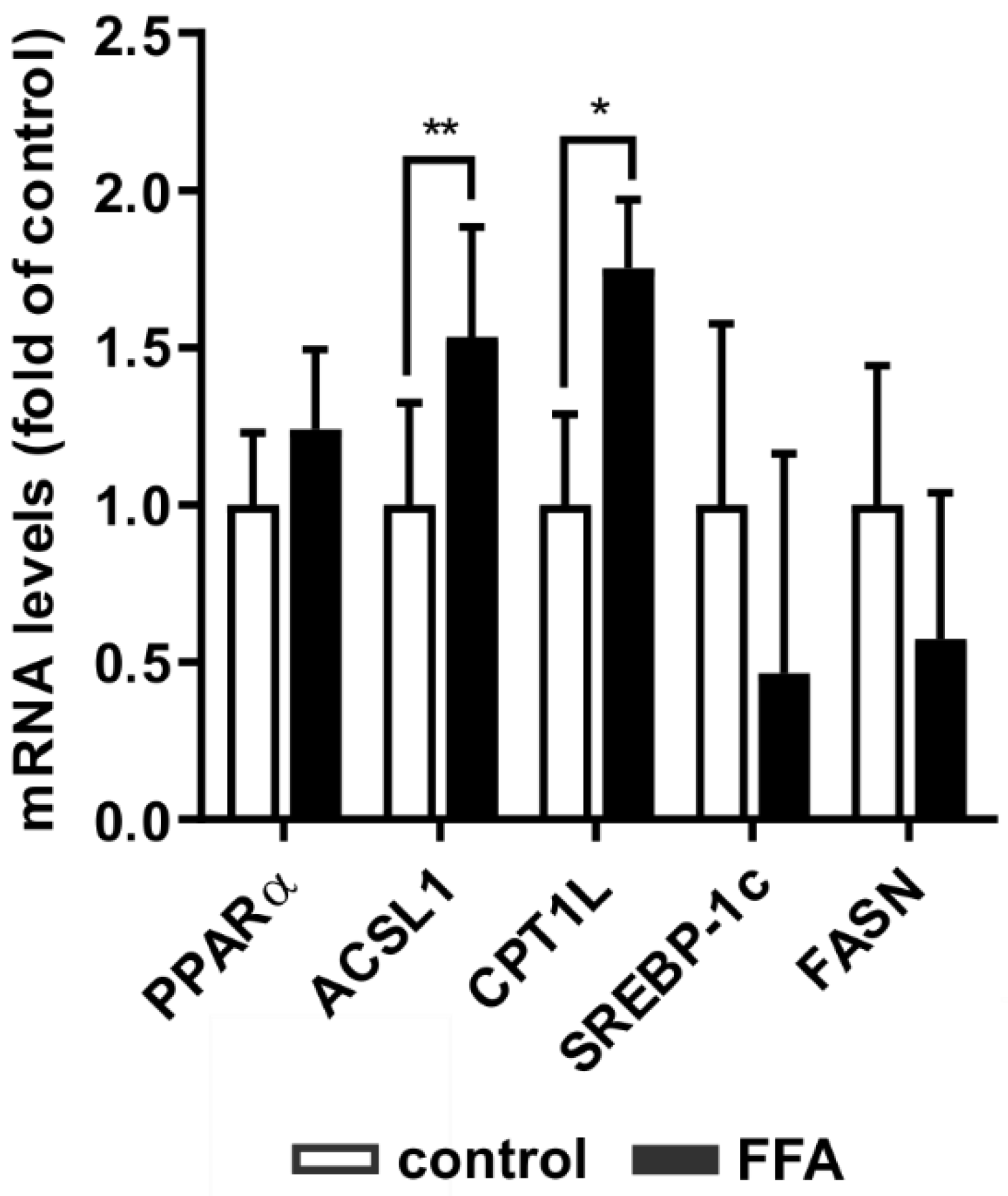
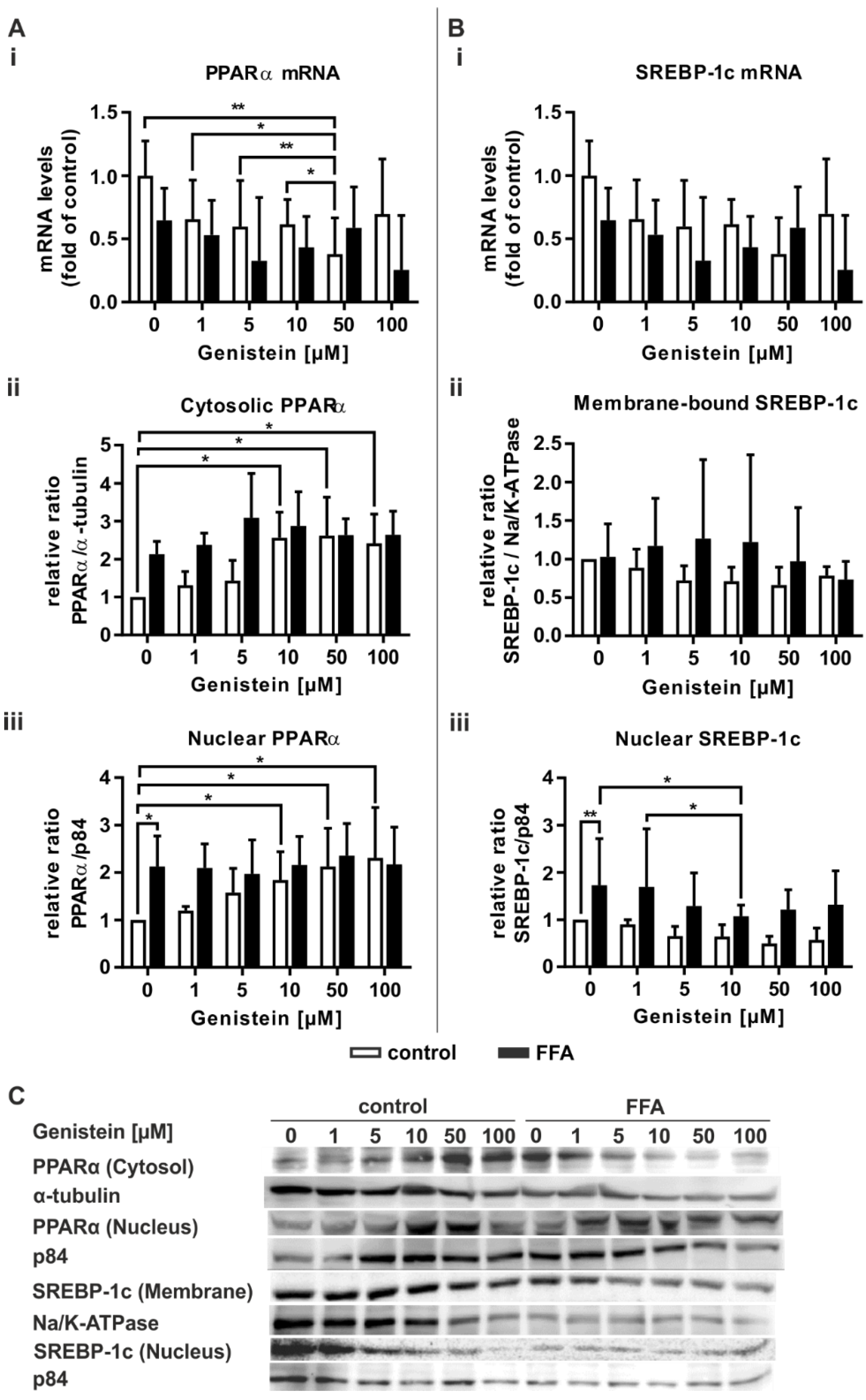
2.3. Transcription of PPARα Downstream Signaling Targets Is Increased in Steatosis
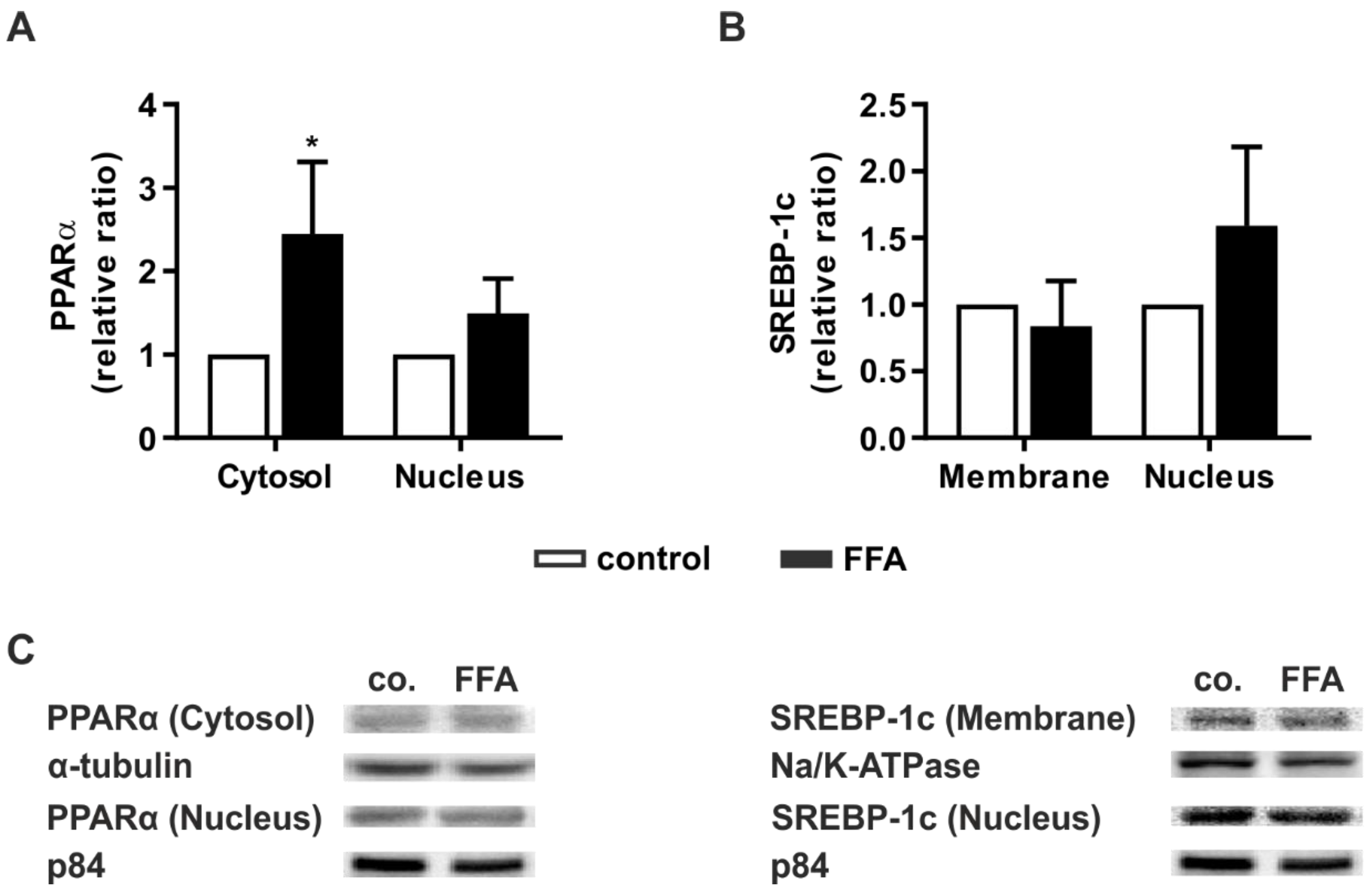
2.4. Steatotic Treatment Increases the Level of Cytosolic PPARα
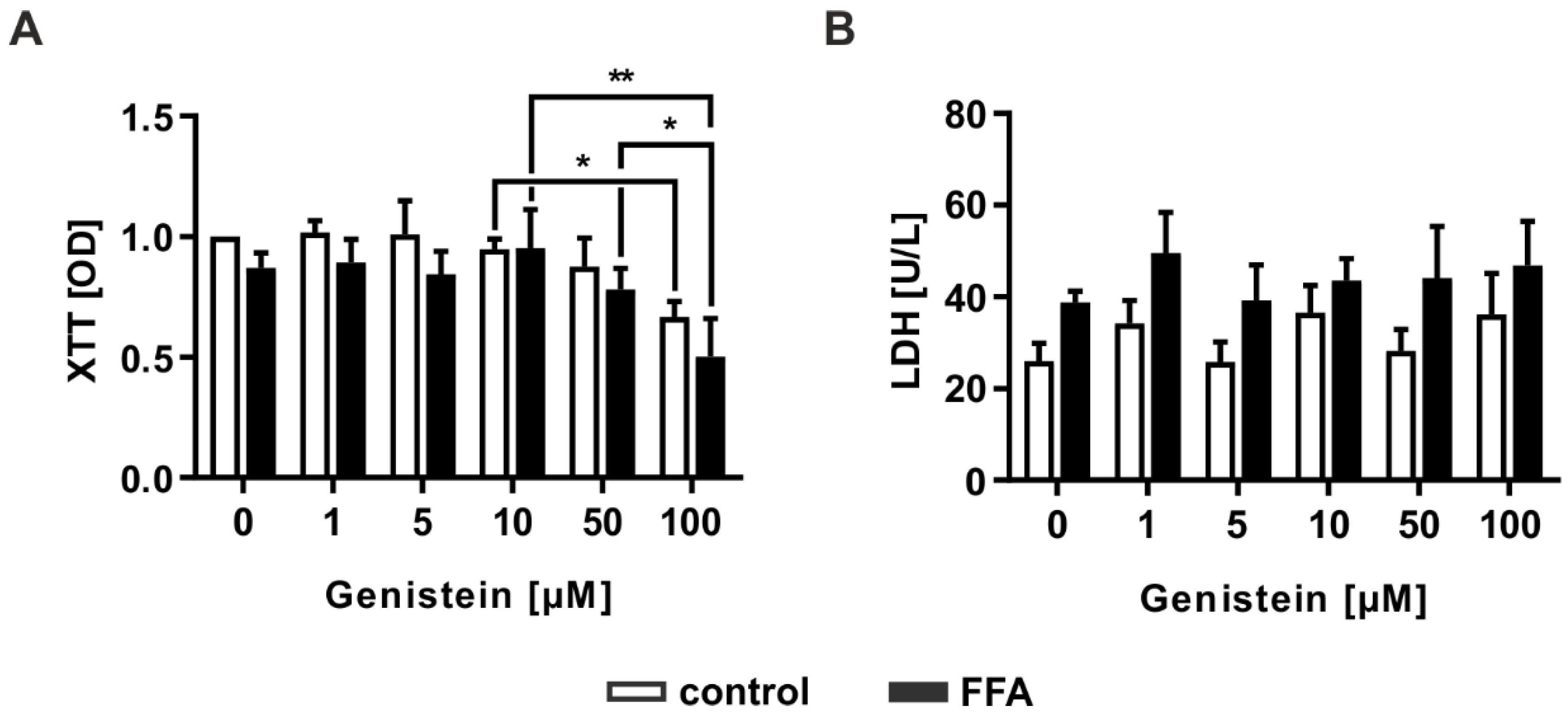
2.5. Genistein Administration Causes Hepatotoxicity Only in High Doses
2.6. Genistein Induces PPARα Activation Only in Control PHHs
3. Discussion
3.1. Induction of Steatosis Is Accompanied by Mild Lipotoxic Effects
3.2. FFA-Treated PHHs Exhibit Changes in Lipid Catabolism and Anabolism Pathways Analogous to the First Stages of NAFLD
3.3. Genistein Shows Hepatotoxic Effects at High Concentrations
3.4. Genistein Acts as a PPARα Agonist Only in Nonsteatotic PHHs
3.5. In Steatotic PHHs, Genistein Leads to the Downregulation of Activated SREBP-1c
3.6. In Steatotic Hepatocytes, Genistein Suppresses De Novo Lipogenesis, While Genistein-Mediated Induction of Hepatic Lipid Degradation Is Abrogated
3.7. Limitations
4. Materials and Methods
4.1. Isolation of Primary Human Hepatocytes
4.2. Cell Culture, In Vitro Induction of Steatosis and Genistein Treatment
4.3. Evaluation of Steatosis by Oil Red O and Sulforhodamine B (SRB) Staining
4.4. XTT Assay
4.5. LDH, AST and Urea Assays
4.6. RT-qPCR Analyses
4.7. Western Blot Analyses
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA carboxylase |
| ACSL1 | long-chain acyl-CoA synthetase 1 |
| acyl-CoA | acyl-coenzyme A |
| ACTB | β-actin gene |
| ANOVA | analysis of variance |
| AST | aspartate transaminase |
| BCA | bicinchoninic acid assay |
| cDNA | complementary deoxyribonucleic acid |
| CPT1A | carnitine palmitoyltransferase 1A |
| CPT1L | carnitine palmitoyltransferase 1L |
| dH2O | distilled water |
| DEPC | diethylpyrocarbonate |
| DMSO | dimethyl sulfoxide |
| EGTA | ethylene glycol tetraacetic acid |
| ER | endoplasmatic reticulum |
| FASN | fatty acid synthase |
| FBS | fetal bovine serum |
| FFA | free fatty acid |
| FGF21 | fibroblast growth factor 21 |
| LDH | lactate dehydrogenase |
| NAFLD | nonalcoholic fatty liver disease |
| miRNA | micro ribonucleic acid |
| mRNA | messenger ribonucleic acid |
| Na+/K+-ATPase | sodium–potassium adenosine triphosphatase |
