4.3. Experimental Procedures
Compounds
2a–
2d were synthesized according to literature reports [
31,
32].
Oxocane-2,8-dione (2a) (white solid, yield 83%): 1H NMR (400 MHz, CDCl3) δ 2.40 (t, J = 7.2 Hz, 4H), 1.62 (t, J = 7.4 Hz, 4H), 1.43–1.29 (m, 2H).
Oxonane-2,9-dione (2b) (white solid, yield 76%): 1H NMR (400 MHz, CDCl3) δ 2.43 (t, J = 7.2 Hz, 4H), 1.58 (t, J = 7.2 Hz, 4H), 1.40–1.31 (m, 4H).
Oxonane-2,10-dione (2c) (white solid, yield 89%): 1H NMR (400 MHz, CDCl3) δ 2.45 (t, J = 7.4 Hz, 4H), 1.68–1.60 (m, 4H), 1.39–1.28 (m, 6H).
Oxonane-2,11-dione (2d) (white solid, yield 85%): 1H NMR (400 MHz, CDCl3) δ 2.46 (t, J = 7.2 Hz, 4H), 1.63–1.57 (m, 4H), 1.42–1.23 (m, 8H).
Compounds
4a–
4d were synthesized according to previous published reports [
33,
34].
7-(Benzyloxy)-7-oxoheptanoic acid (4a) (white solid, yield 75%): 1H NMR (400 MHz, CDCl3) δ 7.41–7.24 (m, 2H), 5.11 (s, 2H), 2.35 (dt, J = 11.8, 7.5 Hz, 4H), 1.74–1.58 (m, 4H), 1.44–1.26 (m, 2H).
8-(Benzyloxy)-8-oxooctanoic acid (4b) (white solid, yield 69%): 1H NMR (400 MHz, DMSO-d6) δ 11.92 (s, 1H), 7.33–7.21 (m, 5H), 5.03 (s, 2H), 2.29 (t, J = 6.8 Hz, 2H), 2.14 (t, J = 7.2 Hz, 2H), 1.50–1.36 (m, 4H), 1.28–1.13 (m, 4H).
9-(Benzyloxy)-9-oxononanoic acid (4c) (white solid, yield 66%): 1H NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H), 7.40–7.32 (m, 5H), 5.09 (s, 2H), 2.34 (t, J = 7.3 Hz, 2H), 2.18 (t, J = 7.3 Hz, 2H), 1.59–1.41 (m, 4H), 1.30–1.17 (m, 6H).
10-(Benzyloxy)-10-oxononanoic acid (4d) (white solid, yield 64%): 1H NMR (400 MHz, DMSO-d6) δ 12.03–11.91 (m, 1H), 7.46–7.27 (m, 5H), 5.20–4.99 (m, 2H), 2.34 (t, J = 7.3 Hz, 2H), 2.18 (t, J = 7.3 Hz, 2H), 1.61–1.41 (m, 4H), 1.28–1.20 (m, 8H).
General procedures for the preparation of compounds 6a–6e. To a solution of 4a–4d (1.0 eq) in anhydrous DMF, HATU (2.0 eq) and DIEA (3.0 eq) were added. After stirring in an ice bath for 0.5 h, 5a or 5b (1.0 eq) was added. The mixture was stirred at room temperature overnight. After the reaction completed, the residue was diluted with H2O and extracted with EtOAc. The combined organic layers were then washed with HCl (0.1 M), saturated brine, dried over anhydrous Na2SO4, and concentrated in vacuo to afford the crude product, which was purified by column chromatography with DCM/ MeOH (30:1–20:1) to produce 6a–6e.
Benzyl 7-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-7-oxoheptanoate (6a). (white solid, yield 82%): 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 9.76 (s, 1H), 7.81 (d, J = 6.8 Hz, 1H), 7.49 (q, J = 7.3 Hz, 2H), 7.35 (s, 5H), 5.14 (dd, J = 13.2, 4.9 Hz, 1H), 5.08 (s, 2H), 4.45–4.28 (m, 2H), 2.90 (dd, J = 21.7, 9.1Hz, 1H), 2.60 (d, J = 17.3 Hz, 1H), 2.43–2.27 (m, 5H), 2.08–1.95 (m, 1H), 1.60 (dd, J = 15.5, 8.0 Hz, 4H), 1.34 (dd, J = 14.8, 7.6 Hz, 2H). 13C NMR (100 MHz, DMSO) δ 172.8, 172.7, 171.2, 171.0, 167.8, 136.2, 133.8, 133.7, 132.6, 128.6, 128.4, 127.9, 127.9, 125.2, 119.0, 65.3, 51.5, 46.5, 35.6, 33.3, 31.2, 29.0, 28.0, 24.7, 24.2, 22.6.
Benzyl 8-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-8-oxoheptanoate (6b) (white solid, yield 85%): 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 9.75 (s, 1H), 7.81 (d, J = 7.0 Hz, 1H), 7.49 (q, J = 7.4 Hz, 2H), 7.41–7.28 (m, 5H), 5.14 (dd, J = 13.2, 5.0 Hz, 1H), 5.08 (s, 2H), 4.43–4.29 (m, 2H), 3.00–2.83(m, 1H), 2.60 (d, J = 16.8 Hz, 1H), 2.35 (dd, J = 13.8, 6.9 Hz, 5H), 2.08–1.95 (m, 1H), 1.68–1.50 (m, 4H), 1.31 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 172.7, 171.3, 171.0, 167.8, 136.3, 133.8, 133.7, 132.6, 128.6, 128.4, 127.9, 127.9, 125.2, 118.9, 65.3, 51.5, 46.5, 40.1, 39.9, 39.7, 39.5, 39.3, 39.1, 38.9, 35.7, 33.4, 31.2, 28.3, 28.1, 24.9, 24.3, 22.6.
Benzyl 9-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-9-oxoheptanoate (6c) (white solid, yield 81%): 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 9.75 (s, 1H), 7.81 (d, J = 6.8 Hz, 1H), 7.49 (q, J = 7.4 Hz, 2H), 7.42–7.27 (m, 5H), 5.14 (dd, J = 13.2, 5.0 Hz, 1H), 5.08 (s, 2H), 4.48–4.27 (m, 2H), 3.07–2.82 (m, 1H), 2.61 (d, J = 17.1 Hz, 1H), 2.35 (t, J = 7.2 Hz, 5H), 2.10–1.94 (m, 1H), 1.56 (dd, J = 15.8, 8.7 Hz, 4H), 1.29 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 172.8, 171.3, 171.0, 167.8, 136.3, 133.8, 133.7, 132.6, 128.6, 128.4, 127.9, 127.9, 125.2, 118.9, 65.3, 51.5, 46.4, 35.7, 33.4, 31.2, 28.5, 28.4, 28.3, 25.0, 24.4, 22.6.
Benzyl 10-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-10-oxoheptanoate (6d) (white solid, yield 75%): 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 9.75 (s, 1H), 7.82 (d, J = 7.0 Hz, 1H), 7.49 (q, J = 7.3 Hz, 2H), 7.41–7.27 (m, 5H), 5.15 (dd, J = 13.2, 4.8 Hz, 1H), 5.08 (s, 2H), 4.45–4.28 (m, 2H), 2.93 (dd, J = 21.6, 8.7 Hz, 1H), 2.61 (d, J = 16.6 Hz, 1H), 2.34 (t, J = 7.1 Hz, 5H), 2.10–1.97 (m, 1H), 1.57 (dd, J = 16.3, 7.0 Hz, 4H), 1.26 (s, 8H). 13C NMR (100 MHz, DMSO-d6) δ 172.9, 172.8, 171.4, 171.0, 167.8, 136.3, 133.8, 133.7, 132.6, 128.6, 128.4, 127.9, 127.9, 125.2, 119.0, 65.2, 51.5, 46.5, 35.8, 33.4, 31.2, 28.6, 28.6, 28.5, 28.3, 25.0, 24.4, 22.6.
