3.2. General Procedure for Preparation of Iodobenzamide 6
Amine was added dropwise at 0 °C to a stirred mixture of iodobenzoyl chloride and triethylamine in dichloromethane. The reaction was stirred for 1 h at 0 °C. After completion of the reaction, water (10–20 mL) was added, and the mixture was extracted with dichloromethane. The organic phase was dried over anhydrous Na2SO4 and concentrated under reduced pressure. Purification by recrystallization (CH2Cl2-hexane) afforded the analytically pure iodobenzamide 6.
2-Iodo-N-isopropylbenzamide 6a [
31]. The reaction with 2-iodobenzoyl chloride (3171 mg, 11.9 mmol), triethylamine (2649 mg, 26.2 mmol), and isopropylamine (774 mg, 13.1 mmol) in dichloromethane (15 mL) according to the general procedure afforded 3325 mg (97%) of product
6a, isolated as a white solid: mp 136.4–137.7 °C (lit. [
31], mp 135–136 °C);
1H NMR (500 MHz, CDCl
3): δ 7.84 (d,
J = 8.0 Hz, 1H), 7.41–7.34 (m, 2H), 7.11–7.05 (m, 1H), 5.57 (brs, 1H), 4.38–4.22 (m, 1H), and 1.29 (d,
J = 7.0 Hz, 1H).
N-Ethyl-2-iodobenzamide 6b [
32]. The reaction with 2-iodobenzoyl chloride (1062 mg, 3.99 mmol), triethylamine (887 mg, 8.77 mmol), and ethylamine (198 mg, 4.38 mmol) in dichloromethane (15 mL) according to the general procedure afforded 1032 mg (94%) of product
6b, isolated as a white solid: mp 114.3–115.5 °C (lit. [
32], 114–116 °C);
1H NMR (500 MHz, CDCl
3): δ 7.85 (d,
J = 8.0 Hz, 1H), 7.41–7.33 (m, 2H), 7.11–7.05 (m, 1H), 5.78 (brs, 1H), 3.62–3.40 (m, 2H), and 1.27 (t,
J = 7.3 Hz, 3H).
N-(tert-Butyl)-2-iodobenzamide 6c [
33]. The reaction with 2-iodobenzoyl chloride (1063 mg, 3.99 mmol), triethylamine (888 mg, 8.78 mmol), and
tert-butylamine (321 mg, 4.39 mmol) in dichloromethane (15 mL) according to the general procedure afforded 1140 mg (94%) of product
6c, isolated as a white solid: mp 122.2–124.1 °C (Lit. [
33], 120–122 °C);
1H NMR (500 MHz, CDCl
3): δ 7.83 (d,
J = 8.0 Hz, 1H), 7.41–7.32 (m, 2H), 7.09–7.03 (m, 1H), 5.55 (brs, 1H), and 1.49 (s, 9H).
N-Cyclohexyl-2-iodobenzamide 6d [
33]. The reaction with 2-iodobenzoyl chloride (1025 mg, 3.85 mmol), triethylamine (857 mg, 8.46 mmol), and cyclohexylamine (420 mg, 4.23 mmol) in dichloromethane (15 mL) according to the general procedure afforded 1201 mg (95%) of product
6d, isolated as a white solid: mp 141.2–142.3 °C (lit. [
33], mp 141–143 °C);
1H NMR (500 MHz, CDCl
3): δ 7.85 (d,
J = 8.0 Hz, 1H), 7.41–7.34 (m, 2H), 7.11–7.05 (m, 1H), 5.62 (brs, 1H), 4.07–3.94 (m, 1H), 2.12–2.04 (m, 2H), 1.80–1.72 (m, 2H), 1.69–1.61 (m, 1H), 1.49–1.38 (m, 2H), and 1.32–1.16 (m, 3H).
2-Iodo-N-phenylbenzamide 6e [
33]. The reaction with 2-iodobenzoyl chloride (1089 mg, 4.87 mmol), triethylamine (908 mg, 9.00 mmol), and aniline (419 mg, 4.50 mmol) in dichloromethane (15 mL) according to the general procedure afforded 1085 mg (82%) of product
6e, isolated as a white solid: mp 144.2–145.0 °C (Lit. [
33], 144–146 °C);
1H NMR (500 MHz, CDCl
3): δ 7.92 (d,
J = 8.0 Hz, 1H), 7.64 (d,
J = 8.5 Hz, 2H), 7.54 (d,
J = 8.0 Hz, 1H), 7.47–7.42 (m, 2H), 7.42–7.35 (m, 2H), and 7.21–7.13 (m, 2H).
2-Iodo-N-isopropyl-5-methylbenzamide 6f [
34]. The reaction with 2-iodo-5-methylbenzoyl chloride (2140 mg, 7.63 mmol), triethylamine (1698 mg, 16.8 mmol), and isopropylamine (496 mg, 8.39 mmol) in dichloromethane (10 mL) according to the general procedure afforded 2220 mg (96%) of product
5f, isolated as a white solid: mp 147.8–148.7 °C (Lit. [
34], 147–149 °C);
1H NMR (500 MHz, CDCl
3): δ 7.68 (d,
J = 8.0 Hz, 1H), 7.20 (s, 1H), 6.90 (d,
J = 8.0 Hz, 1H), 5.62 (brs, 1H), 4.33–4.233 (m, 1H), 2.30 (s, 3H), and 1.28 (d,
J = 7.0 Hz, 6H); HRMS (ESI-positive): calcd for C
11H
15INO ([M + H])
+: 304.0198, found: 304.0196.
2-Iodo-N-isopropyl-4-methylbenzamide 6g. The reaction with 2-iodo-4-methylbenzoyl chloride (1070 mg, 3.81 mmol), triethylamine (849 mg, 8.38 mmol), and isopropylamine (248 mg, 4.19 mmol) in dichloromethane (10 mL) according to the general procedure afforded 1118 mg (97%) of product 5g, isolated as a white solid: mp 117.0–119.0 °C; IR (neat) cm−1: 3305, 3062, 2973, 2927, 1874, 1637, 1598. 1531, 1484, 1464, 1450, 1389, 1369, 1351, 1329, 1289, 1264, 1178, 1127, 1036, 883, 850, 831, 821, 805, 601, and 570; 1H NMR (500 MHz, CDCl3): δ 7.69 (s, 1H), 7.27 (d, J = 8.0 Hz, 1H), 7.15 (d, J = 8.0 Hz, 1H), 5.61 (brs, 1H), 4.33–4.22 (m, 1H), 2.31 (s, 3H), and 1.27 (d, J = 7.0 Hz, 6H); 13C NMR (125 MHz, CDCl3): δ 168.5, 141.4, 140.2, 139.5, 128.9, 128.1, 92.4, 42.2, 22.6, and 20.7; HRMS (ESI-positive): calcd for C11H15INO ([M + H])+: 304.0198, found: 304.0193.
2-Iodo-N-isopropyl-3-methylbenzamide 6h. The reaction with 2-iodo-3-methylbenzoyl chloride (1063 mg, 3.79 mmol), triethylamine (844 mg, 8.34 mmol), and isopropylamine (224 mg, 4.17 mmol) in dichloromethane (10 mL) according to the general procedure afforded 1084 mg (94%) of product 6h, isolated as a white solid: mp 135.6–137.2 °C.; IR (neat) cm−1: 3248, 3069, 2970, 2937, 2875, 1657, 1551, 1459, 1366, 1352, 1331, 1303, 1266, 1204, 1156, 1129, 1012, 919, 858, 805, 780, 738, 718, and 685; 1H NMR (500 MHz, CDCl3): δ 7.33–7.19 (m, 2H), 7.16–7.05 (m, 1H), 5.50 (brs, 1H), 4.41–4.21 (m, 1H), 2.45 (s, 3H), and 1.29 (d, J = 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3): δ 169.9, 144.2, 142.8, 130.2, 128.1, 124.9, 99.3, 42.1, 29.1, and 22.6; HRMS (ESI-positive): calcd for C11H15INO ([M + H])+: 304.0198, found: 304.0190.
5-Bromo-2-iodo-N-isopropylbenzamide 6i. The reaction with 5-bromo-2-iodobenzoyl chloride (1317 mg, 3.81 mmol), triethylamine (848 mg, 8.38 mmol), and isopropylamine (248 mg, 4.19 mmol) in dichloromethane (10 mL) according to the general procedure afforded 1347 mg (96%) of product 6i, isolated as a white solid: mp 187.8–188.5 °C; IR (neat) cm−1: 3255, 3073, 2971, 1642, 1541, 1453, 1083, 1017, 900, 810, and 727; 1H NMR (500 MHz, CDCl3): δ 7.69 (d, J = 8.0 Hz, 1H), 7.52–7.48 (m, 1H), 7.24–7.19 (m, 1H), 5.58 (brs, 1H), 4.36–4.16 (m, 1H), and 1.29 (d, J = 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3): δ 167.0, 144.1, 141.1, 134.0, 131.2, 122.6, 90.4, 42.4, and 22.6; HRMS (APCI-positive): calcd for C10H1279BrINO ([M + H])+: 367.9147, found: 367.9122.
