
Fractionation of Lycopodiaceae Alkaloids and Evaluation of Their Anticholinesterase and Cytotoxic Activities

 , ,
, ,  and
and 
Abstract

1. Introduction
2. Results and Discussion
2.1. Optimization of Alkaloid Fractionation and Isolation
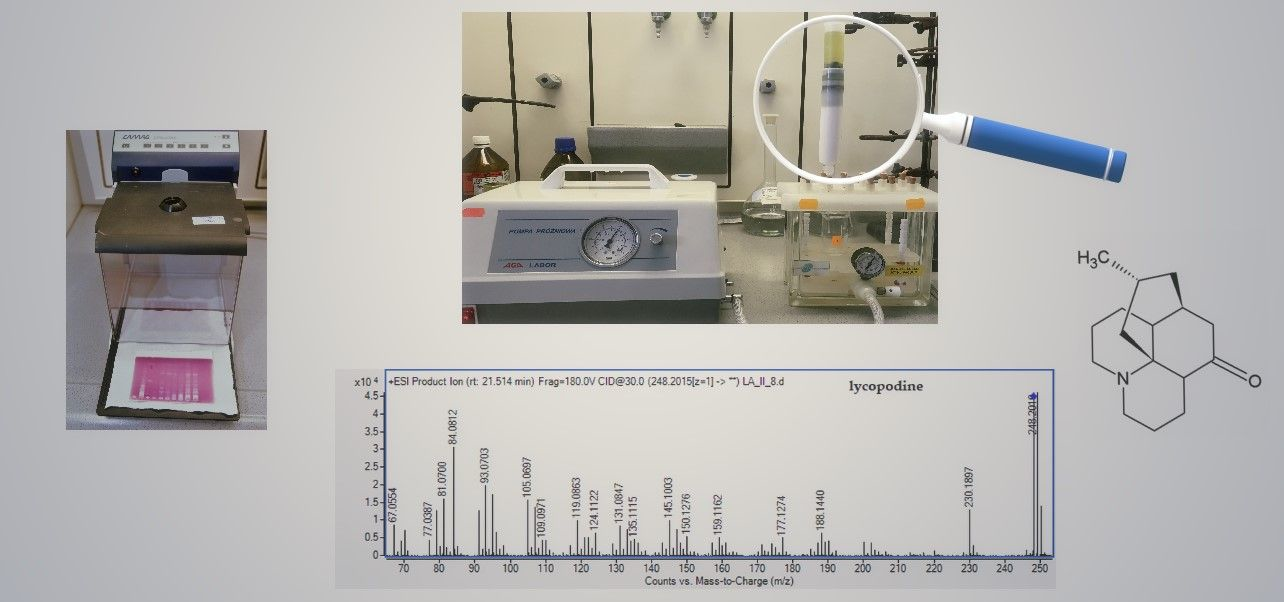
2.2. LC-MS Identification of the Isolated Compounds
2.3. Anticholinesterase Assay
2.4. Cytotoxicity Assay
3. Materials and Methods
3.1. Plant Materials
3.2. Plant Materials Extraction
3.3. Sample Preparation and Purificatoin by VLC
3.4. HPLC/ESI-QTOF-MS Analysis of the Obtained Fractions
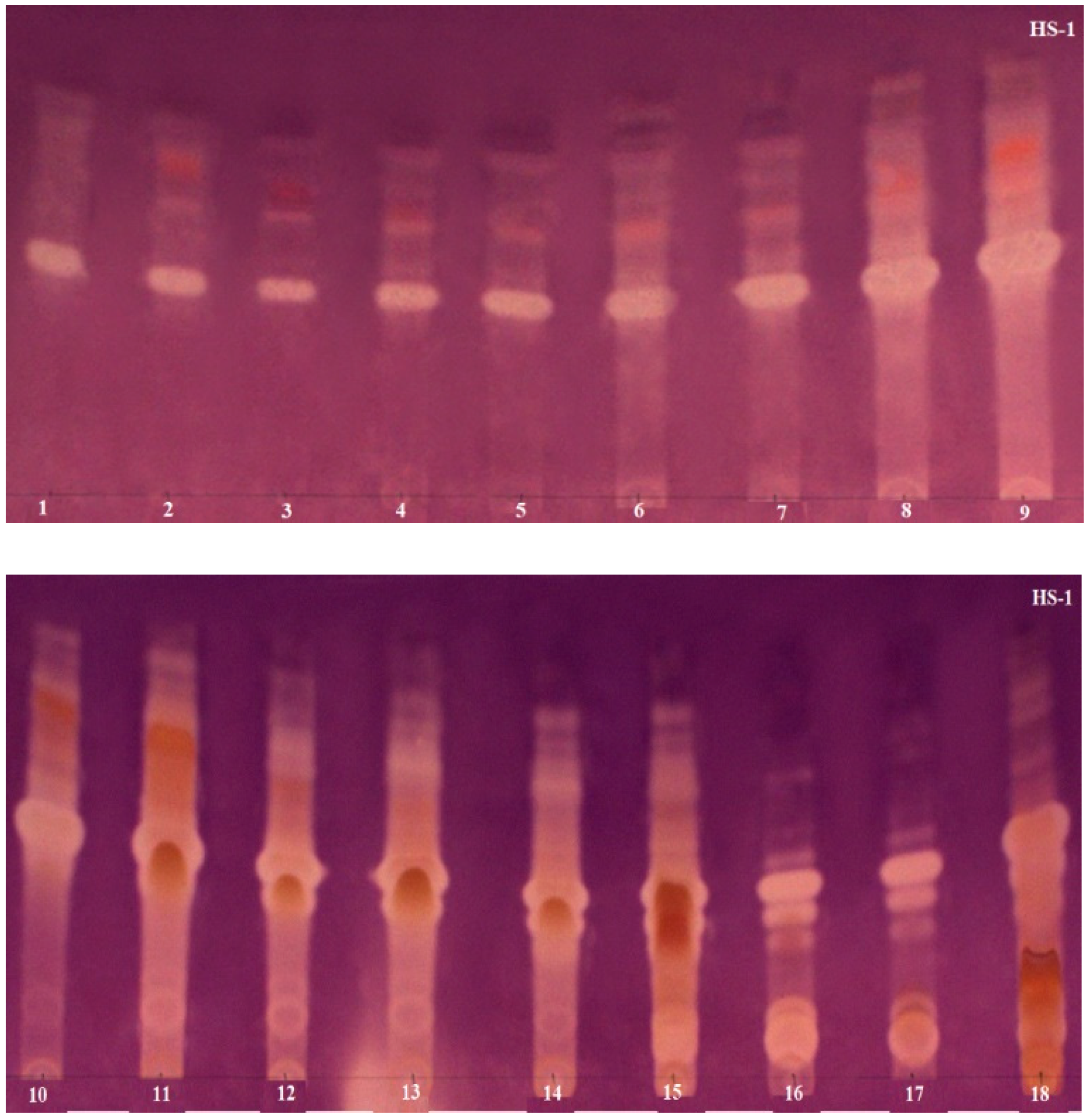
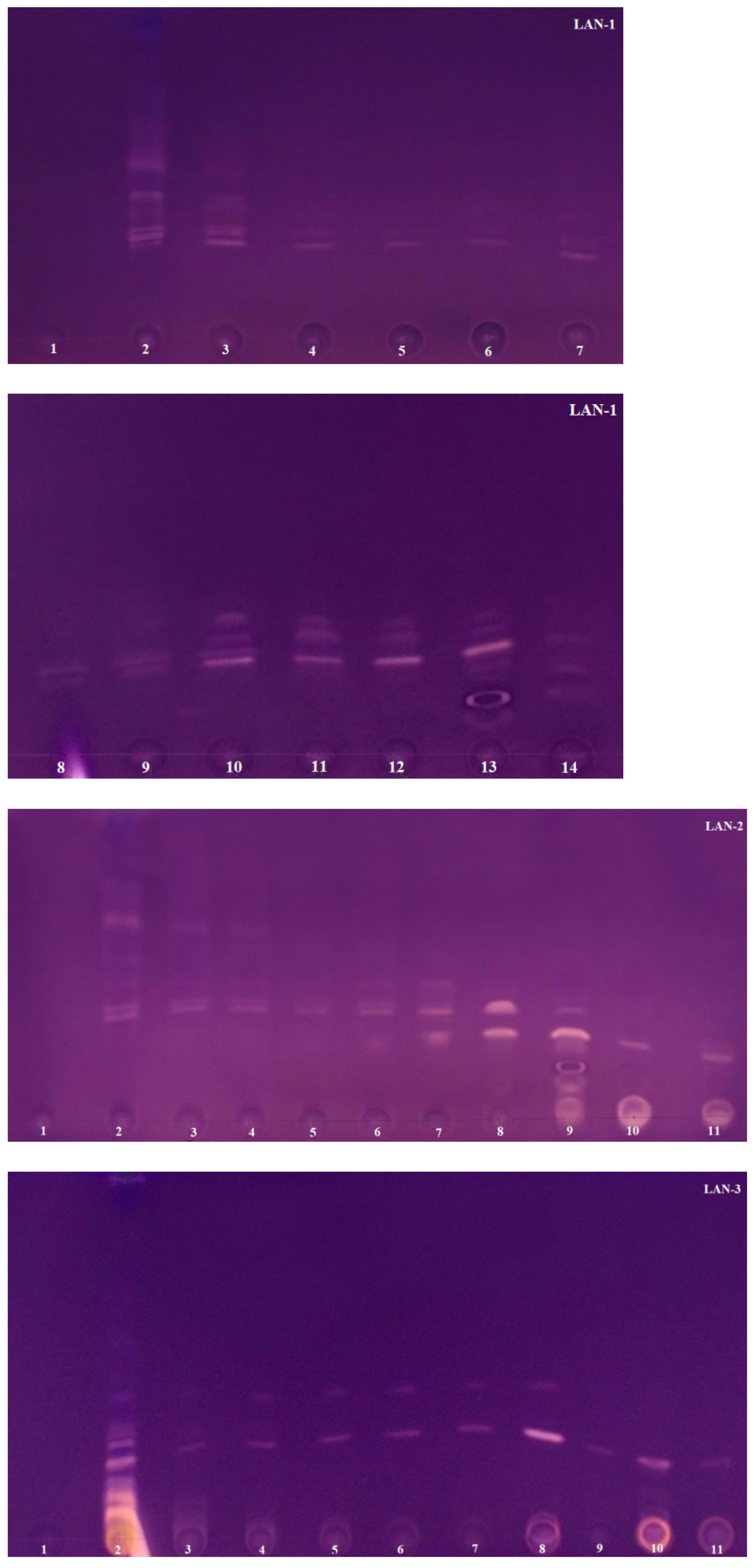
3.5. TLC Bioautography towards AChE Inhibitors
3.6. Cytotoxicity against Human Skin Fibroblasts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Shan, S.-M.; Luo, J.-G.; Pan, K.; Zou, H.-Y.; Kong, L.-Y. Rapid screening and identification of lycodine-type alkaloids in Lycopodiaceae and Huperziaceae plants by ultra-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. Biomed. Chromatogr. 2016, 30, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-C.; Zhang, Z.-J.; Su, J.; Peng, L.-Y.; Pan, L.-T.; Wu, X.-D.; Zhao, Q.-S. Lycodine-type Lycopodium alkaloids from the whole plants of Huperzia serrata. Nat. Prod. Bioprospect. 2017, 7, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Gang, D.R. The Lycopodium alkaloids. Nat. Prod. Rep. 2004, 21, 752–772. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.-Q.; Hu, G.-W.; Guo, M.-Q. Components and anti-HepG2 activity comparison of Lycopodium alkaloids from four geographic origins. Evid. Based Complement. Alternat. Med. 2016, 2016, 1–9. [Google Scholar] [CrossRef]
- Li, X.; Kang, M.; Ma, N.; Pang, T.; Zhang, Y.; Jin, H.; Yang, Z.; Song, L. Identification and analysis of chemical constituents and rat serum metabolites in Lycopodium clavatum using UPLC-Q-TOF/MS combined with multiple data-processing approaches. Evid. Based Complement. Alternat. Med. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Chen, Y.; Qian, Y.; Zhang, Y. Lycopodium japonicum: A comprehensive review on its phytochemicals and biological activities Arab. J. Chem. 2020, 13, 5438–5450. [Google Scholar] [CrossRef]
- Ma, X.; Tan, C.; Zhu, D.; Gang, D.R.; Xiao, P. Huperzine A from Huperzia species-an ethnopharmacolgical review. J. Ethnopharmacol. 2007, 113, 15–34. [Google Scholar] [CrossRef]
- Banerjee, J.; Biswas, S.; Madhu, N.R.; Karmakar, S.R.; Biswas, S.J. A better understanding of pharmacological activities and uses of phytochemicals of Lycopodium clavatum: A review. J. Pharmacogn. Phytochem. 2014, 3, 207–210. [Google Scholar]
- Cummings, J.L.; Vinters, H.V.; Cole, G.M.; Khachaturian, Z.S. Alzheimer’s disease: Etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology 1998, 51, 2–17. [Google Scholar] [CrossRef]
- Szypuła, W.J.; Mistrzak, P.; Olszowska, O. A new and fast method to obtain in vitro cultures of Huperzia selago (Huperziaceae) sporophytes, a club moss which is a source of huperzine A. Acta Soc. Bot. Pol. 2013, 82, 313–320. [Google Scholar] [CrossRef][Green Version]