Benzyl 8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-8-oxooctanoate (6e) (white solid, yield 64%): 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H), 9.69 (s, 1H), 8.47 (d, J = 8.4 Hz, 1H), 7.83 (t, J = 7.9 Hz, 1H), 7.61 (d, J = 7.2 Hz, 1H), 7.46–7.21 (m, 5H), 5.15 (dd, J = 12.7, 5.3 Hz, 1H), 5.08 (s, 2H), 2.98–2.82 (m, 1H), 2.58 (dd, J = 22.6, 10.8 Hz, 2H), 2.45 (t, J = 7.4 Hz, 2H), 2.36 (t, J = 7.3 Hz, 2H), 2.12–2.01 (m, 1H), 1.66–1.49 (m, 4H), 1.32 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.7, 172.7, 171.9, 169.7, 167.7, 166.6, 136.5, 136.3, 136.1, 131.4, 128.4, 127.9, 127.9, 126.3, 118.3, 117.0, 65.3, 48.9, 36.4, 33.4, 30.9, 28.1, 24.6, 24.3, 21.9.
General procedures for the preparation of compounds 7a–7e. To a solution of 6a–6e (1.0 eq) in MeOH, 10% Pd/C (10% eq) was added. The reaction mixture was purged with N2 and then H2 and stirred at room temperature for 2 h. After the reaction completed, the residue was filtered through celite and washed with DCM. The combined organic layers were then washed with saturated brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce 7a–7e.
7-((2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-7-oxoheptanoic acid (7a) (white solid, yield 95%): 1H NMR (400 MHz, DMSO-d6) δ 11.03 (s, 1H), 9.88 (s, 1H), 7.82 (d, J = 6.1 Hz, 1H), 7.49 (d, J = 6.8 Hz, 2H), 5.13 (dd, J = 12.9, 4.3 Hz, 1H), 4.38 (q, J = 17.5 Hz, 2H), 2.99–2.82 (m, 1H), 2.61 (d, J = 16.7 Hz, 1H), 2.44–2.27 (m, 3H), 2.25–2.12 (m, 2H), 2.10–1.96 (m, 1H), 1.67–1.45 (m, 4H), 1.33 (d, J = 6.4 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.8, 172.8, 171.3, 171.0, 167.8, 133.8, 133.7, 132.6, 128.5, 125.3, 118.9, 51.5, 46.6, 40.1, 39.9, 39.7, 39.5, 39.3, 39.1, 38.9, 35.6, 34.0, 31.2, 28.2, 24.8, 24.4, 22.6, −15.0.
8-((2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-8-oxoheptanoic acid (7b) (white solid, yield 93%): 1H NMR (400 MHz, DMSO-d6) δ 11.96 (s, 1H), 11.01 (s, 1H), 9.77 (s, 1H), 7.81 (d, J = 6.9 Hz, 1H), 7.49 (q, J = 7.2 Hz, 2H), 5.14 (dd, J = 13.2, 5.0 Hz, 1H), 4.36 (q, J = 17.5 Hz, 2H), 3.04–2.82 (m, 1H), 2.61 (d, J = 16.0 Hz, 1H), 2.43–2.28 (m, 3H), 2.20 (t, J = 7.3 Hz, 2H), 2.09–1.95 (m, 1H), 1.68–1.44 (m, 4H), 1.31 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 174.5, 172.8, 171.3, 171.0, 167.8, 133.7, 133.7, 132.6, 128.6, 125.2, 118.9, 51.5, 46.5, 35.7, 33.6, 31.2, 28.3, 28.3, 24.9, 24.4, 22.6.
9-((2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-9-oxoheptanoic acid (7c) (white solid, yield 96%): 1H NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H), 11.02 (s, 1H), 9.76 (s, 1H), 7.81 (dd, J = 6.9, 1.1 Hz, 1H), 7.49 (q, J = 7.2 Hz, 2H), 5.14 (dd, J = 13.3, 5.0 Hz, 1H), 4.36 (q, J = 17.5 Hz, 2H), 3.02–2.81 (m, 1H), 2.61 (d, J = 16.6 Hz, 1H), 2.44–2.28 (m, 3H), 2.19 (t, J = 7.3 Hz, 2H), 2.09–1.98 (m, 1H), 1.67–1.43 (m, 4H), 1.30 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 174.5, 172.8, 171.3, 171.0, 167.8, 133.8, 133.7, 132.6, 128.6, 125.2, 118.9, 51.5, 46.4, 35.7, 33.6, 31.2, 28.5, 28.5, 28.4, 25.0, 24.4, 22.6.
10-((2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-10-oxoheptanoic acid (7d) (white solid, yield 90%): 1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1H), 11.02 (s, 1H), 9.75 (s, 1H), 7.81 (d, J = 6.9 Hz, 1H), 7.54–7.43 (m, 2H), 5.15 (dd, J = 13.2, 5.0 Hz, 1H), 4.36 (q, J = 17.5 Hz, 2H), 2.99–2.82 (m, 1H), 2.61 (d, J = 17.3 Hz, 1H), 2.44–2.26 (m, 3H), 2.19 (t, J = 7.3 Hz, 2H), 2.08–1.95 (m, 1H), 1.54 (dd, J = 45.7, 6.5 Hz, 4H), 1.27 (d, J = 3.7 Hz, 8H). 13C NMR (100 MHz, DMSO-d6) δ 174.5, 172.8, 171.3, 171.0, 167.8, 133.8, 133.7, 132.6, 128.6, 125.2, 118.9, 51.5, 46.4, 35.8, 33.6, 31.2, 28.6, 28.5, 25.0, 24.4, 22.6.
8-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-8-oxooctanoic acid (7e) (white solid, yield 89%): 1H NMR (400 MHz, DMSO-d6) δ 12.14 (s, 1H), 11.15 (s, 1H), 9.70 (s, 1H), 8.47 (d, J = 8.4 Hz, 1H), 7.83 (t, J = 7.8 Hz, 1H), 7.61 (d, J = 7.2 Hz, 1H), 5.15 (dd, J = 12.6, 5.3 Hz, 1H), 2.99–2.82 (m, 1H), 2.58 (dd, J = 23.3, 11.1 Hz, 2H), 2.46 (t, J = 7.4 Hz, 2H), 2.19 (t, J = 7.2 Hz, 2H), 2.13–2.00 (m, 1H), 1.69–1.42 (m, 4H), 1.32 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 174.5, 172.7, 172.0, 169.8, 167.6, 166.6, 136.5, 136.1, 131.4, 126.3, 118.25, 117.0, 48.9, 36.4, 33.7, 30.9, 28.3, 28.2, 24.6, 24.4, 21.9.
General procedures for the preparation of compounds 8a–8e. To a solution of 7a–7e (1.0 eq) in anhydrous DMF, HATU (2.0 eq) and DIEA (3.0 eq) were added. After stirring in an ice bath for 0.5 h, O-tritylhydroxylamine (1.0 eq) was added. The mixture was stirred at room temperature overnight. After the reaction completed, the residue was diluted with H2O and extracted with EtOAc. The combined organic layers were then washed saturated brine, dried over anhydrous Na2SO4, and concentrated in vacuo to produce the crude product, which was purified by column chromatography with DCM/ MeOH (30:1–20:1) to produce 8a–8e.