3-Iodo-N-isopropyl-2-naphthamide 6j. The reaction with 3-iodo-2-naphthoyl chloride (158 mg, 0.50 mmol), triethylamine (111 mg, 1.1 mmol), and isopropylamine (32.5 mg, 0.55 mmol) in dichloromethane (5.0 mL) according to the general procedure afforded 105 mg (62%) of product 6j, isolated as a white solid: mp 191.6–193.3 °C; IR (neat) cm−1: 3279, 2970, 1642, 1541, 1124, 954, 908, 895, 880, 805, and 743; 1H NMR (500 MHz, CDCl3): δ 8.38 (s, 1H), 7.86 (s, 1H), 7.84–7.79 (m, 1H), 7.75–7.71 (m, 1H), 7.57–7.50 (m, 2H), 5.68 (brs, 1H), 4.49–4.28 (m, 1H), and 1.33 (d, J = 6.5 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 168.5, 139.3, 139.0, 134.9, 131.9, 128.1, 127.8, 127.4, 127.3, 126.7, 88.8, 42.3, and 22.7; HRMS (ESI-positive): calcd for C14H15INO ([M + H])+: 340.0198, found: 340.0188.
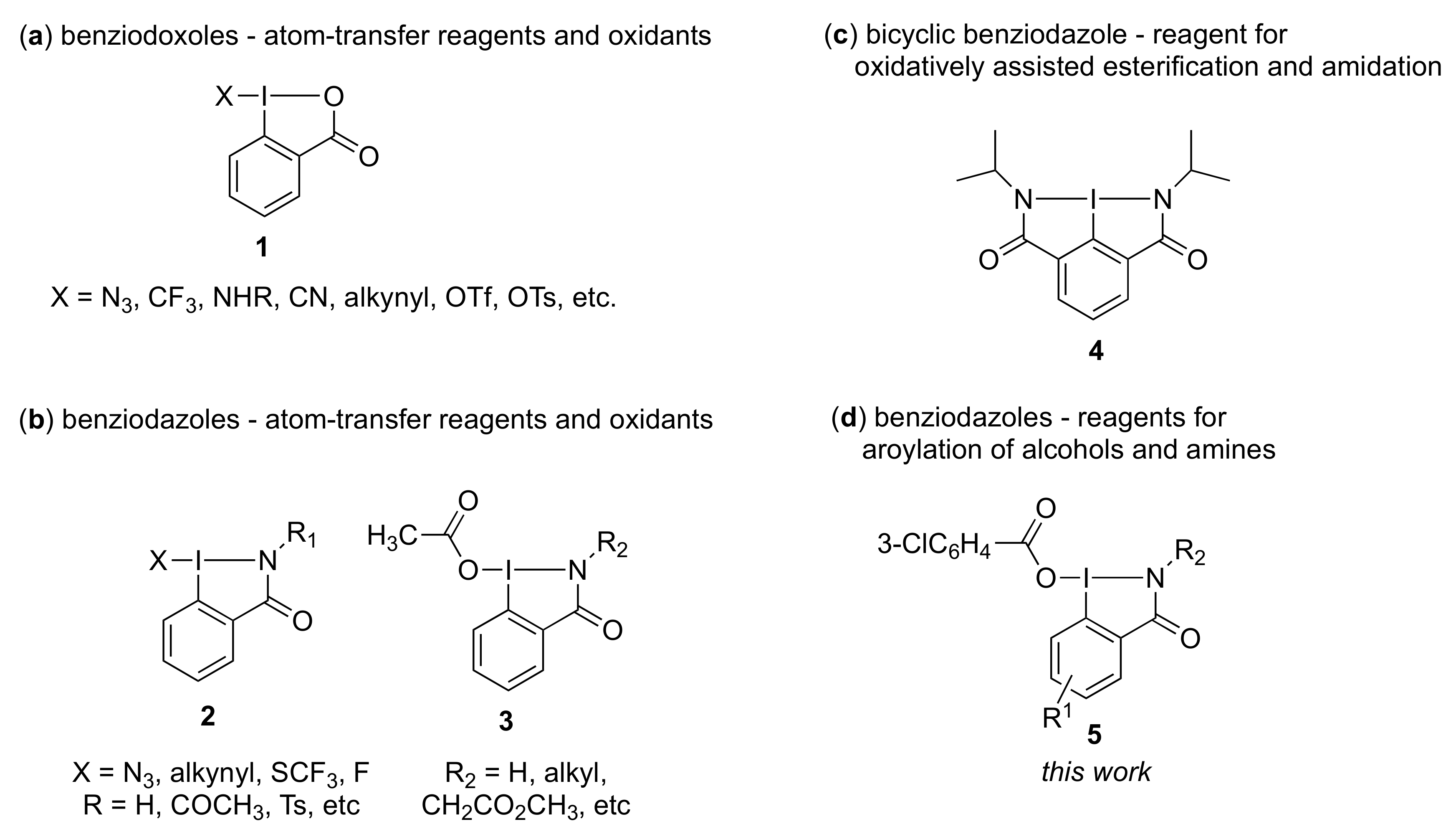