- Konrath, E.L.; Neves, B.M.; Lunardi, P.S.; Passos, C.S.; Simões-Pires, A.; Ortega, M.G.; Gonçalves, C.A.; Cabrera, J.L.; Moreira, J.C.F.; Henriques, A.T. Investigation of the in vitro and ex vivo acetylcholinesterase and antioxidant activities of traditionally used Lycopodium species from South America on alkaloid extracts. J. Ethnopharmacol. 2012, 139, 58–67. [Google Scholar] [CrossRef]
- Konrath, E.L.; Neves, B.M.; Passos, C.S.; Lunardi, P.S.; Ortega, M.G.; Cabrera, J.L.; Gonçalves, C.A.; Henriques, A.T. Huperzia quadrifariata and Huperzia reflexa alkaloids inhibit acetylcholinesterase activity in vivo in mice brain. Phytomedicine 2012, 19, 1321–1324. [Google Scholar] [CrossRef][Green Version]
- Tang, Y.; Li, N.; Zou, Y.; Ai, Y.; Ma, G.-L.; Osman, E.E.A.; Xiong, J.; Li, J.; Jin, Z.-X.; Hu, J.-F. LC-MS guided isolation and dereplication of Lycopodium alkaloids from Lycopodium cernuum var. sikkimense of different geographical origins. Phytochemistry 2019, 160, 25–30. [Google Scholar] [CrossRef]
- MacLean, D.B. Lycopodium alkaloids XIII. Mass spectra of representative alkaloids. Can. J. Chem. 1963, 41, 2654–2670. [Google Scholar] [CrossRef]
- Manske, R.H.; Marion, L. The alkaloids of Lycopodium species; Lycopodium annotinum var. acrifolium, Fern. and the structure of annotinine. J. Am. Chem. Soc. 1947, 9, 2126–2129. [Google Scholar] [CrossRef]
- Burnell, R.H.; Taylor, D.R. Lycopodium alkaloids—VII: Lycofoline and related bases. Tetrahedron 1962, 18, 1467–1469. [Google Scholar] [CrossRef]
- Ayer, W.A. The Lycopodium alkaloids. Nat. Prod. Rep. 1991, 8, 455–463. [Google Scholar] [CrossRef]
- Siengalewicz, P.; Mulzer, J.; Rinner, U. Lycopodium alkaloids—Synthetic highlights and recent developments. Alkaloids Chem. Biol. 2013, 72, 1–151. [Google Scholar] [CrossRef]
- Halldorsdottir, E.S.; Jaroszewski, J.W.; Olafsdottir, E.S. Acetylcholinesterase inhibitory activity of lycopodane-type alkaloids from Icelandic Lycopodium annotinum spp. alpestre. Phytochemistry 2010, 71, 149–157. [Google Scholar] [CrossRef]
- Ortega, M.G.; Agnese, A.M.; Cabrera, J.L. Anticholinesterase activity in an alkaloid extract of Huperzia saururus. Phytomedicine 2004, 11, 539–543. [Google Scholar] [CrossRef]
- Mroczek, T.; Mazurek, J. Pressurized liquid extraction and anticholinesterase activity-based thin-layer chromatography with bioautography of Amaryllidaceae alkaloids. Anal. Chim. Acta 2009, 633, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Mroczek, T. Qualitative and quantitative two-dimensional thin-layer chromatography/high performance liquid chromatography/diode-array/electrospray-ionization-time-of-flight mass spectrometry of cholinesterase inhibitors. J. Pharm. Biomed. Anal. 2016, 129, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Mroczek, T.; Dymek, A.; Widelski, J.; Wojtanowski, K.K. The bioassay-guided fractionation and identification of potent