N1-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-N7-(trityloxy)heptanediamide (8a) (white solid, yield 73%): 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 10.17 (s, 1H), 9.73 (s, 1H), 7.81 (d, J = 6.5 Hz, 1H), 7.57–7.44 (m, 2H), 7.32 (s, 15H), 5.14 (dd, J = 13.1, 4.6 Hz, 1H), 4.45–4.27 (m, 2H), 2.99–2.81 (m, 1H), 2.60 (d, J = 17.0 Hz, 1H), 2.41–2.18 (m, 3H), 2.08–1.94 (m, 1H), 1.80 (s, 2H), 1.47 (s, 2H), 1.24 (s, 5H), 1.04 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 171.2, 171.0, 170.2, 167.8, 142.4, 133.8, 133.6, 132.6, 128.9, 128.6, 127.5, 127.4, 125.2, 118.9, 91.7, 51.5, 46.5, 35.6, 31.8, 31.2, 28.0, 24.7, 24.5, 22.6.
N1-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-N8-(trityloxy)octanediamide (8b) (white solid, yield 79%): 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 10.15 (s, 1H), 9.73 (s, 1H), 7.82 (d, J = 7.0 Hz, 1H), 7.49 (q, J = 7.4 Hz, 2H), 7.32 (s, 15H), 5.14 (dd, J = 13.2, 4.9 Hz, 1H), 4.46–4.28 (m, 2H), 3.00–2.83 (m, 1H), 2.60 (d, J = 17.3 Hz, 1H), 2.34 (dt, J = 14.7, 8.3 Hz, 3H), 2.09–1.95 (m, 1H), 1.78 (s, 2H), 1.60–1.41 (m, 2H), 1.19 (d, J = 5.8 Hz, 4H), 1.02 (d, J = 5.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 171.3, 171.0, 170.3, 167.8, 142.4, 133.8, 133.6, 132.6, 128.9, 128.6, 127.5, 127.4, 125.2, 119.0, 91.7, 64.9, 51.5, 46.5, 40.1, 39.9, 39.7, 39.5, 39.3, 39.1, 38.9, 35.7, 31.9, 31.2, 28.3, 28.1, 24.9, 24.6, 22.6, 15.1.
N1-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-N9-(trityloxy)nonanediamide (8c) (white solid, yield 75%): 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 10.14 (s, 1H), 9.75 (s, 1H), 7.82 (d, J = 7.0 Hz, 1H), 7.49 (q, J = 7.3 Hz, 2H), 7.32 (s, 15H), 5.14 (dd, J = 13.2, 5.0 Hz, 1H), 4.44–4.21 (m, 2H), 2.97–2.84 (m, 1H), 2.60 (d, J = 18.3 Hz, 1H), 2.42–2.28 (m, 3H), 2.07–1.94 (m, 1H), 1.77 (s, 2H), 1.63–1.50 (m, 2H), 1.20 (dd, J = 16.3, 9.7 Hz, 6H), 1.03–0.92 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 171.3, 171.0, 170.4, 167.8, 142.4, 133.8, 133.6, 132.6, 128.9, 128.6, 127.5, 127.4, 125.2, 118.9, 91.7, 51.5, 46.4, 35.8, 32.0, 31.2, 28.5, 28.2, 26.3, 25.0, 24.7, 22.6.
N1-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-N10-(trityloxy)decanediamide (8d) (white solid, yield 74%): 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 10.13 (s, 1H), 9.75 (s, 1H), 7.82 (d, J = 6.5 Hz, 1H), 7.49 (q, J = 7.5 Hz, 2H), 7.32 (s, 15H), 5.14 (dd, J = 13.2, 5.0 Hz, 1H), 4.45–4.28 (m, 2H), 2.99–2.84 (m, 1H), 2.60 (d, J = 18.4 Hz, 1H), 2.34 (t, J = 7.2 Hz, 3H), 2.07–1.95 (m, 1H), 1.77 (s, 2H), 1.65–1.54 (m, 2H), 1.23 (dd, J = 20.0, 7.1 Hz, 8H), 0.96 (dd, J = 12.2, 6.4 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 171.4, 171.0, 170.4, 167.8, 142.4, 133.8, 133.6, 132.6, 128.9, 128.6, 127.5, 127.4, 125.2, 118.9, 91.7, 51.5, 46.4, 35.8, 32.0, 31.2, 28.6, 28.3, 25.1, 24.7, 22.60, 18.8.
N1-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-N8-(trityloxy)octanediamide (8e) (white solid, yield 62%): 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H), 10.16 (s, 1H), 9.68 (s, 1H), 8.48 (d, J = 8.4 Hz, 1H), 7.83 (t, J = 7.9 Hz, 1H), 7.62 (d, J = 7.3 Hz, 1H), 7.33 (s, 15H), 5.15 (dd, J = 12.6, 5.3 Hz, 1H), 2.97–2.82 (m, 1H), 2.58 (dd, J = 22.7, 10.9 Hz, 2H), 2.41 (t, J = 7.4 Hz, 2H), 2.12–2.03 (m, 1H), 1.78 (t, J = 6.6 Hz, 2H), 1.53 (dt, J = 14.6, 7.4 Hz, 2H), 1.19 (s, 4H), 1.05–0.94 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.7, 172.0, 169.8, 167.7, 166.6, 142.4, 136.5, 136.1, 131.4, 128.9, 127.5, 127.4, 126.2, 118.3, 116.9, 91.5, 48.9, 48.6, 36.5, 30.9, 28.2, 28.0, 24.6, 21.9.
General procedures for the preparation of compounds 9a–9e. To a solution of 8a–8e (200 mg) in TFA (0.3 mL) and DCM (6 mL), triethylsilane (0.15·mL) was added dropwise. The mixture was stirred at room temperature for 30 min. After the reaction completed, the mixture was concentrated in vacuo to create the crude product, which was purified by column chromatography with DCM/ MeOH (20:1–15:1) to produce 9a–9e.
N1-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-N7-hydroxyheptanediamide (9a) (white solid, yield 83%): 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 10.37 (s, 1H), 9.82 (s, 1H), 8.69 (s, 1H), 7.82 (d, J = 6.6 Hz, 1H), 7.58–7.41 (m, 2H), 5.14 (dd, J = 13.2, 4.9 Hz, 1H), 4.37 (q, J = 17.6 Hz, 2H), 3.01–2.82 (m, 1H), 2.61 (d, J = 17.6 Hz, 1H), 2.35 (t, J = 7.3 Hz, 3H), 2.08–1.89 (m, 3H), 1.55 (ddd, J = 29.6, 14.6, 7.3 Hz, 4H), 1.36–1.26 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.9, 171.3, 171.1, 169.1, 167.8, 133.8, 133.7, 132.6, 128.6, 125.2, 118.9, 51.5, 46.5, 35.6, 34.2, 32.1, 31.2, 28.2, 26.3, 24.9, 24.8, 22.6. HRMS (ESI) m/z calculated for C20H24N4O6Na (M + Na)+: 439.1607, found: 439.1588. AlogP: 0.84; H-bond acceptor: 6; H-bond donor: 4; rotatable bonds: 8.