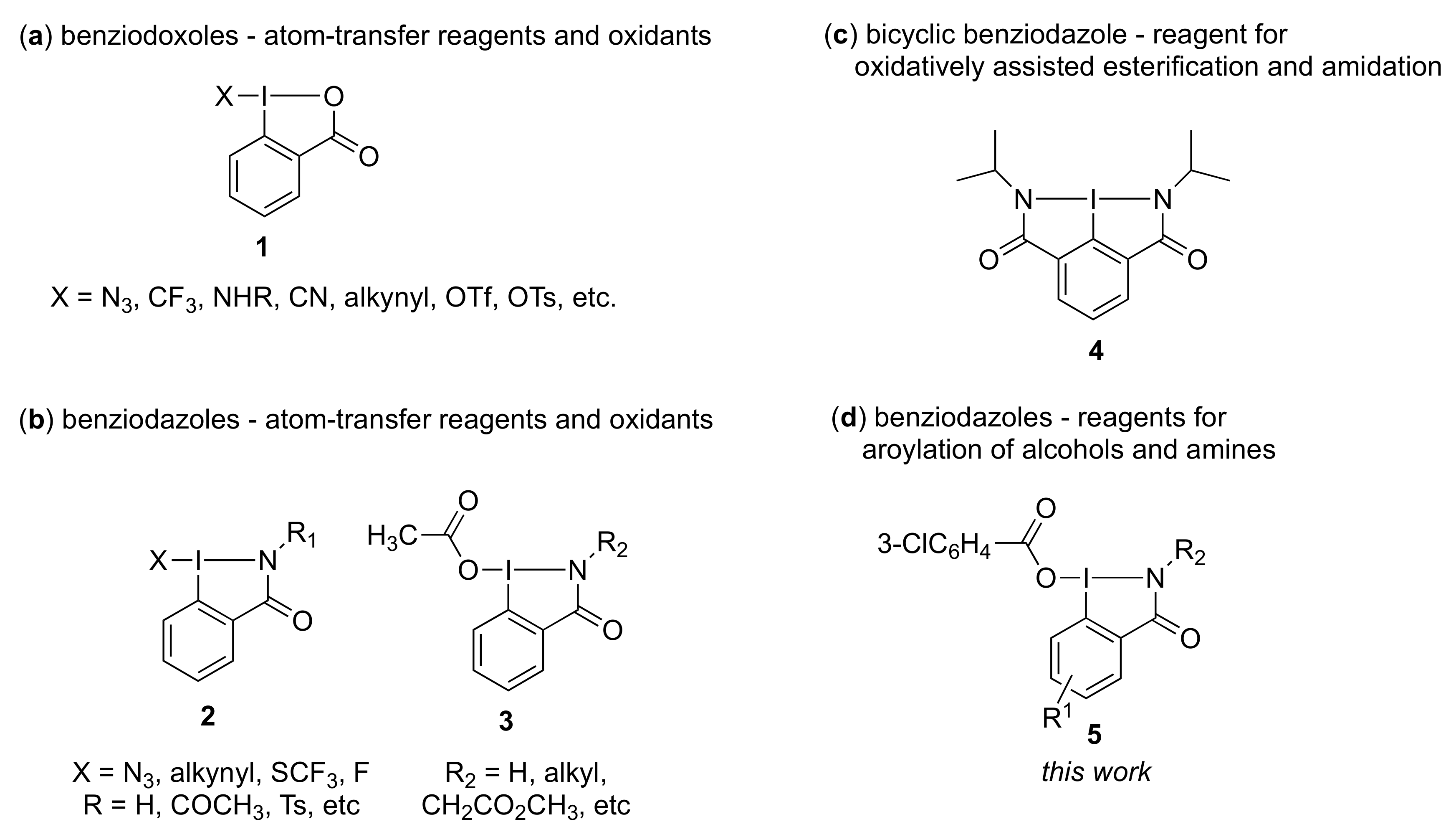
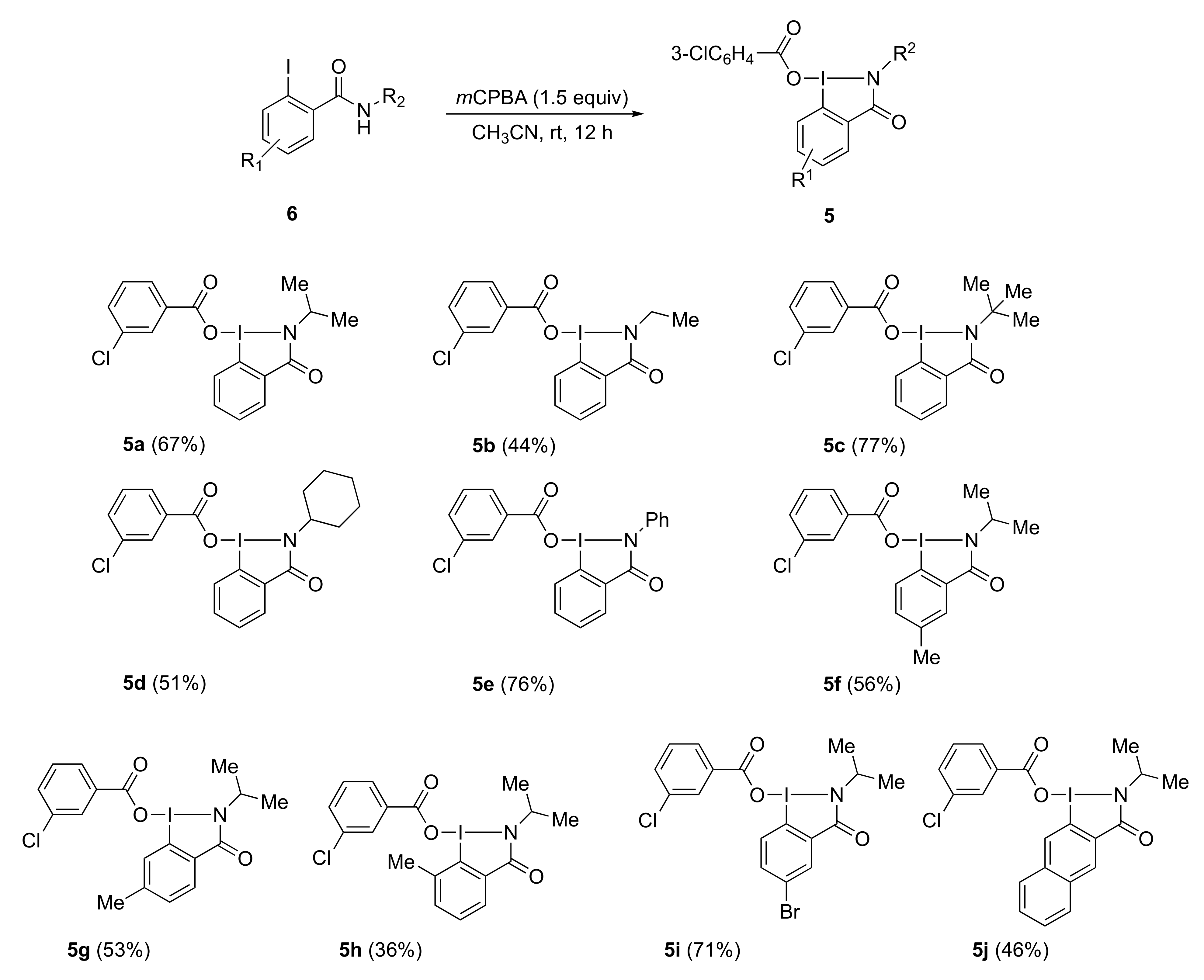
3.3. General Procedure for Preparation of 3-Chlorobenzoyloxybenziodazolone 5
Iodobenzamide 6 was added at 0 °C to a stirred mixture of m-CPBA in acetonitrile. The reaction was stirred for 12 h at room temperature. After completion of the reaction, the solvent was removed under reduced pressure to give solid residue. Then diethyl ether was added to the solid residue to prepare the suspended solution, which was filtered, and dried in a vacuum to give the desired 3-chlorobenzoyloxybenziodazolone 5.
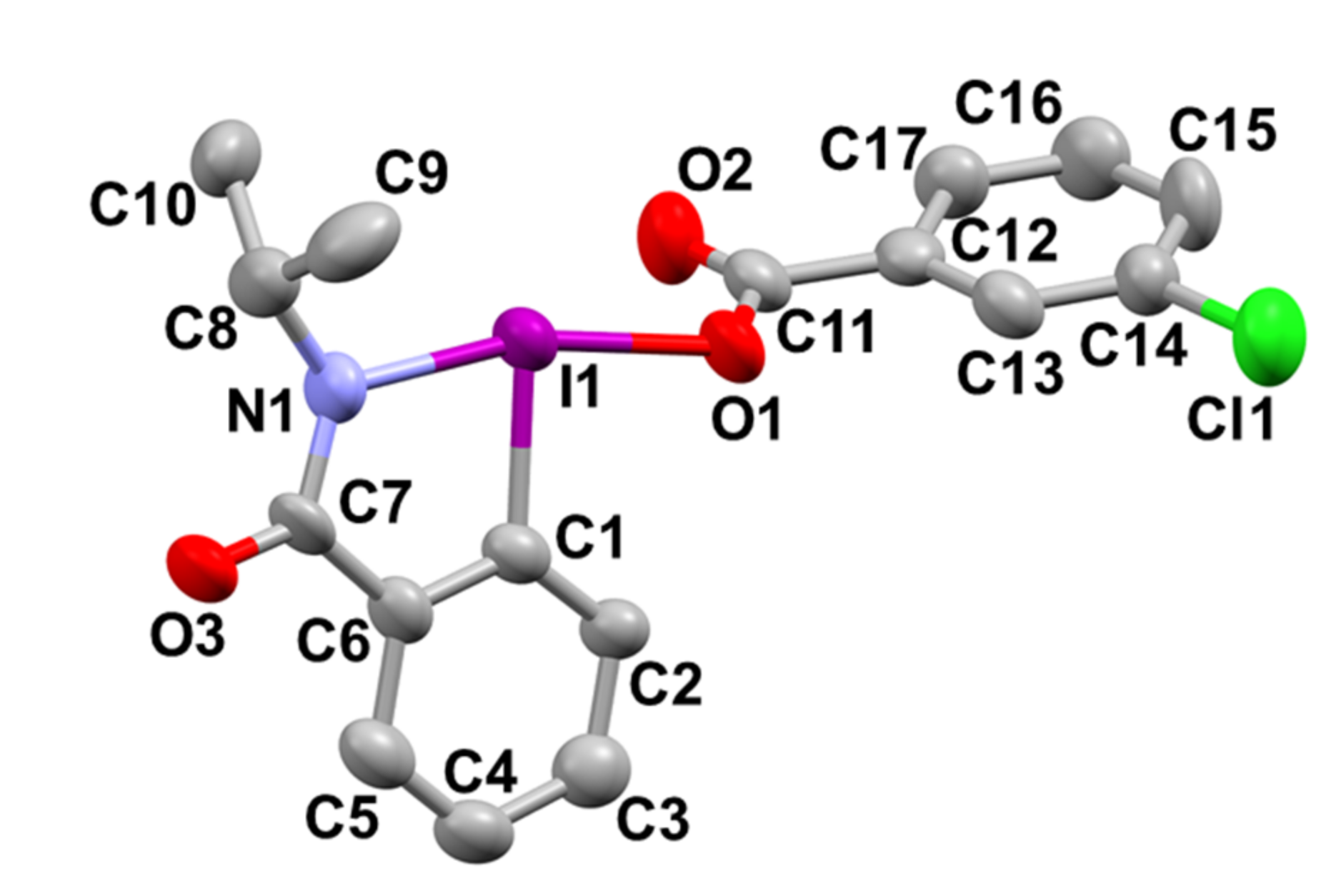
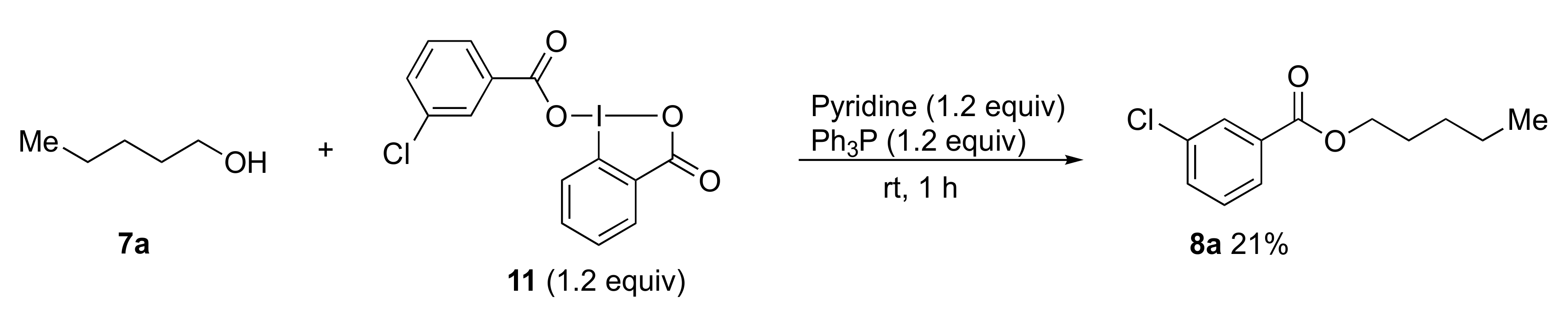
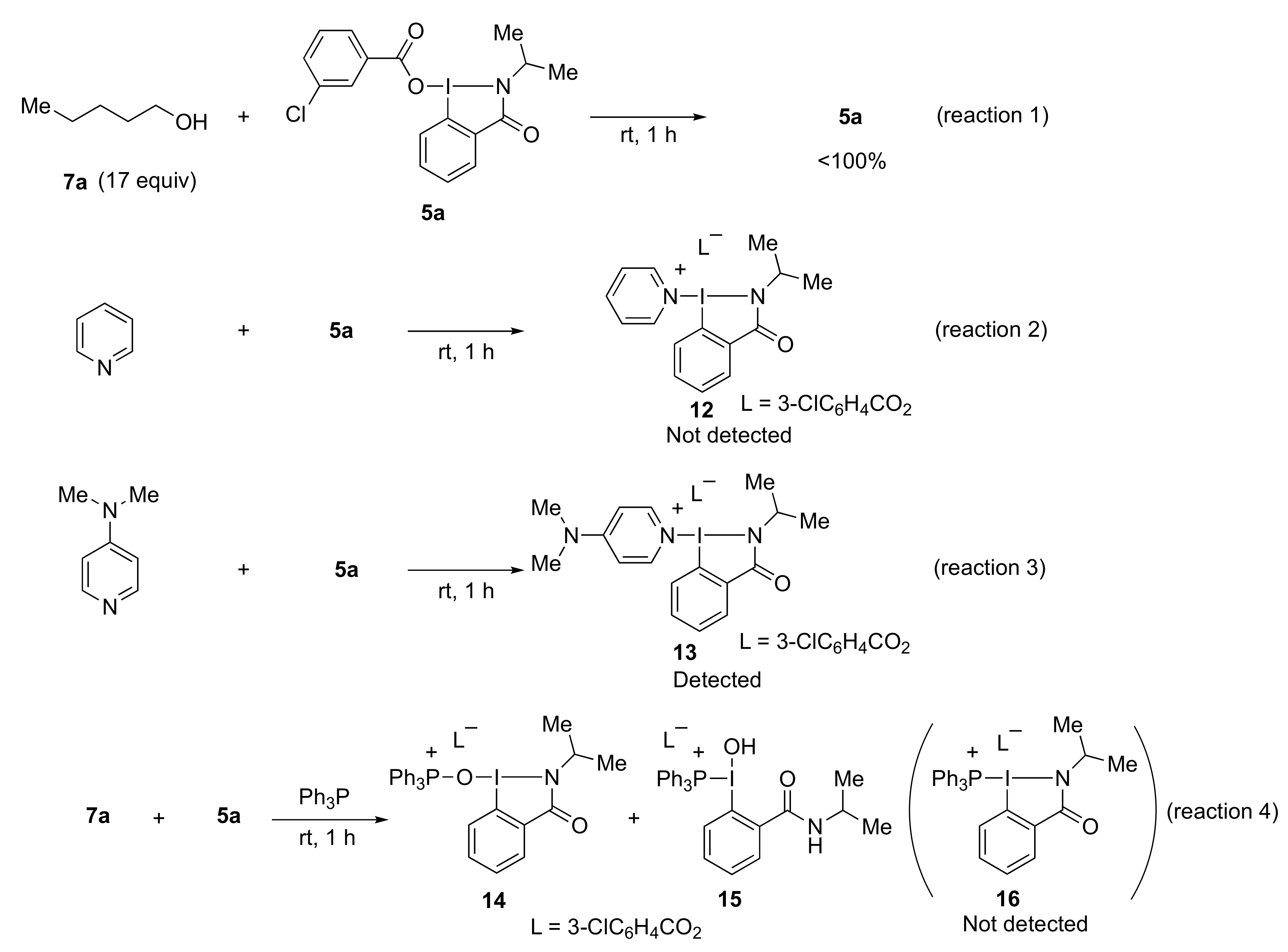
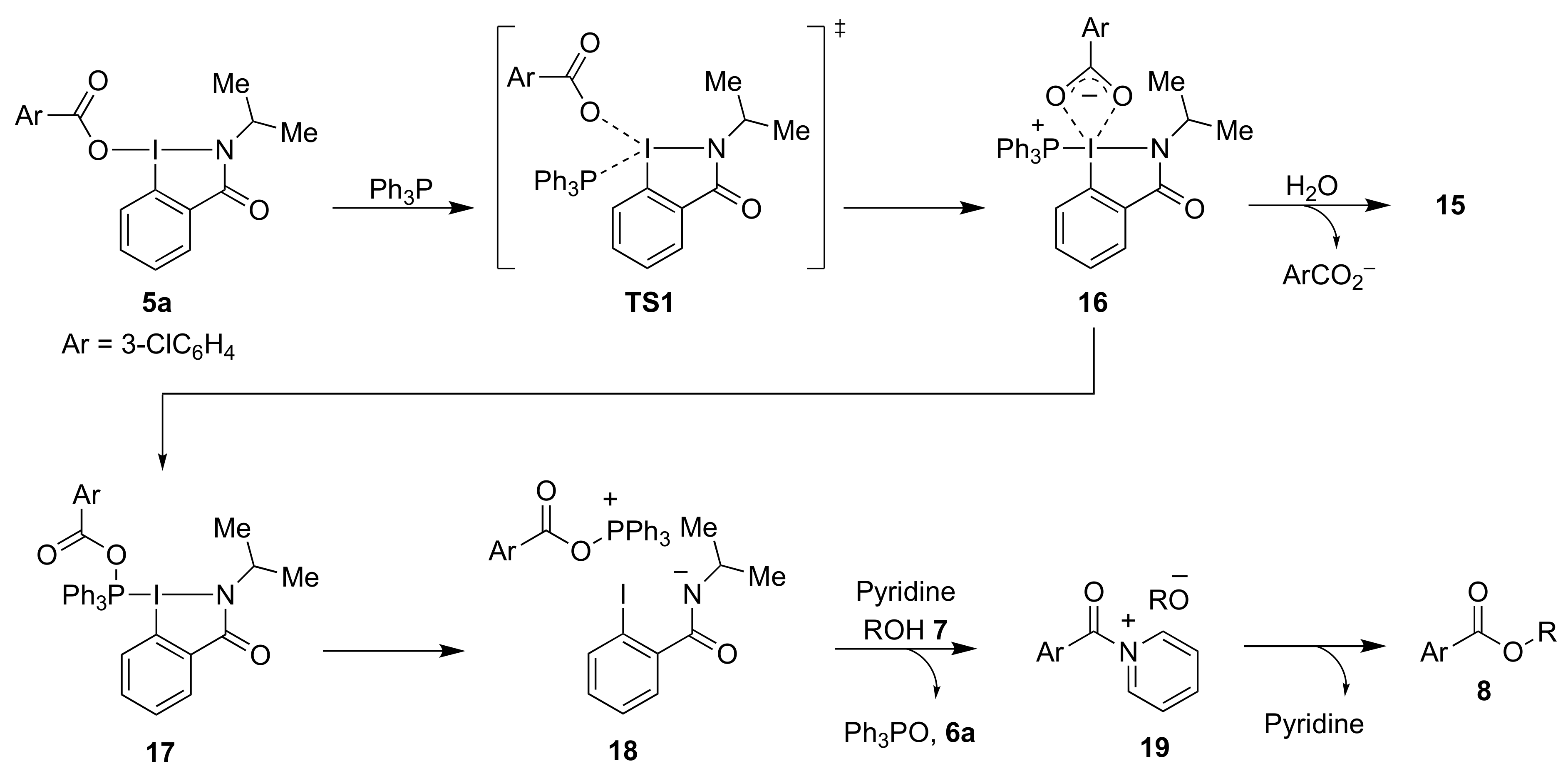