acetylcholinesterase inhibitors form Narcissus c.v. ‘Hawera’ using optimized vacuum liquid chromatography, high resolution mass spectrometry and bioautography. Metabolites 2020, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Szypuła, W.J.; Pietrosiuk, A. Production of Cholinesterase-Inhibiting Compounds in In Vitro Cultures of Club Mosses. In Plant Cell and Tissue Differentiation and Secondary Metabolites; Ramawat, K.G., Ekiert, H.M., Goyal, S., Eds.; Springer: Cham, Switzerland, 2020; pp. 1–40. [Google Scholar] [CrossRef]
- Szypuła, W.J.; Wilenska, B.; Misicka, A.; Pietrosiuk, A. Huperzine A and Huperzine B Production by Prothallus Cultures of Huperzia selago (L.) Bernh. ex Schrank et Mart. Molecules 2020, 25, 3262. [Google Scholar] [CrossRef] [PubMed]
- Dymek, A.; Widelski, J.; Wojtanowski, K.K.; Płoszaj, P.; Zhuravchak, R.; Mroczek, T. Optimization of Pressurized Liquid Extraction of Lycopodiaceae Alkaloids Obtained from Two Lycopodium Species. Molecules 2021, 26, 1626. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Dong, L.B.; Zhang, Z.J.; Fan, M.; Zhu, Q.F.; Qi, Y.Y.; Liu, Y.C.; Peng, L.Y.; Wu, X.D.; Zhao, Q.S. Three new Lycopodium alkaloids from Lycopodium japonicum. J. Asian Nat. Prod. Res. 2019, 21, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.-T.; Tan, C.-H.; Ma, X.-Q.; Jiang, S.-H.; Zhu, D.-Y. Lycopodium alkaloids from Huperzia Miyoshiana. Nat. Prod. Res. 2003, 15, 383–386. [Google Scholar]
- Liu, Y.; Li, J.; Li, D.; Li, X.-M.; Li, D.; Zhou, G.; Xu, K.P.; Kang, F.-H.; Zou, Z.-X.; Xu, P.-S.; et al. Anti-cholinesterase activities of constituents isolated from Lycopodiastrum casuarinoides. Fitoterapia 2019, 139, 104366. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, H.-W.; Mei, Z.-N.; Yang, G.-Z. Lycopodium alkaloids from Lycopodium obscurum L. Helv. Chim. Acta 2014, 97, 519–523. [Google Scholar] [CrossRef]
- He, J.; Wu, X.-D.; Liu, F.; Liu, Y.-C.; Peng, L.-Y.; Zhao, Y.; Cheng, X.; Luo, H.-R.; Zhao, Q.-S. Lycopodine-Type Alkaloids from Lycopodium japonicum. Nat. Prod. Bioprospect. 2014, 4, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Pongpamorn, P.; Wan-erlor, S.; Ruchirawat, S.; Thasana, N. Lycoclavatumide and 8β,11α-dihydroxylycopodine, a new fawcettimine and lycopodine-type alkaloid from Lycopodium clavatum. Tetrahedron 2016, 72, 7065–7069. [Google Scholar] [CrossRef]
- Wang, X.-J.; Li, L.; Si, Y.-K.; Yu, S.-S.; Ma, S.-G.; Bao, X.-Q.; Zhang, D.; Qu, J.; Liu, Y.-B.; Li, Y. Nine new lycopodine-type alkaloids from Lycopodium japonicum Thunb. Tetrahedron 2013, 69, 6234–6240. [Google Scholar] [CrossRef]
- Orhan, I.; Kupeli, E.; Sener, B.; Yesilada, E. Appraisal of anti-inflammatory potential of the clubmoss, Lycopodium clavatum L. J. Ethnopharmacol. 2007, 109, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Li, L.; Yu, S.-S.; Ma, S.-G.; Qu, J.; Liu, Y.B.; Li, Y.; Wang, Y.; Tang, W. Five new fawcettimine-related alkaloids from Lycopodium japonicum Thunb. Fitoterapia 2013, 91, 74–81. [Google Scholar] [CrossRef]