| NASH | nonalcoholic steatohepatitis |
| PBS | phosphate buffered saline |
| PHH | primary human hepatocyte |
| PPARa | peroxisome proliferator activated receptor alpha |
| qPCR | quantitative polymerase chain reaction |
| RT | room temperature |
| RT-qPCR | reverse transcription-quantitative polymerase chain reaction |
| SD | standard deviation |
| SDS | sodium dodecyl sulfate |
| SRB | sulforhodamine B |
| SREBF1 | sterol regulatory element binding transcription factor 1 |
| SREBP-1c | sterol regulatory element-binding protein 1c |
| TBS | tris-buffered saline |
| Tris | tris(hydroxymethyl)aminomethane |
| UNG | uracil-N-glycosylase |
| XTT | 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilid |
References
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Barritt, A.S.; Calmus, Y.; Scatton, O.; Runge, T.; Lebray, P.; Poynard, T.; Ratziu, V.; Conti, F. NAFLD and liver transplantation: Current burden and expected challenges. J. Hepatol. 2016, 65. [Google Scholar] [CrossRef]
- Heimbach, J. Debate: A bridge too far--liver transplantation for nonalcoholic steatohepatitis will overwhelm the organ supply. Liver Transpl. 2014, 20 (Suppl. S2), S32–S37. [Google Scholar] [CrossRef] [PubMed]
- Wallstab, C.; Eleftheriadou, D.; Schulz, T.; Damm, G.; Seehofer, D.; Borlak, J.; Holzhütter, H.-G.; Berndt, N. A unifying mathematical model of lipid droplet metabolism reveals key molecular players in the development of hepatic steatosis. FEBS J. 2017, 284, 3245–3261. [Google Scholar] [CrossRef] [PubMed]
- Sanders, F.W.B.; Acharjee, A.; Walker, C.; Marney, L.; Roberts, L.D.; Imamura, F.; Jenkins, B.; Case, J.; Ray, S.; Virtue, S.; et al. Hepatic steatosis risk is partly driven by increased de novo lipogenesis following carbohydrate consumption. Genome Biol. 2018, 19, 79. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Buyl, K.; Rogiers, V.; de Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Human-based systems: Mechanistic NASH modelling just around the corner? Pharmacol. Res. 2018, 134, 257–267. [Google Scholar] [CrossRef]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Yang, X.-F.; Liu, H.-L.; Fu, N.; Ouyang, Y.; Qing, K. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: An update. World J. Gastroenterol. 2015, 21, 3492–3498. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Hooiveld, G.; Müller, M.; Kersten, S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS ONE 2009, 4, e6796. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G. Hepatic fatty acid trafficking: Multiple forks in the road. Adv. Nutr. 2013, 4, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, B.D.; Bobard, A.; Hainault, I.; Ferré, P.; Bossard, P.; Foufelle, F. Distinct roles of insulin and liver X receptor in the induction and cleavage of sterol regulatory element-binding protein-1c. Proc. Natl. Acad. Sci. USA 2005, 102, 791–796. [Google Scholar] [CrossRef]
- Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Hasty, A.H.; Osuga, J.; Tamura, Y.; Shionoiri, F.; Iizuka, Y.; Ohashi, K.; Harada, K.; et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J. Biol. Chem. 1999, 274, 35832–35839. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, N.; Kato, M.; Shundo, Y.; Tajiri, H.; Tanaka, M.; Yamashita, N.; Kohjima, M.; Kotoh, K.; Nakamuta, M.; Takayanagi, R.; et al. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol. Res. 2008, 38, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef]
- Yanagitani, A.; Yamada, S.; Yasui, S.; Shimomura, T.; Murai, R.; Murawaki, Y.; Hashiguchi, K.; Kanbe, T.; Saeki, T.; Ichiba, M.; et al. Retinoic acid receptor alpha dominant negative form causes steatohepatitis and liver tumors in transgenic mice. Hepatology 2004, 40, 366–375. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Stienstra, R.; Mandard, S.; Patsouris, D.; Maass, C.; Kersten, S.; Müller, M. Peroxisome proliferator-activated receptor alpha protects against obesity-induced hepatic inflammation. Endocrinology 2007, 148, 2753–2763. [Google Scholar] [CrossRef] [PubMed]
- Gervois, P.; Kleemann, R.; Pilon, A.; Percevault, F.; Koenig, W.; Staels, B.; Kooistra, T. Global suppression of IL-6-induced acute phase response gene expression after chronic in vivo treatment with the peroxisome proliferator-activated receptor-alpha activator fenofibrate. J. Biol. Chem. 2004, 279, 16154–16160. [Google Scholar] [CrossRef] [PubMed]