2-Isopropyl-3-oxo-2,3-dihydro-1H-1λ3-benzo[d][1,2]iodazol-1-yl 3-chlorobenzoate 5a. The reaction with 2-iodo-N-isopropylbenzamide 6a (289 mg, 1.00 mmol) and m-CPBA (259 mg, 1.50 mmol) in acetonitrile (10 mL) according to the general procedure afforded 296 mg (67%) of product 5a, isolated as a white solid: mp 142.0–142.8 °C; IR (neat) cm−1: 3067, 2967, 2932, 2875, 1627, 1588, 1570, 1296, 1257, and 739; 1H NMR (500 MHz, CDCl3): δ 8.28 (d, J = 8.0 Hz, 1H), 8.22 (dd, J = 7.8, 1.8 Hz, 1H), 8.04 (s, 1H), 7.96 (d, J = 7.5 Hz, 1H), 7.88–7.80 (m, 1H), 7.76–7.69 (m, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.45–7.37 (m, 1H), 4.52 (sept., J = 6.5 Hz, 1H), and 1.47 (d, J = 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3): δ 171.3, 166.0, 134.6, 133.7, 133.6, 132.3, 131.8, 131.0, 130.0, 129.8, 129.6, 127.9, 116.6, 46.7, and 24.4; HRMS (APCI-positive): calcd for C17H16ClINO3 ([M + H])+: 443.9863, found: 443.9877.