- Zhang, H.; Liang, H.; Kuang, P.; Yuan, Q.; Wang, Y. Simultaneously preparative purification of Huperzine A and Huperzine B from Huperzia serrata by macroporous resin and preparative high performance liquid chromatography. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 904, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Marston, A.; Kissling, J.; Hostettmann, K. A rapid TLC bioautographic method for the detection of acetylcholinesterase and butyrylcholinesterase inhibitors in plants. Phytochem. Anal. 2002, 13, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Borloz, A.; Marston, A.; Hostettmann, K. The determination of huperzine A in European Lycopodiaceae species by HPLC-UV-MS. Phytochem. Anal. 2006, 17, 332–336. [Google Scholar] [CrossRef]
- Thorroad, S.; Worawittayanont, P.; Khunnawutmanotham, N.; Chimnoi, N.; Jumruksa, A.; Ruchirawat, S.; Thasana, N. Three new Lycopodium alkaloids from Huperzia carinata and Huperzia squarrosa. Tetrahedron 2014, 70, 8017–8022. [Google Scholar] [CrossRef]
- Ho, R.; Marsousi, N.; Eugster, P.; Bianchini, J.-P.; Raharivelomanana, P. Detection by UPLC/ESI-TOF-MS of alkaloids in three Lycopodiaceae species from French Polynesia and their anticholinesterase activity. Nat. Prod. Commun. 2009, 4, 1349–1352. [Google Scholar] [CrossRef]
- Orhan, I.; Terzioglu, S.; Sener, B. Aplha-onocerin: An acetylcholinesterase inhibitor from Lycopodium clavatum. Planta Med. 2003, 69, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Calderon, A.I.; Simithy-Williams, J.; Sanchez, R.; Espinosa, A.; Valdespino, I.; Gupta, M.P. Lycopodiaceae from Panama: A new source of acetylcholinesterase inhibitors. Nat. Prod. Res. 2013, 27, 500–505. [Google Scholar] [CrossRef]
- Mroczek, T. Highly Efficient, selective and sensitive molecular screening of acetylcholinesterase inhibitors of natural origin by solid-phase extraction-liquid chromatography/electrospray ionisation-octopole-orthogonal acceleration time-of-flight-mass spectrometry and novel thin-layer chromatography-based bioautography. J. Chromatogr. A 2009, 1216, 2519–2528. [Google Scholar] [CrossRef]
- Tori, M.; Shimoji, T.; Takaoka, S.; Nakashima, K.; Sono, M.; Ayer, W.A. Structure of oxolucidine A, a lycopodium alkaloid. Tetrahedron. Lett. 1999, 40, 323–324. [Google Scholar] [CrossRef]
- Tori, M.; Shimoji, T.; Shimura, S.E.; Takaoka, S.; Nakashima, K.; Sono, M.; Ayer, W.A. Four alkaloids, lucidine B, oxolucidine A, lucidine A, and lucidulinone from Lycopodium lucidulum. Phytochemistry 2000, 53, 503–509. [Google Scholar] [CrossRef]
- Rollinger, J.M.; Ewelt, J.; Seger, C.; Sturm, S.; Ellmerer, E.P.; Stuppner, H. New insights into the acetylcholinesterase inhibitory activity of Lycopodium clavatum. Planta Med. 2005, 71, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Przekora, A.; Czechowska, J.; Pijocha, D.; Ślósarczyk, A.; Ginalska, G. Do novel cement-type biomaterials reveal ion reactivity that affects cell viability in vitro? Cent. Eur. J. Biol. 2014, 9, 277–289. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Number of Experiments | 1 | 2 | 3 | 4 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Solvent gradient system used | 95:5:0.2 (v/v/v) chloroform: methanol: 25% aqueous ammonia solution | 60% aqueous methanol solution with 2 drops of 10% aqueous tartaric acid 85% aqueous methanol solution with 2 drops of 10% aqueous tartaric acid | |||||||