N1-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-N8-hydroxyoctanediamide (9b) (white solid, yield 82%): 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 10.33 (s, 1H), 9.77 (s, 1H), 8.65 (s, 1H), 7.81 (d, J = 6.7 Hz, 1H), 7.59–7.39 (m, 2H), 5.15 (dd, J = 13.2, 5.0 Hz, 1H), 4.37 (q, J = 17.5 Hz, 2H), 3.01–2.82 (m, 1H), 2.61 (d, J = 17.1 Hz, 1H), 2.35 (t, J = 7.3 Hz, 3H), 2.09–1.98 (m, 1H), 1.95 (t, J = 7.3 Hz, 2H), 1.66–1.41 (m, 4H), 1.29 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 171.3, 171.0, 169.0, 167.7, 133.8, 133.7, 132.6, 128.6, 125.2, 118.9, 51.5, 46.5, 35.7, 32.2, 31.2, 28.4, 28.3, 25.0, 24.9, 22.6. HRMS (ESI) m/z calculated for C21H26N4O6Na (M + Na)+: 453.1762, found: 453.1745. AlogP: 1.23; H-bond acceptor: 6; H-bond donor: 4; rotatable bonds: 9.
N1-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-N9-hydroxynonanediamide (9c) (white solid, yield 71%): 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 10.32 (s, 1H), 9.76 (s, 1H), 8.64 (s, 1H), 7.81 (d, J = 6.9 Hz, 1H), 7.57–7.41 (m, 2H), 5.14 (dd, J = 13.3, 5.0 Hz, 1H), 4.36 (q, J = 17.5 Hz, 2H), 3.03–2.82 (m, 1H), 2.61 (d, J = 16.9 Hz, 1H), 2.43–2.28 (m, 3H), 2.09–1.99 (m, 1H), 1.94 (t, J = 7.3 Hz, 2H), 1.51 (dd, J = 29.9, 22.9 Hz, 4H), 1.30 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 171.3, 171.1, 169.1, 167.8, 133.8, 133.7, 132.6, 128.6, 125.2, 118.9, 51.5, 46.5, 35.8, 32.2, 31.2, 28.5, 28.5, 28.4, 25.1, 25.0, 22.6. HRMS (ESI) m/z calculated for C22H28N4O6Na (M + Na)+: 467.1913, found: 467.1901. AlogP: 1.62; H-bond acceptor: 6; H-bond donor: 4; rotatable bonds: 10.
N1-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-N10-hydroxydecanediamide (9d) (white solid, yield 84%): 1H NMR (400 MHz, DMSO-d6) δ 11.00 (s, 1H), 10.31 (s, 1H), 9.75 (s, 1H), 8.63 (s, 1H), 7.81 (d, J = 6.6 Hz, 1H), 7.50 (t, J = 6.7 Hz, 2H), 5.14 (dd, J = 13.2, 4.8 Hz, 1H), 4.36 (q, J = 17.4 Hz, 2H), 3.01–2.84 (m, 1H), 2.61 (d, J = 17.0 Hz, 1H), 2.41–2.29 (m, 3H), 2.09–1.99 (m, 1H), 1.93 (t, J = 7.2 Hz, 2H), 1.63–1.44 (m, 4H), 1.29 (s, 8H). 13C NMR (100 MHz, DMSO-d6) δ 172.9, 171.4, 171.0, 169.1, 167.8, 133.8, 133.7, 132.6, 128.6, 125.2, 118.9, 51.5, 46.4, 35.8, 32.2, 31.2, 28.7, 28.6, 28.5, 25.1, 22.6. HRMS (ESI) m/z calculated for C23H30N4O6Na (M + Na)+: 481.2079, found: 481.2058. AlogP: 2.01; H-bond acceptor: 6; H-bond donor: 4; rotatable bonds: 11.
N1-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-N8-hydroxyoctanediamide (9e) (white solid, yield 84%): 1H NMR (400 MHz, DMSO-d6) δ 11.14 (s, 1H), 10.32 (s, 1H), 9.69 (s, 1H), 8.65 (s, 1H), 8.47 (d, J = 8.3 Hz, 1H), 7.83 (t, J = 7.7 Hz, 1H), 7.61 (d, J = 7.1 Hz, 1H), 5.15 (dd, J = 12.6, 5.1 Hz, 1H), 2.99–2.80 (m, 1H), 2.58 (dd, J = 24.1, 10.9 Hz, 2H), 2.46 (t, J = 7.4 Hz, 2H), 2.11–2.01 (m, 1H), 1.94 (t, J = 7.1 Hz, 2H), 1.67–1.54 (m, 2H), 1.54–1.42 (m, 2H), 1.38–1.19 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.7, 172.0, 169.8, 169.1, 167.7, 166.6, 136.5, 136.1, 131.4, 126.3, 118.3, 117.0, 48.9, 48.6, 36.5, 32.2, 30.9, 28.3, 28.2, 25.0, 24.7, 21.9. HRMS (ESI) m/z calculated for C21H24N4O7Na (M + Na)+: 467.1566, found: 467.1537. AlogP: 0.87; H-bond acceptor: 7; H-bond donor: 4; rotatable bonds: 9.
General procedures for the preparation of compounds 11a–11b. To a solution of 5a or 5b (1.0 eq) in THF, 10 (1.5 eq) was added. The mixture was stirred at reflux for 4 h. After the reaction completed, the mixture was cooled to room temperature, filtered and washed with THF to produce 11a–11b.
4-Nitrophenyl (2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)carbamate (11a) (yellow solid, yield 74%): 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 10.39 (s, 1H), 8.33 (d, J = 9.0 Hz, 2H), 7.81 (dd, J = 6.5, 2.0 Hz, 1H), 7.63–7.48 (m, 4H), 5.16 (dd, J = 13.2, 5.0 Hz, 1H), 4.49 (dd, J = 37.2, 17.7 Hz, 2H), 3.04–2.83 (m, 1H), 2.63 (d, J = 17.0 Hz, 1H), 2.46–2.29 (m, 1H), 2.13–1.96 (m, 1H).
4-Nitrophenyl (2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)carbamate (11b) (white solid, yield 69%): 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 10.28 (s, 1H), 8.17 (d, J = 5.3 Hz, 2H), 7.51 (dd, J = 5.5, 1.7 Hz, 1H), 7.43–7.32 (m, 4H), 5.12 (d, J = 5.0 Hz, 1H), 3.14–2.98 (m, 1H), 2.59 (d, J = 12.0 Hz, 1H), 2.41–2.23 (m, 1H), 2.10–1.95 (m, 1H).
General procedures for the preparation of compounds 13a–13d. To a solution of 11a or 11b (1.0 eq) in DMF, 12a–12c (1.5 eq) was added. The mixture was stirred at room temperature overnight. After the reaction completed, was diluted with H2O and extracted with EtOAc. The combined organic layers were then washed saturated brine, dried over anhydrous Na2SO4, and concentrated in vacuo to create the crude product, which was purified by column chromatography with DCM/ MeOH (15:1–10:1) to produce 13a–13d.
6-(3-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ureido)hexanoic acid (13a) (white solid, yield 74%): 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 8.68 (s, 1H), 8.04 (d, J = 7.9 Hz, 1H), 7.39 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.3 Hz, 1H), 6.74 (s, 1H), 5.13 (dd, J = 13.1, 4.9 Hz, 1H), 4.32 (dd, J = 53.0, 17.1 Hz, 2H), 3.08 (d, J = 5.8 Hz, 2H), 2.99–2.84 (m, 1H), 2.61 (d, J = 16.7 Hz, 1H), 2.32 (dd, J = 21.9, 12.8 Hz, 1H), 2.07 (t, J = 7.1 Hz, 3H), 1.46 (ddd, J = 30.3, 13.0, 5.7 Hz, 4H), 1.33–1.22 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.84 171.1, 168.2, 155.0, 135.8, 132.2, 130.4, 128.6, 121.5, 116.0, 51.5, 46.1, 36.3, 31.2, 29.6, 26.3, 25.1, 22.7.