- Tomic, D.; Kemp, W.W.; Roberts, S.K. Nonalcoholic fatty liver disease: Current concepts, epidemiology and management strategies. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Messina, M. Soy foods, isoflavones, and the health of postmenopausal women. Am. J. Clin. Nutr. 2014, 100 (Suppl. S1), 423S–430S. [Google Scholar] [CrossRef]
- Merritt, J.C. Metabolic syndrome: Soybean foods and serum lipids. J. Natl. Med. Assoc. 2004, 96, 1032–1041. [Google Scholar]
- Akhlaghi, M. Non-alcoholic Fatty Liver Disease: Beneficial Effects of Flavonoids. Phytother. Res. 2016, 30, 1559–1571. [Google Scholar] [CrossRef]
- Amanat, S.; Eftekhari, M.H.; Fararouei, M.; Bagheri Lankarani, K.; Massoumi, S.J. Genistein supplementation improves insulin resistance and inflammatory state in non-alcoholic fatty liver patients: A randomized, controlled trial. Clin. Nutr. 2018, 37, 1210–1215. [Google Scholar] [CrossRef]
- Zhong, H.; Liu, H.; Jiang, Z. Genistein Ameliorates Fat Accumulation through AMPK Activation in Fatty Acid-Induced BRL Cells. J. Food Sci. 2017, 82, 2719–2725. [Google Scholar] [CrossRef]
- Yalniz, M.; Bahcecioglu, I.H.; Kuzu, N.; Poyrazoglu, O.K.; Bulmus, O.; Celebi, S.; Ustundag, B.; Ozercan, I.H.; Sahin, K. Preventive role of genistein in an experimental non-alcoholic steatohepatitis model. J. Gastroenterol. Hepatol. 2007, 22, 2009–2014. [Google Scholar] [CrossRef]
- Huang, C.; Qiao, X.; Dong, B. Neonatal exposure to genistein ameliorates high-fat diet-induced non-alcoholic steatohepatitis in rats. Br. J. Nutr. 2011, 106, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Farruggio, S.; Cocomazzi, G.; Marotta, P.; Romito, R.; Surico, D.; Calamita, G.; Bellan, M.; Pirisi, M.; Grossini, E. Genistein and 17β-Estradiol Protect Hepatocytes from Fatty Degeneration by Mechanisms Involving Mitochondria, Inflammasome and Kinases Activation. Cell. Physiol. Biochem. 2020, 54, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Fujiki, Y. Ligand-dependent nucleo-cytoplasmic shuttling of peroxisome proliferator-activated receptors, PPARα and PPARγ. Genes Cells 2012, 17, 576–596. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Sato, R. New insights into the activation of sterol regulatory element-binding proteins by proteolytic processing. Biomol. Concepts 2013, 4, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.P.; Barnes, S. Isoflavones and PPAR Signaling: A Critical Target in Cardiovascular, Metastatic, and Metabolic Disease. PPAR Res. 2010, 2010, 153252. [Google Scholar] [CrossRef]