| Sorbent filling ratio | 1:3 Al2O3 (17 g):silica gel (25 g) | 3:1 Al2O3 (57 g):silica gel (8 g) | silica gel 60 H silanized (26 g) | silica gel 60 H silanized (26 g) | |||||
| Plant materials | LCL | LAN | HS | LCL | LAN | HS | LCL | LAN | LCL |
| Number of fractions obtained | 6 | 14 | 18 | 10 | 11 | 16 | 7 | 11 | 11 |
| Isolated Compounds | Fraction Number | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |
| Acrifoline | - | - | - | - | - | 4.5 | - | - | 12.6 | 16.0 | 20.7 | 22.7 | 21.6 | 6.4 |
| Lycopodine | - | - | - | - | - | 22.1 | 28.6 | 22.9 | 16.8 | 23.5 | 17.3 | 21.0 | 8.1 | - |
| Annotinine | - | 85.7 | 52.6 | 9.8 | - | - | - | - | - | - | - | - | - | - |
| Dihydroannotine | - | - | - | - | - | - | - | - | - | 15.0 | 26.0 | 29.1 | 12.5 | - |
| Lycodine | - | 1.6 | 14.6 | 28.5 | 32.2 | 25.9 | 28.4 | 7.0 | 3.1 | 0.7 | - | - | - | - |
| Lyconnotine | - | - | - | 6.1 | 7.8 | - | - | - | - | - | - | - | 41.9 | - |
| Annotine N-oxide | - | - | 16.8 | 41.3 | 14.3 | 14.6 | 12.4 | - | - | - | - | - | - | 22.3 |
| Huperzinine | - | - | - | 2.1 | 1.7 | 1.6 | 1.8 | - | - | - | - | - | - | - |
| Lyconadine A | - | - | - | 2.3 | 1.9 | 2.5 | 2.8 | 1.2 | 0.9 | - | - | - | - | - |
| Lycopodine N-oxide | - | - | - | - | - | 59.8 | 56.8 | 38.5 | 31.9 | 23.5 | 3.1 | |||
| Acetylfawcettiine | - | - | - | - | - | - | - | - | - | - | - | - | - | 4.8 |
| 8-β,11-α Aihydroxylycopodine | - | - | - | - | - | - | - | - | - | - | - | - | - | 43.4 |
| Des-N-methyl-α-obscurine | - | - | - | - | - | - | - | - | - | - | - | - | 4.5 | 5.5 |
| % SUM | - | 87.3 | 84.0 | 90.2 | 57.9 | 71.1 | 73.8 | 90.9 | 90.3 | 93.7 | 95.8 | 96.2 | 88.5 | 85.4 |
| Isolated Compounds | Fraction Number | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
| Acrifoline | - | - | 1.9 | - | - | 2.0 | 17.4 | 25.1 | 18.9 | 14.5 | 3.2 |
| Lycopodine | - | - | 14.5 | - | 12.5 | 73.9 | 41.1 | 12.0 | - | - | - |
| Annotinine | 85.2 | 89.0 | 71.3 | 87.9 | 61.7 | - | - | - | - | 9.2 | 5.6 |
| Fawcettimine | - | - | - | - | - | - | - | 7.2 | 17.5 | 8.3 | - |
| Annotine | - | - | - | - | - | - | - | 14.4 | - | - | - |
| Dihydroannotine | - | - | - | - | - | - | - | 8.1 | 16.6 | - | 4.5 |
| Dihydroannotinol | - | - | - | - | - | - | - | 6.5 | 11.6 | - | 4.7 |
| N-Methyl dihydrolycodine | - | - | - | - | - | - | - | 6.9 | 6.9 | 4.7 | 8.0 |
| Lycodine | 1.4 | 1.4 | 3.4 | 1.2 | 3.6 | 5.8 | 3.5 | - | - | - | - |
| Dihydrolycopodine | - | - | - | - | - | - | - | - | - | 18.1 | 7.0 |
| Deacetyllycofawcine | - | - | - | - | - | - | - | - | - | - | 3.2 |