7-(3-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ureido)heptanoic acid (13b) (white solid, yield 81%): 1H NMR (400 MHz, DMSO-d6) δ 11.03 (s, 1H), 8.38 (s, 1H), 8.01 (t, J = 8.2 Hz, 1H), 7.45–7.38 (m, 1H), 7.32 (d, J = 7.3 Hz, 1H), 6.46 (s, 1H), 5.14 (dd, J = 13.2, 5.1 Hz, 1H), 4.31 (dd, J = 47.4, 17.0 Hz, 2H), 3.09 (dd, J = 12.3, 6.3 Hz, 2H), 2.99–2.86 (m, 1H), 2.61 (d, J = 16.3 Hz, 1H), 2.41–2.26 (m, 1H), 2.15 (t, J = 7.2 Hz, 1H), 2.03 (dd, J = 10.9, 6.0 Hz, 1H), 1.55–1.38 (m, 3H), 1.29 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 172.9, 171.1, 168.1, 154.9, 135.7, 132.2, 130.5, 128.7, 123.1, 121.6, 116.1, 51.5, 45.9, 31.2, 29.5, 28.4, 26.2, 24.8, 22.7.
9-(3-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ureido)nonanoic acid (13c) (white solid, yield 71%): 1H NMR (400 MHz, DMSO-d6) δ 11.95 (s, 1H), 11.03 (s, 1H), 8.27 (s, 1H), 8.03 (d, J = 7.8 Hz, 1H), 7.42 (dd, J = 16.5, 8.8 Hz, 1H), 7.32 (d, J = 7.3 Hz, 1H), 6.36 (s, 1H), 5.15 (dd, J = 13.1, 4.6 Hz, 1H), 4.30 (dd, J = 43.5, 16.9 Hz, 2H), 3.09 (d, J = 5.8 Hz, 2H), 2.93 (t, J = 12.9 Hz, 1H), 2.61 (d, J = 16.7 Hz, 1H), 2.33 (dd, J = 22.3, 12.9 Hz, 1H), 2.18 (t, J = 7.1 Hz, 2H), 2.10–1.94 (m, 1H), 1.46 (dd, J = 16.1, 6.3 Hz, 4H), 1.27 (s, 8H). 13C NMR (100 MHz, DMSO-d6) δ 174.6, 172.8, 171.1, 168.1, 154.8, 135.7, 132.3, 130.5, 128.7, 121.6, 116.1, 51.5, 45.9, 33.7, 31.2, 29.6, 28.7, 28.6, 28.5, 26.3, 24.5, 22.7.
6-(3-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)ureido)hexanoic acid (13d) (white solid, yield 77%): 1H NMR (400 MHz, DMSO-d6) δ 12.00 (s, 1H), 11.15 (s, 1H), 8.78 (s, 1H), 8.61 (d, J = 8.6 Hz, 1H), 7.72 (t, J = 7.7 Hz, 2H), 7.42 (d, J = 7.1 Hz, 1H), 5.12 (dd, J = 12.6, 5.2 Hz, 1H), 3.10 (dd, J = 11.6, 5.9 Hz, 2H), 2.97–2.82 (m, 1H), 2.61 (d, J = 18.1 Hz, 1H), 2.22 (t, J = 7.2 Hz, 2H), 2.14–2.03 (m, 1H), 1.57–1.38 (m, 4H), 2.50–2.38 (m, 4H), 1.32 (dd, J = 14.5, 7.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.4, 172.8, 169.9, 168.3, 166.9, 154.2, 139.1, 135.8, 131.3, 124.1, 115.7, 113.5, 48.8, 33.6, 30.9, 29.0, 25.9, 24.2, 22.1.
General procedures for the preparation of compounds 14a–14d. To a solution of 13a–13d (1.0 eq) in anhydrous DMF, HATU (2.0 eq) and DIEA (3.0 eq) were added. After stirring in an ice bath for 0.5 h, O-tritylhydroxylamine (1.0 eq) was added. The mixture was stirred at room temperature overnight. After the reaction completed, the residue was diluted with H2O and extracted with EtOAc. The combined organic layers were then washed saturated brine, dried over anhydrous Na2SO4, and concentrated in vacuo to create the crude product, which was purified by column chromatography with DCM/ MeOH (30:1–20:1) to produce 14a–14d.
6-(3-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ureido)-N-(trityloxy)hexanamide (14a) (white solid, yield 81%): 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 10.17 (s, 1H), 8.23 (s, 1H), 8.02 (d, J = 7.8 Hz, 1H), 7.50–7.18 (m, 16H), 6.29 (s, 1H), 5.14 (dd, J = 13.0, 4.3 Hz, 1H), 4.30 (dd, J = 41.6, 17.0 Hz, 2H), 2.96 (dd, J = 40.9, 9.1 Hz, 3H), 2.61 (d, J = 16.6 Hz, 1H), 2.32 (dd, J = 25.1, 15.5 Hz, 1H), 2.11–1.95 (m, 1H), 1.79 (s, 2H), 1.26 (dd, J = 26.0, 7.1 Hz, 4H), 1.01 (s, 2H). 13C NMR (100 MHz, DMSO) δ 174.4, 172.8, 169.9, 168.3, 166.9, 154.2, 139.1, 135.8, 131.3, 124.1, 115.7, 113.5, 48.8, 33.6, 30.9, 29.0, 25.9, 24.2, 22.1.
7-(3-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ureido)-N-(trityloxy)heptanamide (14b) (white solid, yield 80%): 1H NMR (400 MHz, DMSO-d6) δ 11.03 (s, 1H), 10.16 (s, 1H), 8.24 (s, 1H), 8.03 (d, J = 7.8 Hz, 1H), 7.44–7.17 (m, 16H), 6.33 (s, 1H), 5.15 (dd, J = 13.1, 4.5 Hz, 1H), 4.30 (dd, J = 42.3, 17.0 Hz, 2H), 3.05 (d, J = 5.7 Hz, 2H), 2.99–2.85 (m, 1H), 2.61 (d, J = 16.8 Hz, 1H), 2.41–2.23 (m, 1H), 2.14–1.95 (m, 1H), 1.78 (s, 2H), 1.40–1.30 (m, 2H), 1.23–1.10 (m, 4H), 0.98 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 171.1, 170.3, 168.1, 154.8, 142.4, 135.6, 132.3, 128.9, 128.7, 127.5, 127.4, 121.7, 116.1, 91.7, 51.5, 45.9, 31.9, 31.2, 29.5, 28.1, 26.1, 24.7, 22.7.