- Dufour, J.-F.; Caussy, C.; Loomba, R. Combination therapy for non-alcoholic steatohepatitis: Rationale, opportunities and challenges. Gut 2020, 69, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Hedrington, M.S.; Davis, S.N. Peroxisome proliferator-activated receptor alpha-mediated drug toxicity in the liver. Expert Opin. Drug Metab. Toxicol. 2018, 14, 671–677. [Google Scholar] [CrossRef]
- Zhang, S.; Kumari, S.; Gu, Y.; Wu, X.; Li, X.; Meng, G.; Zhang, Q.; Liu, L.; Wu, H.; Wang, Y.; et al. Soy Food Intake Is Inversely Associated with Newly Diagnosed Nonalcoholic Fatty Liver Disease in the TCLSIH Cohort Study. J. Nutr. 2020. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Martínez-Romero, A.; Jiménez, N.; Castell, J.V.; O’Connor, J.-E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Rennert, C.; Heil, T.; Schicht, G.; Stilkerich, A.; Seidemann, L.; Kegel-Hübner, V.; Seehofer, D.; Damm, G. Prolonged Lipid Accumulation in Cultured Primary Human Hepatocytes Rather Leads to ER Stress than Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 7097. [Google Scholar] [CrossRef]
- Borlak, J.; Chougule, A.; Singh, P.K. How useful are clinical liver function tests in in vitro human hepatotoxicity assays? Toxicol. In Vitro 2014, 28, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [PubMed]
- Stellavato, A.; Pirozzi, A.V.A.; de Novellis, F.; Scognamiglio, I.; Vassallo, V.; Giori, A.M.; de Rosa, M.; Schiraldi, C. In vitro assessment of nutraceutical compounds and novel nutraceutical formulations in a liver-steatosis-based model. Lipids Health Dis. 2018, 17, 24. [Google Scholar] [CrossRef]
- Rogue, A.; Anthérieu, S.; Vluggens, A.; Umbdenstock, T.; Claude, N.; de La Moureyre-Spire, C.; Weaver, R.J.; Guillouzo, A. PPAR agonists reduce steatosis in oleic acid-overloaded HepaRG cells. Toxicol. Appl. Pharmacol. 2014, 276, 73–81. [Google Scholar] [CrossRef]
- Jump, D.B. Dietary polyunsaturated fatty acids and regulation of gene transcription. Curr. Opin. Lipidol. 2002, 13, 155–164. [Google Scholar] [CrossRef]