| Lycopodine N-oxide | - | - | - | - | 4.6 | 5.8 | 28.0 | 9.1 | 18.4 | - | - |
| Deacetylfawcettiine | - | - | - | - | - | - | - | - | - | 9.2 | 26.3 |
| α-Obscurine | - | - | - | - | - | - | - | - | 1.5 | 4.1 | 2.2 |
| Lyconnotine | 8.2 | 2.6 | 3.8 | 6.2 | 4.7 | 6.1 | 8.4 | 7.2 | 7.9 | - | |
| Unidentified 474.3794 C26H52NO6 | - | - | - | - | - | - | - | - | - | 22.7 | - |
| Unidentified 459.2649 C29H35N2O3 | - | - | 4.7 | 6.1 | 6.2 | 6.4 | 2.1 | - | - | - | - |
| % SUM | 86.6 | 98.6 | 98.4 | 99.0 | 94.9 | 98.5 | 98.1 | 97.6 | 98.5 | 98.8 | 64.6 |
| Isolated Compounds | Fraction Number | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
| Acrifoline | - | 16.2 | 17.8 | 21.6 | 28.2 | 23.5 | 28.3 | 63.6 | 30.4 | - | 12.4 |
| Lycopodine | 0.9 | 15.8 | - | - | - | - | - | - | - | - | - |
| Annotinine | - | 27.4 | 43.4 | 34.3 | 30.2 | 36.6 | 31.1 | - | - | - | - |
| Annotine | - | - | - | - | - | 1.99? | 1.9 | - | - | - | - |
| Dihydroannotine | - | - | - | - | - | - | - | - | 7.6 | 18.8 | 11.8 |
| Fawcettimine | - | 9.9 | 8.7 | 13.7 | 11.4 | 13.5 | 15.8 | 13.7 | 34.1 | 11.5 | 6.7 |
| β-Obscurine | - | - | 1.4 | 2.3 | 1.1 | 1.3 | 1.7 | 1.2 | - | - | 2.8 |
| Lycopodine N-oxide | - | - | - | - | - | - | - | 4.1 | 7.4 | 9.2 | - |
| Lycodine | - | 3.0 | 1.5 | 1.6 | - | - | - | - | - | - | - |
| Deacetylfawcettiine | 14.0 | - | - | - | - | - | - | 5.3 | 1.8 | 14.9 | 4.5 |
| Lyconnotine | - | 15.8 | 19.3 | 18.5 | 21.0 | 16.1 | 11.6 | 3.2 | - | - | - |
| Huperzinine | - | - | - | - | - | - | - | - | 2.5 | 3.9 | 7.4 |
| N-Methyl lycodine | - | - | - | - | - | - | - | - | - | 5.5 | 14.1 |
| Flabelline | - | 3.5 | 2.3 | 4.2 | 2.1 | 2.0 | 1.5 | - | - | - | - |
| Unidentified 474.3794 C26H52NO6 | 12.6 | - | - | - | - | - | - | 4.6 | 11.8 | 14.2 | - |
| % SUM | 27.4 | 91.7 | 94.4 | 96.2 | 94.0 | 93.0 | 91.9 | 95.7 | 95.5 | 78.1 | 59.7 |
| Isolated Compounds | Fraction Number | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | |
| Selagoline | - | - | - | - | - | - | - | - | - | 6.1 | 18.8 | 14.7 | 26.7 | 29.1 | 22.9 | 4.1 | 18.1 | |
| Lycopodine N-oxide | - | - | - | - | - | - | - | - | - | 12.0 | 24.7 | 38.7 | 27.0 | 26.4 | 22.0 | 4.1 | 20.6 | 35.6 |
| Anhydrolycodoline | 1.1 | 1.2 | 1.0 | 0.9 | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 8-β,11-α Dihydroxy-lycopodine | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 4.2 | 18.0 | 30.2 | |
| Lycoposerramine B | - | - | 2.2 | 1.8 | 1.2 | 1.9 | 1.8 | 1.7 | 3.2 | 1.2 | - | - | - | - | - | - | - | - |
| Huperzine A | - | - | - | - | - | - | - | - | - | - | 5.2 | 5.6 | 4.7 | 2.3 | 1.7 | - | - | - |
| 16-Hydroxyhuperzine B | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.8 | 2.2 | 1.3 | - |
| Huperzine B | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 4.5 | - | - | - |
| Lycodine | - | - | - | - | 3.1 | 5.3 | 5.6 | 4.8 | 4.2 | 4.1 | 2.2 | - | - | - | - | - | - | - |
| Des-N-methyl- β-obscurine | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 5.4 | 4.6 | - |