9-(3-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ureido)-N-(trityloxy)nonanamide (14c) (white solid, yield 88%): 1H NMR (400 MHz, DMSO-d6) δ 11.03 (s, 1H), 10.14 (s, 1H), 8.24 (s, 1H), 8.03 (d, J = 8.0 Hz, 1H), 7.47–7.17 (m, 16H), 6.33 (t, J = 5.2 Hz, 1H), 5.15 (dd, J = 13.2, 4.9 Hz, 1H), 4.30 (dd, J = 41.8, 16.9 Hz, 2H), 3.09 (dd, J = 12.3, 6.4 Hz, 2H), 3.00–2.85 (m, 1H), 2.61 (d, J = 15.6 Hz, 1H), 2.32 (dt, J = 13.1, 9.1 Hz, 1H), 2.04 (d, J = 5.8 Hz, 1H), 1.77 (s, 2H), 1.24 (s, 4H), 1.20–1.07 (m, 6H), 0.96 (dd, J = 9.2, 6.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 171.1, 170.5, 168.1, 154.8, 142.4, 135.6, 132.3, 128.9, 128.7, 127.5, 127.4, 121.6, 116.0, 91.8, 51.5, 45.9, 32.0, 31.2, 29.6, 28.7, 28.6, 26.3, 24.7, 22.7.
6-(3-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)ureido)-N-(trityloxy)hexanamide (14d) (white solid, yield 73%): 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H), 10.18 (s, 1H), 8.77 (s, 1H), 8.61 (d, J = 8.6 Hz, 1H), 7.71 (dd, J = 19.2, 11.4 Hz, 2H), 7.42 (d, J = 7.2 Hz, 1H), 7.33 (s, 15H), 5.12 (dd, J = 12.7, 5.3 Hz, 1H), 3.01 (dd, J = 12.0, 6.2 Hz, 2H), 2.96–2.83 (m, 1H), 2.61 (d, J = 18.0 Hz, 1H), 2.51–2.39(m, 1H), 2.15–2.03 (m, 1H), 1.79 (t, J = 6.3 Hz, 2H), 1.35–1.15 (m, 4H), 1.03 (d, J = 6.5 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 170.2, 169.9, 168.3, 166.8, 154.1, 142.4, 139.1, 135.8, 131.3, 128.9, 127.5, 127.4, 124.1, 115.7, 113.5, 91.7, 48.8, 31.9, 30.9, 29.0, 26.3, 25.7, 24.5, 22.1.
General procedures for the preparation of compounds 15a–15d. To a solution of 14a–14d (200 mg) in TFA (0.3 mL) and DCM (6 mL), triethylsilane (0.15 mL) was added dropwise. The mixture was stirred at room temperature for 30 min. After the reaction completed, the mixture was concentrated in vacuo to afford the crude product, which was purified by column chromatography with DCM/ MeOH (20:1–15:1) to produce 15a–15d.
6-(3-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ureido)-N-hydroxyhexanamide (15a) (white solid, yield 75%): 1H NMR (400 MHz, DMSO-d6) δ 10.97 (s, 1H), 10.36 (s, 1H), 8.68 (s, 1H), 8.45 (s, 1H), 8.04 (d, J = 7.8 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H), 7.32 (d, J = 7.3 Hz, 1H), 6.52 (s, 1H), 5.14 (dd, J = 13.0, 4.5 Hz, 1H), 4.31 (dd, J = 48.0, 17.0 Hz, 2H), 3.09 (d, J = 5.4 Hz, 2H), 3.00–2.84 (m, 1H), 2.62 (d, J = 16.8 Hz, 1H), 2.32 (dt, J = 13.3, 10.5 Hz, 1H), 2.09–2.00 (m, 1H), 1.96 (t, J = 7.0 Hz, 2H), 1.60–1.37 (m, 4H), 1.34–1.20 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 171.0, 169.0, 168.1, 154.9, 135.7, 132.2, 130.4, 128.6, 121.5, 116.1, 51.5, 45.9, 38.9, 32.2, 31.2, 29.3, 25.9, 24.9, 22.7. HRMS (ESI) m/z calculated for C20H25N5O6Na (M + Na)+: 454.1712, found: 454.1697. AlogP: 0.63; H-bond acceptor: 6; H-bond donor: 5; rotatable bonds: 8.
7-(3-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ureido)-N-hydroxyheptanamide (15b) (white solid, yield 80%): 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 10.35 (s, 1H), 8.66 (s, 1H), 8.35 (s, 1H), 8.03 (d, J = 7.7 Hz, 1H), 7.41 (t, J = 7.4 Hz, 1H), 7.32 (d, J = 7.0 Hz, 1H), 6.44 (s, 1H), 5.14 (dd, J = 12.9, 4.1 Hz, 1H), 4.31 (dd, J = 46.1, 16.9 Hz, 2H), 3.09 (d, J = 5.0 Hz, 2H), 3.01–2.85 (m, 1H), 2.62 (d, J = 17.2 Hz, 1H), 2.33 (dd, J = 25.8, 15.2 Hz, 1H), 2.10–2.00 (m, 1H), 1.95 (t, J = 6.5 Hz, 2H), 1.46 (d, J = 26.7 Hz, 4H), 1.27 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.9, 171.1, 169.0, 168.1, 154.9, 135.7, 132.3, 130.5, 128.7, 121.6, 116.1, 51.5, 46.0, 32.2, 31.2, 29.5, 28.3, 26.1, 25.1, 22.7. HRMS (ESI) m/z calculated for C21H27N5O6Na (M + Na)+: 468.1870, found: 468.1854. AlogP: 1.03; H-bond acceptor: 6; H-bond donor: 5; rotatable bonds: 9.
9-(3-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ureido)-N-hydroxynonanamide (15c) (white solid, yield 80%): 1H NMR (400 MHz, DMSO-d6) δ 11.03 (s, 1H), 10.49 (s, 1H), 8.68 (s, 1H), 8.32 (s, 1H), 8.03 (d, J = 7.9 Hz, 1H), 7.41 (t, J = 7.5 Hz, 1H), 7.32 (d, J = 7.3 Hz, 1H), 6.42 (s, 1H), 5.14 (d, J = 8.6 Hz, 1H), 4.30 (dd, J = 46.1, 16.9 Hz, 2H), 3.09 (d, J = 4.6 Hz, 2H), 3.01–2.83 (m, 1H), 2.62 (d, J = 16.8 Hz, 1H), 2.33 (dd, J = 24.7, 14.0 Hz, 1H), 2.13–2.02 (m, 1H), 1.95 (t, J = 6.5 Hz, 2H), 1.45 (dd, J = 13.4, 5.6 Hz, 4H), 1.27 (s, 8H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 171.1, 168.1, 154.9, 135.7, 132.3, 130.5, 128.7, 121.6, 116.1, 51.5, 45.9, 32.0, 31.2, 29.6, 28.7, 28.6, 28.5, 26.3, 25.1, 22.7. HRMS (ESI) m/z calculated for C23H31N5O6Na (M + Na)+: 496.2191, found: 496.2167. AlogP: 1.81; H-bond acceptor: 6; H-bond donor: 5; rotatable bonds: 11.
6-(3-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)ureido)-N-hydroxyhexanamide (15d) (white solid, yield 53%): 1H NMR (400 MHz, DMSO-d6) δ 11.14 (s, 1H), 10.37 (s, 1H), 8.78 (s, 1H), 8.60 (d, J = 8.5 Hz, 1H), 7.82–7.65 (m, 2H), 7.41 (d, J = 7.0 Hz, 1H), 5.12 (dd, J = 12.6, 4.5 Hz, 1H), 3.09 (d, J = 4.7 Hz, 2H), 3.02–2.82 (m, 1H), 2.61 (d, J = 18.1 Hz, 1H), 2.20 (d, J = 21.8 Hz, 1H), 2.12–2.02 (m, 1H), 1.96 (t, J = 6.0 Hz, 2H), 1.58–1.38 (m, 4H), 1.34–1.20 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 169.9, 169.0, 168.3, 166.9, 154.2, 139.1, 135.8, 131.3, 124.1, 115.7, 113.5, 48.8, 48.5, 32.2, 30.9, 29.0, 26.0, 24.8, 22.0. HRMS (ESI) m/z calculated for C20H23N5O7Na (M + Na)+: 468.2683, found: 468.2672. AlogP: 0.28; H-bond acceptor: 7; H-bond donor: 5; rotatable bonds: 8.