Single crystals of product 5a suitable for X-ray crystallographic analysis were obtained by slow crystallization from the acetonitrile solution. X-ray diffraction data for 5a were collected on Rigaku RAPID II Image Plate system using graphite-monochromated CuKα radiation (λ = 1.54187 Å) at 173 K. The structure was solved by Sir 2011 and refined on F2 using ShelXle. Crystal data for 5a C17H15ClINO3 are as follows: monoclinic, space group P21/c, a = 12.6913(3), b = 14.8285(4), c = 18.3997(13) Å, α =90°, β = 104.695(7)°, γ = 90°, V = 3349.4(3) Å3, Z = 8, 22,784 reflections measured, and 5815 unique (4560 I > 2σ/(I)); Rint = 0.0780, Rsigma = 0.0834, R1 (I > 2σ/(I)) = 0.0595, R1 = 0.0696, wR2all = 0.1700, and S = 1.086; please see the cif for more detailed information: CCDC-2122170.
2-Ethyl-3-oxo-2,3-dihydro-1H-1λ3-benzo[d][1,2]iodazol-1-yl 3-chlorobenzoate 5b. The reaction with N-ethyl-2-iodobenzamide 6b (550 mg, 2.00 mmol) and m-CPBA (518 mg, 3.00 mmol) in acetonitrile (15 mL) according to the general procedure afforded 380 mg (44%) of product 5b, isolated as a white solid: mp 76.0–78.0 °C; IR (neat) cm−1: 3074, 2969, 2935, 2876, 1627, 1590, 1571, 1442, 1319, 1263, 756, and 739; 1H NMR (500 MHz, CDCl3): δ 8.29 (d, J = 8.0 Hz, 1H), 8.23 (d, J = 7.5 Hz, 1H), 8.04 (s, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.90–7.83 (m, 1H), 7.73 (t, J = 8.0 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.44–7.37 (m, 1H), 3.76 (q, J = 7.3 Hz, 2H), and 1.37 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 171.3, 166.3, 134.9, 134.4, 133.6, 132.5, 132.4, 131.9, 131.0, 130.0, 129.8, 129.6, 127.9, 116.9, 38.3, and 16.2; HRMS (ESI-positive): calcd for C9H11INO2 ([M-3ClC6H4CO2+H])+: 291.9834, found: 291.9817.
2-(tert-Butyl)-3-oxo-2,3-dihydro-1H-1λ3-benzo[d][1,2]iodazol-1-yl 3-chlorobenzoate 5c. The reaction with N-(tert-butyl)-2-iodobenzamide 6c (606 mg, 2.00 mmol) and m-CPBA (518 mg, 3.00 mmol) in acetonitrile (15 mL) according to the general procedure afforded 705 mg (77%) of product 5c, isolated as a white solid: mp 170.8–172.0 °C; IR (neat) cm−1: 3070, 2966, 1628, 1585, 1570, 1438, 1317, 1262, 930, 756, and 740; 1H NMR (500 MHz, CDCl3): δ 8.26 (d, J = 8.5 Hz, 1H), 8.15 (d, J = 7.5 Hz, 1H), 8.03 (s, 1H), 7.95 (d, J = 7.5 Hz, 1H), 7.84 (t, J = 7.5 Hz, 1H), 7.73–7.67 (m, 1H), 7.52 (d, J = 7.5 Hz, 1H), 7.42–7.37 (m, 1H), and 1.71 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 171.3, 165.8, 135.0, 134.5, 134.4, 133.7, 132.3, 131.8, 130.9, 129.6, 129.5, 127.9, 114.8, 58.2, and 30.2; HRMS (ESI-positive): calcd for C11H15INO2 ([M-3ClC6H4CO2+H])+: 320.0147, found: 320.0137.
2-Cyclohexyl-3-oxo-2,3-dihydro-1H-1λ3-benzo[d][1,2]iodazol-1-yl 3-chlorobenzoate 5d. The reaction with N-cyclohexyl-2-iodobenzamide 6d (750 mg, 2.28 mmol) and m-CPBA (590 mg, 3.42 mmol) in acetonitrile (17.1 mL) according to the general procedure afforded 633 mg (51%) of product 5d, isolated as a white solid: mp 159.0–160.3 °C; IR (neat) cm−1: 3071, 2929, 2854, 1629, 1589, 1566, 1439, 1323, 1294, 1621, 1067, 967, 756, 739, and 659; 1H NMR (500 MHz, CDCl3): δ 8.29 (d, J = 8.5 Hz, 1H), 8.22 (d, J = 7.8 Hz, 1H), 8.04 (s, 1H), 7.96 (d, J = 7.5 Hz, 1H), 7.83 (t, J = 7.8 Hz, 1H), 7.75–7.69 (m, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.43–7.38 (m, 1H), 4.30–4.06 (m, 1H), 2.34–2.13 (m, 2H), 1.95–1.85 (m, 2H), 1.78–1.71 (m, 1H), 1.54–1.43 (m, 4H), and 1.30–1.20 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 171.4, 165.9, 134.5, 134.4, 133.7, 133.6, 132.3, 131.8, 131.0, 130.0, 129.8, 129.6, 127.8, 116.9, 54.2, 35.5, 25.7, and 25.3; HRMS (ESI-positive): calcd for C13H17INO2 ([M-3ClC6H4CO2+H])+: 346.0304, found: 346.0286.
3-Oxo-2-phenyl-2,3-dihydro-1H-1λ3-benzo[d][1,2]iodazol-1-yl 3-chlorobenzoate 5e. The reaction with 2-iodo-N-phenylbenzamide 6e (750 mg, 2.32 mmol) and m-CPBA (601 mg, 3.48 mmol) in acetonitrile (17.4 mL) according to the general procedure afforded 841 mg (76%) of product 5e, isolated as a white solid: mp 154.8 °C (decomp.); IR (neat) cm−1: 3067, 3033, 1637, 1586, 1569, 1488, 1441, 1507, 1262, 1125, 754, 741, and 659; 1H NMR (500 MHz, CDCl3): δ 8.34 (d, J = 6.0 Hz, 1H), 8.27 (d, J = 8.5 Hz, 1H), 8.03 (s, 1H), 7.97–7.90 (m, 2H), 7.81–7.74 (m, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.50–7.39 (m, 5H), and 7.35–7.28 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 170.9, 164.6, 138.1, 135.3, 134.5, 132.9, 132.8, 132.6, 132.6, 131.2, 129.9, 129.8, 129.7, 129.7, 128.0, 127.2, 126.4, and 117.2; HRMS (ESI-positive): calcd for C13H11INO2 ([M-3ClC6H4CO2+H])+: 339.9834, found: 339.9807.
2-Isopropyl-5-methyl-3-oxo-2,3-dihydro-1H-1λ3-benzo[d][1,2]iodazol-1-yl 3-chlorobenzoate 5f. The reaction with 2-iodo-N-isopropyl-5-methylbenzamide 6f (606 mg, 2.00 mmol) and m-CPBA (518 mg, 3.00 mmol) in acetonitrile (15 mL) according to the general procedure afforded 513 mg (56%) of product 5f, isolated as a white solid: mp 148.0–15.0 °C; IR (neat) cm−1: 3067, 2965, 2927, 2874, 1630, 1574, 1456, 1307, 1260, 1143, 756, and 740; 1H NMR (500 MHz, CDCl3): δ 8.11 (d, J = 9.5 Hz, 1H), 8.02 (s, 2H), 7.95 (d, J = 7.5 Hz, 1H), 7.66–7.61 (m, 1H), 7.54–7.50 (m, 1H), 7.43–7.37 (m, 1H), 4.61–4.42 (m, 1H), 2.54 (s, 3H), and 1.45 (d, J = 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3): δ 171.2, 166.1, 141.9, 135.6, 134.3, 133.8, 133.4, 132.3, 132.2, 129.8, 129.6, 129.6, 127.9, 112.8, 46.7, 24.3, and 20.9; HRMS (ESI-positive): calcd for C11H15INO2 ([M-3ClC6H4CO2+H])+: 320.0147, found: 320.0141.
2-Isopropyl-6-methyl-3-oxo-2,3-dihydro-1H-1λ3-benzo[d][1,2]iodazol-1-yl 3-chlorobenzoate 5g. The reaction with 2-iodo-N-isopropyl-4-methylbenzamide 6g (606 mg, 2.00 mmol) and m-CPBA (518 mg, 3.00 mmol) in acetonitrile (15 mL) according to the general procedure afforded 486 mg (53%) of product 6g, isolated as a white solid: mp 150.2–151.5 °C; IR (neat) cm−1: 3073, 2964, 2929, 1618, 1569, 1463, 1313, 1295, 1260, 1143, 757, 740, and 667; 1H NMR (500 MHz, CDCl3): δ 8.07 (d, J = 7.5 Hz, 1H), 8.06–8.02 (m, 2H), 7.95 (d, J = 7.8 Hz, 1H), 7.55–7.49 (m, 2H), 7.41 (t, J = 7.8 Hz, 1H), 4.60–4.41 (m, 1H), 2.56 (s, 3H), and 1.45 (d, J = 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3): δ 171.3, 166.1, 146.0, 134.4, 133.8, 132.3, 132.1, 131.5, 131.0, 129.9, 129.8, 129.6, 127.8, 116.8, 46.6, 24.4, and 22.3; HRMS (ESI-positive): calcd for C11H15INO2 ([M-3ClC6H4CO2+H])+: 320.0147, found: 320.0140.