| Dehydrooxolucidine B | 19.8 | 51.4 | 33.5 | 57.6 | 55.8 | 49.6 | 54.2 | 45.2 | 14.4 | 3.2 | 6.2 | 5.5 | 3.2 | 4.4 | - | - | - | |
| Serratidine | 2.0 | 4.6 | 4.3 | 3.9 | 2.1 | 0.9 | 1.1 | 1.3 | 1.2 | - | - | - | - | - | - | 1.3 | 1.5 | - |
| Lucidine B | 3.3 | 28.8 | 16.6 | 21.6 | 15.2 | 17.2 | 14.3 | 18.1 | 23.2 | 39.2 | 31.2 | 24.2 | 22.6 | 31.8 | 11.6 | 3.3 | 3.1 | |
| Dehydrolucidine B | - | - | 3.1 | 3.8 | 4.7 | 3.6 | 4.5 | 5.4 | 5.7 | 4.7 | 2.5 | 3.2 | 2.5 | 4.2 | 14.3 | 41.7 | 24.5 | 5.6 |
| Oxolucidine B | 61.2 | 18.6 | 12.3 | 11.7 | 9.7 | 11.5 | 8.7 | 6.6 | 7.1 | 3.7 | 2.1 | 3.1 | 2.3 | 1.9 | - | - | - | - |
| Unidentified 440.3637 C28H46N3O | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 5.7 | 10.8 | 2.8 | 17.9 |
| Unidentified 456.3588 C28H46N3O2 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 1.2 | 2.9 | 3.3 | - |
| SUM % | 67.5 | 72.9 | 90.8 | 77.1 | 93.5 | 96.1 | 85.6 | 91.9 | 89.7 | 85.2 | 89.8 | 95.6 | 91.2 | 98.7 | 93.1 | 93.7 | 88.8 | 80.3 |
| Isolated Compounds | Fraction Number | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | |
| Luciduline | 3.5 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Serratidine | 2.2 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Selagoline | - | 2.9 | 7.3 | 19.0 | 26.4 | 23.5 | 22.6 | 14.8 | 22.4 | 23.2 | 27.2 | 18.2 | 8.0 | 1.2 | - | - |
| Lycopodine N-oxide | - | 9.5 | 43.6 | 26.8 | 30.5 | 25.7 | 24.1 | 23.8 | 24.1 | 26.8 | 24.1 | 15.1 | 28.8 | 41.6 | 10.9 | - |
| Dehydrooxolucidine B | 67.9 | 50.1 | 18.1 | 25.7 | 11.2 | 18.2 | 19.6 | 20.3 | 29.2 | 12.2 | 6.2 | 9.4 | - | - | 21.1 | 43.1 |
| Lucidine B | 6.6 | 30.1 | 24.0 | 24.2 | 22.6 | 20.2 | 23.1 | 27.1 | 17.2 | 29.2 | 32.3 | 33.2 | 13.6 | - | - | 1.1 |
| Dehydrolucidine B | - | - | - | - | 1.9 | 4.8 | 3.2 | 7.0 | 4.1 | 1.9 | 1.2 | 1.6 | 25.2 | 15.2 | 4.1 | 1.4 |
| Oxolucidine B | 17.7 | 4.1 | 3.0 | 2.7 | 2.6 | 2.2 | 2.1 | 1.1 | 1.4 | 2.0 | 0.2 | 1.1 | 0.8 | - | - | - |
| Lycodine | 0.9 | 0.6 | 0.7 | 0.5 | 0.8 | 0.4 | 0.9 | 0.4 | 0.6 | - | - | - | - | - | - | |
| Lycoposerramine B | 1.1 | 1.1 | 1.1 | - | 0.9 | 1.1 | - | - | - | - | - | - | - | - | - | - |
| Fawcettimine | - | - | - | - | - | - | - | - | - | - | - | - | - | 8.3 | - | - |
| Dihydrolycopodine | - | - | - | - | - | - | - | - | - | - | - | - | 3.2 | 1.5 | 15.2 | 14.2 |
| Deacetyllycoclavine | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 3.1 | 7.1 |
| 8-β,11-α Dihydroxy-lycopodine | - | - | - | - | - | 0.8 | 0.9 | 0.6 | 0.9 | 1.0 | 1.0 | 2.2 | 5.2 | 4.2 | 5.3 | 4.2 |
| Huperzine A | - | - | - | - | - | - | - | - | - | - | - | 8.1 | 6.6 | 0.9 | ||
| Unidentified 474.3796 C27H48N5O2 or C26H52NO6 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 8.2 | 10.1 |
| Unidentified 426.3843 C28H48N3 or C30H46O2 | - | - | - | - | - | - | - | - | - | - | - | - | - | 6.5 | - | - |
| Unidentified 440.3637 C28H46N3O | - | - | - | - | - | - | - | - | - | - | - | - | 6.1 | 9.9 | 2.3 | 3.8 |
| Unidentified 456.3588 C28H46N3O2 | - | - | - | - | - | - | - | - | - | - | - | - | 2.4 | 1.4 | 6.1 | - |