General procedures for the preparation of compounds 18a–18e. To a solution of 16a or 16b (1.0 eq) in DMF, 17 (1.1 eq) and K2CO3 (2.0 eq) were added. The mixture was stirred at 50 °C overnight. After the reaction completed, the residue was diluted with H2O and extracted with EtOAc. The combined organic layers were then washed saturated brine, dried over anhydrous Na2SO4, and concentrated in vacuo to create the crude product, which was purified by column chromatography with DCM/ MeOH (50:1–45:1) to produce 18a–18b.
Tert-Butyl 4-(((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oxy)methyl)benzoate (18a) (white solid, yield 62%): 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 7.84 (d, J = 8.2 Hz, 2H), 7.35 (dd, J = 13.4, 7.9 Hz, 3H), 7.19 (d, J = 7.4 Hz, 1H), 7.03 (d, J = 7.9 Hz, 1H), 5.29 (dd, J = 13.4, 4.9 Hz, 1H), 4.97–4.85 (m, 2H), 4.27 (dd, J = 76.0, 17.1 Hz, 2H), 3.20–3.01 (m, 1H), 2.82 (d, J = 17.0 Hz, 1H), 2.14–2.05 (m, 1H), 1.54 (s, 9H). 13C NMR (100 MHz, DMSO-d6) δ 171.8, 170.6, 168.3, 164.7, 152.6, 142.3, 133.3, 130.0, 129.5, 129.0, 127.9, 127.2, 118.0, 113.8, 80.6, 52.3, 45.2, 42.6, 31.4, 27.7, 24.9, 21.6.
Tert-Butyl 4-(((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)methyl)benzoate (18b) (white solid, yield 59%): 1H NMR (400 MHz, DMSO-d6) δ 11.12 (s, 1H), 7.95 (d, J = 8.1 Hz, 2H), 7.83 (t, J = 7.9 Hz, 1H), 7.63 (d, J= 8.1 Hz, 2H), 7.57 (d, J = 8.5 Hz, 1H), 7.49 (d, J = 7.2 Hz, 1H), 5.47 (s, 2H), 5.11 (dd, J = 12.9, 5.3 Hz,1H), 2.88 (d, J = 11.8 Hz, 1H), 2.71–2.53 (m, 2H), 2.06 (s, 1H), 1.55 (s, 9H). 13C NMR (100 MHz, DMSO-d6) δ 172.8, 169.9, 166.8, 165.3, 164.7, 155.2, 141.2, 137.0, 133.3, 130.8, 129.2, 126.9, 120.2, 116.7, 115.7, 80.7, 69.4, 48.8, 30.9, 27.8, 22.0.
General procedures for the preparation of compounds 21a–21b. To a solution of 19a or 19b (1.0 eq) in anhydrous DMF, HATU (2.0 eq) and DIEA (3.0 eq) were added. After stirring in an ice bath for 0.5 h, 20 (1.0 eq) was added. The mixture was stirred at room temperature overnight. After the reaction completed, the residue was diluted with H2O and extracted with EtOAc. The combined organic layers were then washed saturated brine, dried over anhydrous Na2SO4, and concentrated in vacuo to create the crude product, which was purified by column chromatography with DCM/ MeOH (30:1–20:1) to produce 21a–21b.
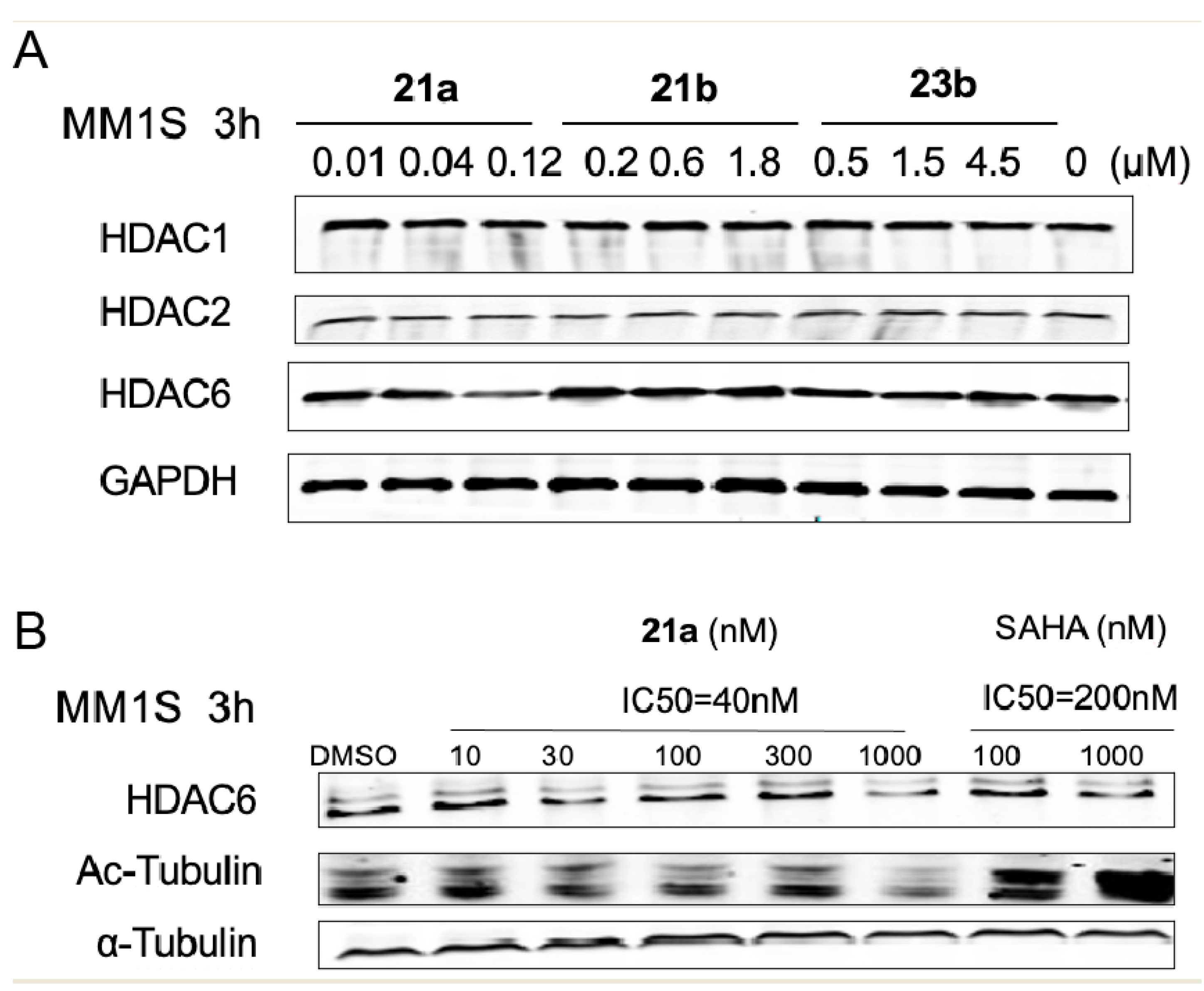
N-(2-Aminophenyl)-4-(((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oxy)methyl)benzamide (21a) (white solid, yield 68%): 1H NMR (400 MHz, DMSO-d6) δ 10.98 (s, 1H), 9.66 (s, 1H), 8.00 (d, J = 7.7 Hz, 2H), 7.62 (d, J = 7.8 Hz, 2H), 7.49 (t, J = 7.7 Hz, 1H), 7.33 (t, J = 7.3 Hz, 2H), 7.17 (d, J = 7.5 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 6.78 (d, J = 7.8 Hz, 1H), 6.60 (t, J = 7.3 Hz, 1H), 5.36 (s, 2H), 5.12 (dd, J = 13.1, 4.7 Hz, 1H), 4.90 (s, 2H), 4.39 (dd, J = 63.0, 17.4 Hz, 2H), 2.91 (dd, J = 21.6, 9.3 Hz, 1H), 2.59 (d, J = 17.4 Hz, 1H), 2.51–2.40(m, 1H), 2.08–1.92 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 172.9, 171.0, 168.0, 165.0, 153.3, 143.1, 140.0, 134.2, 133.4, 130.0, 129.8, 128.0, 127.2, 126.7, 126.5, 123.2, 116.2, 116.1, 115.4, 115.1, 68.9, 51.6, 45.1, 31.2, 22.4. HRMS (ESI) m/z calculated for C27H24N4O5Na (M + Na)+: 507.1660, found: 507.1639. AlogP: 2.86; H-bond acceptor: 6; H-bond donor: 3; rotatable bonds: 6.