- Gori, M.; Barbaro, B.; Arciello, M.; Maggio, R.; Viscomi, C.; Longo, A.; Balsano, C. Protective effect of the Y220C mutant p53 against steatosis: Good news? J. Cell. Physiol. 2014, 229, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Ferré, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 2010, 12 (Suppl. S2), 83–92. [Google Scholar] [CrossRef]
- Einer, C.; Hohenester, S.; Wimmer, R.; Wottke, L.; Artmann, R.; Schulz, S.; Gosmann, C.; Simmons, A.; Leitzinger, C.; Eberhagen, C.; et al. Mitochondrial adaptation in steatotic mice. Mitochondrion 2018, 40, 1–12. [Google Scholar] [CrossRef]
- Lee, J.; Park, J.-S.; Roh, Y.S. Molecular insights into the role of mitochondria in non-alcoholic fatty liver disease. Arch. Pharm. Res. 2019, 42, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Pool-Zobel, B.L.; Adlercreutz, H.; Glei, M.; Liegibel, U.M.; Sittlingon, J.; Rowland, I.; Wähälä, K.; Rechkemmer, G. Isoflavonoids and lignans have different potentials to modulate oxidative genetic damage in human colon cells. Carcinogenesis 2000, 21, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Shin, H.-J.; Kim, S.Y.; Kim, J.H.; Lee, Y.S.; Kim, D.-H.; Lee, M.-O. Genistein enhances expression of genes involved in fatty acid catabolism through activation of PPARalpha. Mol. Cell. Endocrinol. 2004, 220, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Lepri, S.R.; Sartori, D.; Semprebon, S.C.; Baranoski, A.; Coatti, G.C.; Mantovani, M.S. Genistein Affects Expression of Cytochrome P450 (CYP450) Genes in Hepatocellular Carcinoma (HEPG2/C3A) Cell Line. Drug Metab. Lett. 2018, 12, 138–144. [Google Scholar] [CrossRef]
- Lu, R.; Zheng, Z.; Yin, Y.; Jiang, Z. Effect of Genistein on Cholesterol Metabolism-Related Genes in HepG2 Cell. J. Food Sci. 2019, 84, 2330–2336. [Google Scholar] [CrossRef]
- Xin, X.; Chen, C.; Hu, Y.-Y.; Feng, Q. Protective effect of genistein on nonalcoholic fatty liver disease (NAFLD). Biomed. Pharmacother. 2019, 117, 109047. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Choi, J.S.; Kim, M.H.; Jung, M.H.; Lee, Y.S.; Song, J. Effects of dietary genistein on hepatic lipid metabolism and mitochondrial function in mice fed high-fat diets. Nutrition 2006, 22, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Asrih, M.; Montessuit, C.; Philippe, J.; Jornayvaz, F.R. Free Fatty Acids Impair FGF21 Action in HepG2 Cells. Cell. Physiol. Biochem. 