2-Isopropyl-7-methyl-3-oxo-2,3-dihydro-1H-1λ3-benzo[d][1,2]iodazol-1-yl 3-chlorobenzoate 5h. The reaction with 2-iodo-N-isopropyl-3-methylbenzamide 6h (736 mg, 2.00 mmol) and m-CPBA (518 mg, 1.50 mmol) in acetonitrile (15 mL) according to the general procedure afforded 738 mg (71%) of product 5h, isolated as a white solid: mp 117.2–117.8 °C; IR (KBr) cm−1: 3127, 2960, 2909, 1648, 1608, 1569, 1384, 1317, 1139, and 745; 1H NMR (400 MHz, CDCl3): δ 8.08–8.02 (m, 1H), 7.97 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.64–7.56 (m, 2H), 7.49 (t, J = 8.0 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 4.57–4.44 (m, 1H), 2.80 (s, 3H), and 1.49 (d, J = 6.4 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 172.1, 166.7, 140.2, 138.2, 134.9, 134.4, 134.4, 132.1, 130.9, 129.8, 129.6, 127.7, 118.9, 47.1, 24.4, and 23.6; HRMS (ESI-positive): calcd for C11H15INO2 ([M-3ClC6H4CO2+H])+: 320.0147, found: 320.0146.
5-Bromo-2-isopropyl-3-oxo-2,3-dihydro-1H-1λ3-benzo[d][1,2]iodazol-1-yl 3-chlorobenzoate 5i. The reaction with 5-bromo-2-iodo-N-isopropylbenzamide 6i (736 mg, 2.00 mmol) and m-CPBA (518 mg, 1.50 mmol) in acetonitrile (15 mL) according to the general procedure afforded 738 mg (71%) of product 5i, isolated as a white solid: mp 151.6–152.6 °C; IR (neat) cm−1: 3103, 3066, 2968, 2931, 1636, 1617, 1568, 1557, 1464, 1443, 1405, 1305, 1260, 1141, 1070, 959, 903, 813, 755, and 742; 1H NMR (500 MHz, CDCl3): δ 8.35–8.33 (m, 1H), 8.13 (d, J = 9.0 Hz, 2H), 8.02–8.00 (m, 1H), 7.95–7.91 (m, 2H), 7.56–7.52 (m, 1H), 7.43–7.39 (m, 1H), 4.56–4.41 (m, 1H), and 1.46 (d, J = 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3): δ 171.3, 164.5, 137.4, 135.4, 134.7, 134.4, 133.3, 132.5, 131.4, 129.8, 129.7, 127.8, 126.4, 114.5, 47.0, and 24.3; HRMS (ESI-positive): calcd for C10H1279BrINO2 ([M-3ClC6H4CO2+H])+: 383.9096, found: 383.9101.
2-Isopropyl-3-oxo-2,3-dihydro-1H-1λ3-naphtho[2,3-d][1,2]iodazol-1-yl 3-chlorobenzoate 5j. The reaction with 3-iodo-N-isopropyl-2-naphthamide 6j (85 mg, 0.250 mmol) and m-CPBA (65 mg, 0.375 mmol) in acetonitrile (1.9 mL) according to the general procedure afforded 56 mg (46%) of product 5j, isolated as a white solid: mp 153.3–155.0 °C; IR (neat) cm−1: 3058, 2966, 2922, 1636, 1619, 1569, 1296, 1144, 758, and 740; 1H NMR (500 MHz, CDCl3): δ 8.74 (s, 2H), 8.14–8.08 (m, 2H), 8.04–7.98 (m, 2H), 7.77–7.69 (m, 2H), 7.58–7.53 (m, 1H), 7.44 (t, J = 7.8 Hz, 1H), 4.66–4.47 (m, 1H), and 1.49 (d, J = 6.5 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 171.3, 165.9, 136.7, 134.4, 133.8, 133.4, 132.4, 130.6, 129.9, 129.7, 129.3, 129.2, 128.9, 128.7, 128.3, 127.9, 111.1, 46.7, and 24.2; HRMS (ESI-positive): calcd for C14H14INO2 ([M-3ClC6H4CO2+H])+: 356.0147, found: 356.0119.
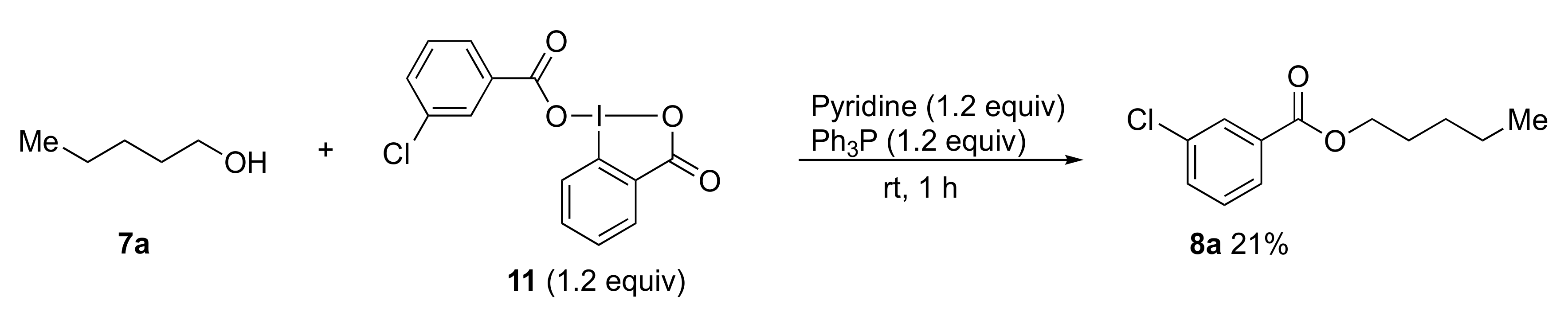
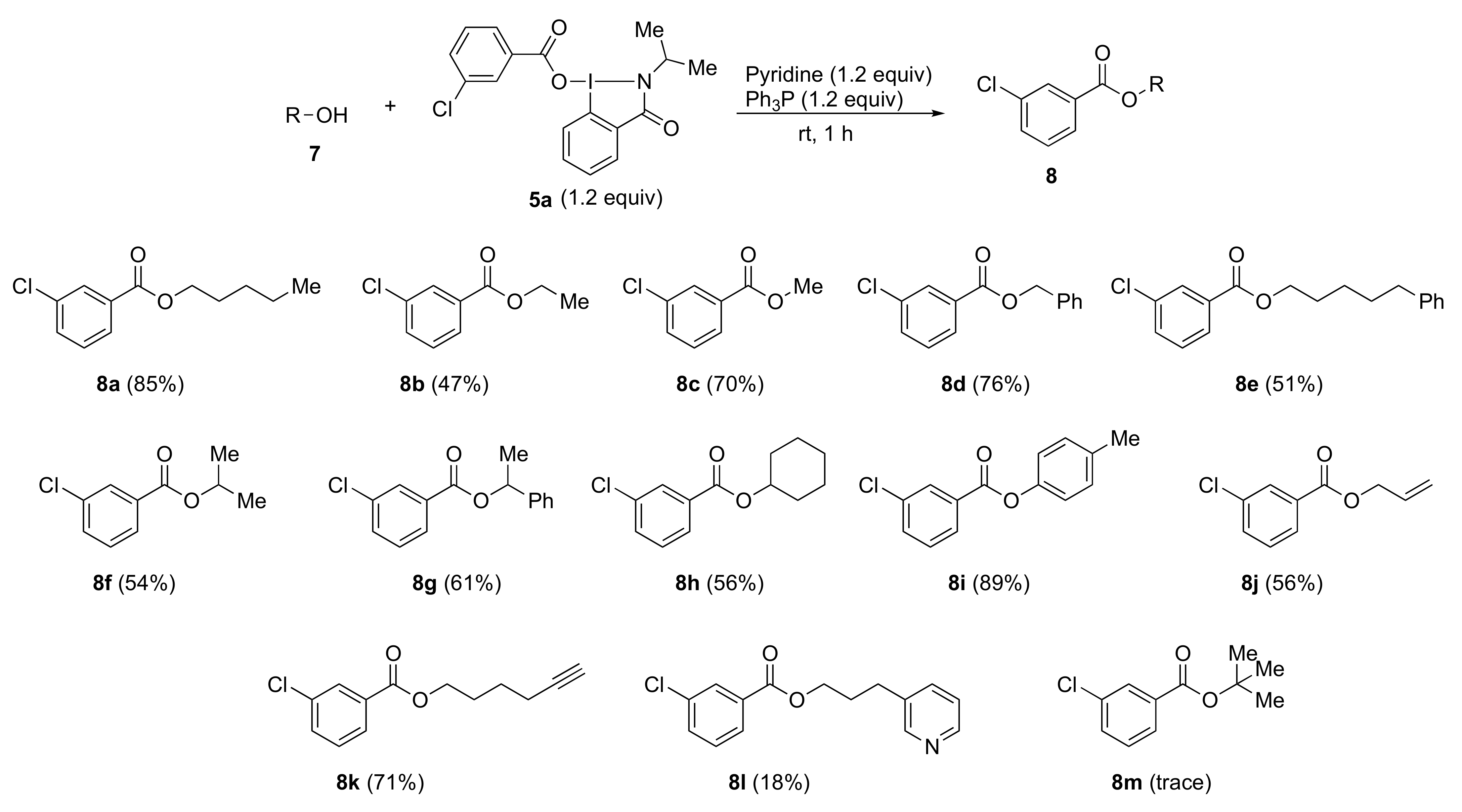
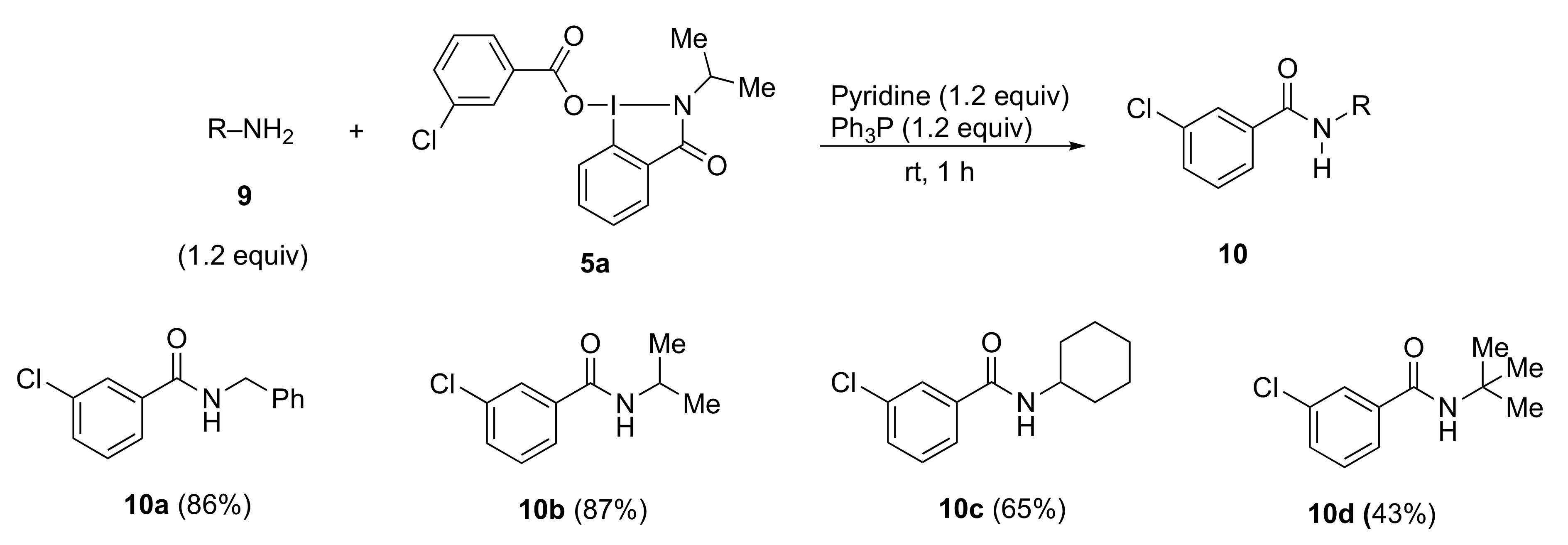
3.6. Typical Procedure for Esterification and Amidation Using Benziodazolones
Triphenylphosphine (47 mg, 0.18 mmol), pyridine (14 mg, 0.18 mmol), and alcohol 7 (0.15 mmol) or amine 9 (0.15 mmol) were added to a test tube containing benziodazolones 5 (0.180 mmol). The mixture was then stirred at room temperature for 1 h. After the reaction was completed, dichloromethane (3.0 mL) was used to transfer the reaction mixture to a separatory funnel. Then saturated NaHCO3 (3.0 mL) was added, and the reaction mixture was extracted with dichloromethane. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. Purification by preparative TLC (hexane:ethyl acetate = 3:1) afforded the analytically pure 8 or 10.