N-(2-Aminophenyl)-4-(((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)methyl)benzamide (21b) (white solid, yield 73%): 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 9.67 (s, 1H), 8.02 (d, J = 7.9 Hz, 2H), 7.84 (t, J = 7.9 Hz, 1H), 7.62 (dd, J = 14.5, 8.3 Hz, 3H), 7.49 (d, J = 7.2 Hz, 1H), 7.18 (d, J = 7.6 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.79 (d, J = 7.9 Hz, 1H), 6.61 (t, J = 7.5 Hz, 1H), 5.48 (s, 2H), 5.11 (dd, J = 12.9, 5.3 Hz, 1H), 4.96 (s, 1H), 2.98–2.82 (m, 1H), 2.58 (dd, J = 19.5, 10.4 Hz, 1H), 2.11–2.01 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 172., 169.9, 166.8, 165.3, 165.0, 155.3, 143.1, 139.5, 137.0, 134.2, 133.3, 128.0, 126.9, 126.7, 126.5, 123.2, 120.2, 116.7, 116.2, 116.1, 115.7, 69.5, 53.6, 48.8, 30.9, 22.0, 18.0. HRMS (ESI) m/z calculated for C27H22N4O6Na (M + Na)+: 521.1443, found: 521.1432. AlogP: 2.50; H-bond acceptor: 7; H-bond donor: 3; rotatable bonds: 6.
General procedures for the preparation of compounds 22a–22b. To a solution of 19a or 19b (1.0 eq) in anhydrous DMF, HATU (2.0 eq) and DIEA (3.0 eq) were added. After stirring in an ice bath for 0.5 h, O-tritylhydroxylamine (1.0 eq) was added. The mixture was stirred at room temperature overnight. After the reaction completed, the residue was diluted with H2O and extracted with EtOAc. The combined organic layers were then washed with saturated brine, dried over anhydrous Na2SO4, and concentrated in vacuo to create the crude product, which was purified by column chromatography with DCM/ MeOH (50:1–40:1) to produce 22a–22b.
4-(((2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oxy)methyl)-N-(trityloxy)benzamide (22a) (white solid, yield 77%): 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.24 (s, 1H), 7.49 (d, J = 5.9 Hz, 6H), 7.37–7.13 (m, 15H), 6.96 (d, J = 7.9 Hz, 1H), 5.07 (dd, J = 13.3, 4.8 Hz, 1H), 4.87–4.76 (m, 2H), 4.30–4.16 (m, 2H), 3.08–2.65 (m, 2H), 2.32–2.10 (m, 1H), 2.07 (dd, J = 5.9, 4.1 Hz, 1H).
4-(((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)methyl)-N-(trityloxy)benzamide (22b) (white solid, yield 72%): 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 7.98 (s, 1H), 7.62 (t, J = 7.8 Hz, 2H), 7.51 (s, 4H), 7.43–7.28 (m, 15H), 7.15 (d, J = 8.4 Hz, 1H), 5.28 (s, 2H), 4.96 (dd, J = 12.0, 5.2 Hz, 1H), 2.80 (d, J = 11.0 Hz, 3H), 2.12 (dt, J = 10.4, 6.3 Hz, 1H).
General procedures for the preparation of compounds 23a–23b. To a solution of 22a or 22b (200 mg) in TFA (0.3 mL) and DCM (6 mL), triethylsilane (0.15 mL) was added dropwise. The mixture was stirred at room temperature for 30 min. After the reaction completed, the mixture was concentrated in vacuo to create the crude product, which was purified by column chromatography with DCM/ MeOH (20:1–15:1) to produce 23a–23b.
4-(((2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oxy)methyl)-N-hydroxybenzamide (23a) (white solid, yield 67%): 1H NMR (400 MHz, DMSO-d6) δ 11.17 (s, 1H), 10.13 (s, 1H), 9.00 (s, 1H), 7.68 (d, J = 8.2 Hz, 2H), 7.33 (dd, J = 14.0, 7.8 Hz, 2H), 7.19 (d, J = 7.3 Hz, 1H), 7.02 (d, J = 7.9 Hz, 1H), 5.29 (dd, J = 13.3, 4.9 Hz, 1H), 4.96–4.78 (m, 2H), 4.27 (dd, J = 74.7, 17.1 Hz, 2H), 3.20–3.05 (m, 1H), 2.82 (d, J = 17.5 Hz, 1H),2.50–2.42(m, 1H), 2.14–2.03 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.9, 170.7, 168.3, 164.0, 152.6, 140.3, 133.3, 131.5, 129.5, 128.0, 127.0, 126.9, 118.0, 113.8, 54.9, 52.3, 45.3, 42.6, 31.4, 21.7. HRMS (ESI) m/z calculated for C21H19N3O6Na (M + Na)+: 432.1178, found: 432.1166. AlogP: 1.15; H-bond acceptor: 6; H-bond donor: 3; rotatable bonds: 5.
4-(((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)methyl)-N-hydroxybenzamide (23b) (white solid, yield 71%): 1H NMR (400 MHz, DMSO-d6) δ 11.24 (s, 1H), 11.12 (s, 1H), 9.06 (s, 1H), 7.92–7.75 (m, 3H), 7.58 (d, J = 7.6 Hz, 3H), 7.49 (d, J = 7.0 Hz, 1H), 5.43 (s, 2H), 5.11 (dd, J = 12.8, 5.0 Hz, 1H), 2.96–2.83 (m, 1H), 2.67–2.53 (m, 2H), 2.11–1.98 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 172.7, 169.9, 166.7, 165.3, 163.8, 155.3, 139.3, 137.0, 133.2, 132.3, 127.0, 126.9, 120.1, 116.6, 115.6, 69.4, 48.7, 30.9, 21.9. HRMS (ESI) m/z calculated for C21H17N3O7Na (M + Na)+: 446.2820, found: 446.2801. AlogP: 0.79; H-bond acceptor: 7; H-bond donor: 3; rotatable bonds: 5.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