2015, 37, 1767–1778. [Google Scholar] [CrossRef]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; de Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Lv, G.-c.; Sheng, J.; Yang, Y.-d. Effect of miRNA-10b in regulating cellular steatosis level by targeting PPAR-alpha expression, a novel mechanism for the pathogenesis of NAFLD. J. Gastroenterol. Hepatol. 2010, 25, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Blanquart, C.; Barbier, O.; Fruchart, J.-C.; Staels, B.; Glineur, C. Peroxisome proliferator-activated receptor alpha (PPARalpha) turnover by the ubiquitin-proteasome system controls the ligand-induced expression level of its target genes. J. Biol. Chem. 2002, 277, 37254–37259. [Google Scholar] [CrossRef]
- Liu, H.; Zhong, H.; Yin, Y.; Jiang, Z. Genistein has beneficial effects on hepatic steatosis in high fat-high sucrose diet-treated rats. Biomed. Pharmacother. 2017, 91, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.S.; Lee, H.H.; Cho, S.Y.; Park, H.W.; Lee, S.J.; Lee, T.R. Genistein downregulates SREBP-1 regulated gene expression by inhibiting site-1 protease expression in HepG2 cells. J. Nutr. 2007, 137, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-R.; Park, K.I.; Ma, J.Y. Leonurus japonicus Houtt Attenuates Nonalcoholic Fatty Liver Disease in Free Fatty Acid-Induced HepG2 Cells and Mice Fed a High-Fat Diet. Nutrients 2017, 10, 20. [Google Scholar] [CrossRef]
- Kegel, V.; Deharde, D.; Pfeiffer, E.; Zeilinger, K.; Seehofer, D.; Damm, G. Protocol for Isolation of Primary Human Hepatocytes and Corresponding Major Populations of Non-parenchymal Liver Cells. J. Vis. Exp. 2016, e53069. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, E.; Kegel, V.; Zeilinger, K.; Hengstler, J.G.; Nüssler, A.K.; Seehofer, D.; Damm, G. Featured Article: Isolation, characterization, and cultivation of human hepatocytes and non-parenchymal liver cells. Exp. Biol. Med. 2015, 240, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Rajan, N.; Habermehl, J.; Coté, M.-F.; Doillon, C.J.; Mantovani, D. Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nat. Protoc. 2006, 1, 2753–2758. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Temperature ( °C) | Time (min) |
|---|---|---|
| Annealing | 65 | 5 |