Ethyl 3-chlorobenzoate 8b [
36]. The reaction of ethanol
7b (7 mg, 0.150 mmol) according to the general procedure afforded 13 mg (47%) of product
8b, isolated as a colorless oil; IR (neat) cm
−1: 2924, 1727, 1573, 1466, 1370, 1293, 1281, 1256, 915, and 749;
1H NMR (500 MHz, CDCl
3): δ 8.03 (s, 1H), 7.94 (d,
J = 7.5 Hz, 1H), 7.56–7.52 (m, 1H), 7.39 (t,
J = 7.5 Hz, 1H), 4.39 (q,
J = 7.0 Hz, 2H), and 1.41 (t,
J = 7.0 Hz, 3H); HRMS (APCI-positive): calcd for C
9H
10ClO
2 ([M + H])
+: 185.0369, found: 185.0389.
Methyl 3-chlorobenzoate 8c [
37]. The reaction of methanol
7c (5 mg, 0.150 mmol) according to the general procedure afforded 18 mg (70%) of product
8c, isolated as a colorless oil;
1H NMR (500 MHz, CDCl
3): δ 8.02 (s, 1H), 7.93 (d,
J = 7.8 Hz, 1H), 7.56–7.51 (m, 1H), 7.39 (t,
J = 7.8 Hz, 1H), and 3.93 (s, 3H);
13C NMR (125 MHz, CDCl
3): δ 165.9, 134.5, 133.0, 131.9, 130.0, 129.7, 127.7, and 52.4.
Benzyl 3-chlorobenzoate 8d [
38]. The reaction of benzyl alcohol
7d (16 mg, 0.150 mmol) according to the general procedure afforded 28 mg (76%) of product
8d, isolated as a colorless oil; IR (neat) cm
−1: 2955, 1721, 1576, 1429, 1290, 1278, 1124, 955, and 659;
1H NMR (500 MHz, CDCl
3): δ 8.04 (s, 1H), 7.95 (d,
J = 8.0 Hz, 1H), 7.54–7.50 (m, 1H), 7.47–7.42 (m, 2H), 7.42–7.33 (m, 4H), and 5.36 (s, 2H);
13C NMR (125 MHz, CDCl
3): δ 165.2, 135.6, 134.5, 133.1, 131.9, 129.7, 129.7, 128.7, 128.4, 128.3, 127.8, and 67.1; HRMS (ESI-positive): calcd for C
14H
11ClO
2Na ([M + Na])
+: 269.0345, found: 269.0341.
5-Phenylpentyl 3-chlorobenzoate 8e. The reaction of 5-phenyl-1-pentyl alcohol 7e (25 mg, 0.150 mmol) according to the general procedure afforded 23 mg (51%) of product 8e, isolated as a colorless oil; IR (neat) cm−1: 2937, 1724, 1576, 1429, 1293, 1256, 1124, 958, 699, 675, and 659; 1H NMR (500 MHz, CDCl3): δ 7.99 (s, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.39–7.33 (m, 1H), 7.30–7.22 (m, 2H), 7.20–7.13 (m, 3H), 4.31 (t, J = 6.5 Hz, 2H), 2.75–2.53 (m, 2H), 1.83–1.75 (m, 2H), 1.73–1.65 (m, 2H), and 1.52–1.43 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 165.4, 142.3, 134.5, 132.8, 132.2, 129.7, 129.6, 128.4, 128.3, 127.7, 125.7, 65.4, 35.8, 31.0, 28.5, and 25.6; HRMS (APCI-positive): calcd for C18H20ClO2 ([M + H])+: 303.1152, found: 303.1173.
Isopropyl 3-chlorobenzoate 8f [
39]. The reaction of isopropanol
7f (9 mg, 0.150 mmol) according to the general procedure afforded 16 mg (54%) of product
8f, isolated as a colorless oil;
1H NMR (500 MHz, CDCl
3): δ 8.01 (s, 1H), 7.92 (d,
J = 7.9 Hz, 1H), 7.54–7.50 (m, 1H), 7.37 (d,
J = 7.9 Hz, 1H), 5.25 (sept,
J = 6.5 Hz, 1H), and 1.37 (d,
J = 6.5 Hz, 6H).
1-Phenylethyl 3-chlorobenzoate 8g [
40]. The reaction of 1-phenylethyl alcohol
7g (18 mg, 0.150 mmol) according to the general procedure afforded 24 mg (61%) of product
8g, isolated as a colorless oil; IR (neat) cm
−1: 2985, 1723, 1574, 1496, 1456, 1428, 1256, 1129, and 697;
1H NMR (500 MHz, CDCl
3): δ 8.05 (s, 1H), 7.97 (d,
J = 8.0 Hz, 1H), 7.56–7.52 (m, 1H), 7.45 (d,
J = 8.0 Hz, 1H), 7.42–7.36 (m, 3H), 7.36–7.30 (m, 1H), 6.14 (q,
J = 6.9 Hz, 1H), and 1.69 (d,
J = 6.9 Hz, 3H);
13C NMR (125 MHz, CDCl
3): δ 164.6, 141.4, 134.5, 133.0, 132.3, 129.7, 128.7, 128.1, 127.8, 126.1, 73.5, and 22.3; HRMS (ESI-positive): calcd for C
15H
13ClO
2Na ([M + Na])
+: 283.0502, found: 283.0489.
Cyclohexyl 3-chlorobenzoate 8h [
40]. The reaction of cyclohexanol
7h (15 mg, 0.150 mmol) according to the general procedure afforded 20 mg (56%) of product
8h, isolated as a colorless oil; IR (neat) cm
−1: 2937, 1721, 1576, 1469, 1290, 1253, 1124, 945, 749, and 678;
1H NMR (500 MHz, CDCl
3): δ 8.03–8.00 (m, 1H), 7.95–7.91 (m, 1H), 7.54–7.50 (m, 1H), 7.38 (t,
J = 8.0 Hz, 1H), 5.09–4.94 (m, 1H), 1.99–1.91 (m, 2H), 1.84–1.75 (m, 2H), 1.64–1.53 (m, 3H), 1.50–1.40 (m, 2H), and 1.40–1.30 (m, 1H);
13C NMR (125 MHz, CDCl
3): δ 164.8, 134.4, 132.8, 132.7, 129.6, 127.7, 73.7, 31.6, 25.4, and 23.7; HRMS (APCI-positive): calcd for C
13H
16ClO
2 ([M + H])
+: 239.0839, found: 239.0843.
p-Tolyl 3-chlorobenzoate 8i [
41]. The reaction of
p-cresol
7i (16 mg, 0.150 mmol) according to the general procedure afforded 33 mg (89%) of product
8i, isolated as a white solid, mp 75.4–76.0 °C; IR (neat) cm
−1: 2925, 1742, 1578, 1474, 1288, 1251, 1197, 1165, 1106, 811, and 743;
1H NMR (500 MHz, CDCl
3): δ 8.17 (s, 1H), 8.07 (d,
J = 8.0 Hz, 1H), 7.61–7.56 (m, 1H), 7.43 (t,
J = 8.0 Hz, 1H), 7.22 (d,
J = 8.5 Hz, 2H), 7.08 (d,
J = 8.5 Hz, 2H), and 2.37 (s, 3H);
13C NMR (125 MHz, CDCl
3): δ 164.2, 148.5, 135.8, 134.7, 133.5, 131.5, 130.2, 130.1, 129.9, 128.3, 121.2, and 20.9; HRMS (ESI-positive): calcd for C
14H
12ClO
2 ([M + H])
+: 247.0526, found: 247.0528.
Allyl 3-chlorobenzoate 8j [
42]. The reaction of allyl alcohol
7j (13 mg, 0.150 mmol) according to the general procedure afforded 16 mg (56%) of product
8j, isolated as a colorless oil; IR (neat) cm
−1: 3077, 2945, 2883, 1726, 1576, 1426, 1289, 1256, 1128, and 748;
1H NMR (400 MHz, CDCl
3): δ 8.07–8.02 (m, 1H), 7.95 (d,
J = 7.8 Hz, 1H), 7.60–7.50 (m, 1H), 7.39 (t,
J = 7.8 Hz, 1H), 5.42 (dd,
J = 17.2, 1.6 Hz, 1H), 5.31 (dd,
J = 10.4, 1.6 Hz, 1H), and 4.83 (d,
J = 5.6 Hz, 2H);
13C NMR (100 MHz, CDCl
3): δ 165.1, 134.6, 133.0, 131.9, 129.7, 127.8, 118.7, 74.2, and 66.0; HRMS (EI): calcd for C
10H
935ClO
2 ([M + H])
+: 196.0291, found: 196.0285.
Hex-5-yn-1-yl 3-chlorobenzoate 8k. The reaction of 5-hexyn-1-ol 7k (15 mg, 0.150 mmol) according to the general procedure afforded 25 mg (71%) of product 8k, isolated as a colorless oil; IR (neat) cm−1: 3298, 3074, 2951, 2870, 2117, 1722, 1575, 1428, 1285, 1260, 1130, 747, and 641; 1H NMR (400 MHz, CDCl3): δ 8.01 (s, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.56–7.50 (m, 1H), 7.38 (d, J = 7.8 Hz, 1H), 4.36 (t, J =6.4 Hz, 2H), 2.36–2.16 (m, 2H), 1.99 (t, J = 2.8 Hz, 1H), 1.96–1.87 (m, 2H), 1.75–and 1.64 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 165.4, 134.5, 132.9, 132.1, 129.7, 129.6, 127.7, 83.8, 68.9, 64.9, 27.7, 25.0, and 18.1; HRMS (CI): calcd for C13H1335ClO2 ([M + H])+: 254.0942, found:254.0934.
3-(Pyridin-3-yl)propyl 3-chlorobenzoate 8l. The reaction of 3-pyridinepropanol 7l (21 mg, 0.150 mmol) according to the general procedure afforded 8 mg (18%) of product 8l, isolated as a colorless oil; IR (neat) cm−1: 3031, 2958, 2909, 2818, 1721, 1575, 1424, 1288, 1257, 1129, 1074, and 749; 1H NMR (400 MHz, CDCl3): δ 8.50 (s, 1H), 8.47 (d, J = 4.8 Hz, 1H), 8.00–7.95 (m, 1H), 7.93–7.87 (m, 1H), 7.54 (d, J = 7.6 Hz, 2H), 7.40 (t, J = 7.6 Hz, 1H), 7.26–7.20 (m, 1H), 4.37 (t, J = 6.4 Hz, 2H), 2.85–2.75 (m, 2H), and 2.18–2.07 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 165.3, 149.9, 147.8, 136.3, 135.8, 134.6, 133.1, 131.9, 129.7, 129.6, 127.7, 123.4, 64.4, 29.9, and 29.6; HRMS (ESI-positive): calcd for C15H1435ClNO2 ([M + H])+: 276.0786, found: 276.0786.
N-Benzyl 3-chlorobenzamide 10a [
43]. The reaction of benzylamine
9a (16 mg, 0.150 mmol) according to the general procedure afforded 32 mg (86%) of product
10a, isolated as a white solid;
1H NMR (500 MHz, CDCl
3): δ 7.77 (s, 1H), 7.65 (d,
J = 7.5 Hz, 1H), 7.51–7.43 (m, 1H), 7.41–7.28 (m, 6H), 6.44 (brs, 1H), and 4.63 (d,
J = 5.5 Hz, 2H);
13C NMR (125 MHz, CDCl
3): δ 166.0, 137.8, 136.2, 134.8, 131.6, 129.9, 128.8, 128.0, 127.8, 127.3, 125.0, and 44.3; HRMS (APCI-positive): calcd for C
14H
13ClNO ([M + H])
+: 246.0686, found: 246.0706.
3-Chloro-N-isopropylbenzamide 10b [
44]. The reaction of isopropylamine
9b (9 mg, 0.150 mmol) according to the general procedure afforded 26 mg (87%) of product
10b, isolated as a white solid;
1H NMR (400 MHz, CDCl
3): δ 7.73 (t,
J = 1.8 Hz, 1H), 7.69–7.59 (m, 1H), 7.49–7.41 (m, 1H), 7.35 (t,
J = 7.8 Hz, 1H), 6.03 (br. s, 1H), 4.44–4.13 (m, 1H), and 1.26 (d,
J = 6.8 Hz, 6H);
13C NMR (100 MHz, CDCl
3): δ 165.4, 136.8, 134.7, 131.3, 129.8, 127.2, 125.0, 42.1, and 22.8; HRMS (ESI-positive): calcd for C
12H
1235ClNO ([M + H])
+: 198.0680, found: 198.0704.
3-Chloro-N-cyclohexylbenzamide 10c [
45]. The reaction of cyclohexylamine
9c (15 mg, 0.150 mmol) according to the general procedure afforded 22 mg (65%) of product
10c, isolated as a white solid; IR (neat) cm
−1: 3322, 3070, 2929, 2853, 1633, 1539, 1327, 1081, 753;
1H NMR (400 MHz, CDCl
3): δ 7.75–7.71 (m, 1H), 7.64–7.60 (m, 1H), 7.47–7.42 (m, 1H), 7.35 (t,
J = 7.8 Hz, 1H), 6.06 (brs, 1H), 4.03–3.89 (m, 1H), 2.07–1.98 (m, 2H), 1.81–1.71 (m, 2H), 1.70–1.62 (m, 1H), 1.49–1.35 (m, 2H), and 1.30–1.17 (m, 3H);
13C NMR (100 MHz, CDCl
3): δ 165.3, 136.9, 134.7, 131.3, 129.8, 127.2, 125.0, 48.9, 33.2, 25.5, and 24.9; HRMS (ESI-positive): calcd for C
13H
16ClNO ([M + H])
+: 238.0993, found: 238.1009.
N-(tert-butyl)-3-chlorobenzamide 10d [
46]. The reaction of
tert-butyl amine
9d (11 mg, 0.150 mmol) according to the general procedure afforded 14 mg (43%) of product
10d, isolated a white solid;
1H NMR (400 MHz, CDCl
3): δ 7.69 (t,
J = 2.0 Hz, 1H), 7.61–7.56 (m, 1H), 7.46–7.42 (m, 1H), 7.35 (t,
J = 7.8 Hz, 1H), 5.92 (brs, 1H), and 1.47 (s, 9H);
13C NMR (100 MHz, CDCl
3): δ 165.5, 137.8, 134.6, 131.1, 129.8, 127.1, 124.9, 51.9, and 28.8; HRMS (ESI-positive): calcd for C
11H
14ClNO ([M + H])
+: 212.0837, found: 212.0853.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