| Reverse transcription | 37 | 60 |
| Inactivation | 70 | 5 |
| Gene Symbol | Gene Name | Assay ID |
|---|---|---|
| PPARA | Peroxisome proliferator activated receptor alpha | Hs00947539_m1 |
| SREBP-1c | Sterol regulatory element binding transcription factor 1 (SREBF1) | Hs01088691_m1 |
| CPT1L | Carnitine palmitoyltransferase 1A (CPT1A) | Hs00912671_m1 |
| ACSL1 | Acyl-CoA synthetase long-chain family member 1 | Hs00242530_m1 |
| FASN | Fatty acid synthase | Hs01005622_m1 |
| ACTB | Actin beta | Hs99999903_m1 |
| Step | Temperature ( °C) | Time (min:s) | Cycles |
|---|---|---|---|
| UNG 1 incubation | 50 | 2:00 | 1 |
| Polymerase activation | 95 | 10:00 | 1 |
| Denaturation | 95 | 0:15 | 40 |
| Annealing/extension | 60 | 1:00 |
| Antibody (Manufacturer) | Dilution | Incubation Time |
|---|---|---|
| Mouse PPARα (Dianova, Hamburg, Germany) | 1:1000 | Overnight |
| Mouse SREBP-1c (Biozol, Eching, Germany) | 1:200 | 2 h |
| Mouse α-tubulin (Sigma-Aldrich, Steinheim, Germany) | 1:2000 | 1 h |
| Mouse p84 (Abcam, Cambridge, UK) | 1:2000 | 1 h |
| Rabbit Na+/K+-ATPase (Cell Signaling, Danvers, MA, USA) | 1:1000 | 1 h |
| Sheep anti-mouse (Amersham, Freiburg, Germany) | 1:4000 | 1 h |
| Donkey anti-rabbit | 1:4000 | 1 h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seidemann, L.; Krüger, A.; Kegel-Hübner, V.; Seehofer, D.; Damm, G. Influence of Genistein on Hepatic Lipid Metabolism in an In Vitro Model of Hepatic Steatosis. Molecules 2021, 26, 1156. https://doi.org/10.3390/molecules26041156
Seidemann L, Krüger A, Kegel-Hübner V, Seehofer D, Damm G. Influence of Genistein on Hepatic Lipid Metabolism in an In Vitro Model of Hepatic Steatosis. Molecules. 2021; 26(4):1156. https://doi.org/10.3390/molecules26041156
Chicago/Turabian StyleSeidemann, Lena, Anne Krüger, Victoria Kegel-Hübner, Daniel Seehofer, and Georg Damm. 2021. "Influence of Genistein on Hepatic Lipid Metabolism in an In Vitro Model of Hepatic Steatosis" Molecules 26, no. 4: 1156. https://doi.org/10.3390/molecules26041156
APA StyleSeidemann, L., Krüger, A., Kegel-Hübner, V., Seehofer, D., & Damm, G. (2021). Influence of Genistein on Hepatic Lipid Metabolism in an In Vitro Model of Hepatic Steatosis. Molecules, 26(4), 1156. https://doi.org/10.3390/molecules26041156