| Unidentified 438.3481 C28H44N3O | - | - | - | - | - | - | - | - | - | - | - | - | - | 4.1 | 4.3 | - |
| SUM % | 98.9 | 98.7 | 97.6 | 99.0 | 96.4 | 97.2 | 96.1 | 95.4 | 99.7 | 96.8 | 92.2 | 88.8 | 99.7 | 94.8 | 80.7 | 84.8 |
| Isolated Compounds | Fraction Number | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Acetyllycofawcine | - | - | - | 10.0 | 13.9 | - |
| Acetylfawcettiine | - | - | - | 12.2 | 10.9 | - |
| Lycopodine N-oxide | - | - | 9.1 | 15.5 | 13.3 | 20.6 |
| Lycopodine | - | - | 7.4 | 7.5 | 8.1 | - |
| Flabelline | - | - | - | 3.9 | 2.6 | - |
| Fawcettimine | - | - | - | 1.5 | 0.8 | 5.7 |
| Deacetyldidehydrolycofawcine | - | - | - | - | 6.9 | - |
| Lycodine | - | 3.9 | 3.9 | 1.4 | - | - |
| 4α,8β,12β-Trihydroxylycopodine | - | - | 4.3 | - | - | - |
| 4α,6α-Dihydroxyanhydrolycodoline | - | - | - | 1.3 | - | - |
| 4,6α-Dihydroxylycopodine or epimer | - | - | - | - | 5.3 | - |
| Lycoposerramine K | 12.3 | 0.8 | - | - | - | - |
| Lycofawcine | - | - | - | - | - | 8.5 |
| α-Lofoline or epimer | - | - | - | - | 0.7 | 5.4 |
| Deacetylfawcettiine | - | - | - | - | - | 2.3 |
| 8β-Hydroxylycoposerramine K | - | - | - | - | - | 5.7 |
| 16-Oxolyclanitin | - | - | - | 5.9 | - | 0.3 |
| Lycoclavanin or epimer | - | - | 4.9 | 7.6 | - | - |
| Serratezomine E | - | - | - | - | - | 4.0 |
| Huperzinine | 0.4 | 2.8 | 3.2 | - | - | - |
| Unidentified 371.2290 C17H31N4O5 or C19H33NO6 | 12.0 | 12.6 | 11.1 | 5.5 | 7.2 | 1.4 |
| Unidentified 415.2532 C19H35N4O6 | 12.5 | 13.2 | 11.7 | 6.6 | 8.4 | 4.9 |
| Unidentified 679.5133 C40H71O8 or C41H67N4O4 | 9.0 | 8.3 | 6.9 | 4.5 | 1.1 | 5.7 |
| SUM % | 46.2 | 41.5 | 62.6 | 83.4 | 79.0 | 64.4 |
| Isolated Compounds | Fraction Number | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Acetyllycofawcine | 7.08 | - | - | 4.39 | 4.4 | 3.29 | 2.48 | 4.97 | 3.17 | 3.19 |
| Acetylfawcettiine | 10.31 | 6.98 | 4.89 | 7.03 | 7.53 | 7.55 | 6.17 | 7.26 | 3.45 | 2.92 |
| Lycopodine N-oxide | 5.57 | 7.03 | 7.28 | 5.16 | 7.46 | 5.26 | 6.71 | 5.03 | 6.88 | 6.24 |
| Lycopodine | - | 2.99 | 2.37 | 3.06 | 3.3 | 5.01 | 8.75 | 6.99 | 6.67 | 4.64 |
| Flabellidine | 0.78 | - | - | - | - | - | 1.13 | 1.01 | 2.14 | 2.03 |
| Fawcettimine | - | - | - | - | - | - | - | - | 1.15 | 6.25 |
| Lycodine | 3.78 | 1.61 | 1.38 | 3.67 | 1.46 | 1.96 | 1.83 | 2.96 | 1.91 | 0.93 |
| Lycoposerramine K | 3.88 | 1.95 | 0.69 | 0.78 | 0.75 | 0.55 | - | - | - | - |
| α-Lofoline or epimer | - | - | - | - | - | - | - | - | 1.1 | 3.3 |
| Serratezomine E | 4.7 | 6.59 | ||||||||
| Deacetylfawcettiine | - | - | - | - | - | - | - | - | - | 2.37 |
| Lycofawcine | 1.12 | 0.89 | ||||||||
| Huperzinine | 2.73 | 0.45 | 0.55 | 2.08 | 2.09 | 2.81 | 2.15 | 2.62 | - | - |
| 16-Oxolyclanitin | 0.77 | 1.24 | ||||||||
| Lycoclavanin or epimer | 3.31 | 2.11 | 3.12 | 3.75 | 2.41 | 3.15 | 2.13 | 0.88 | ||
| Unidentified 371.2290 C17H31N4O5 or C19H33NO6 | 9.74 | 12.05 | 12.1 | 9.48 | 9.48 | 8.06 | 8.55 | 8.07 | 8.29 | 7.09 |
| Unidentified 415.2532 C19H35N4O6 | 10.51 | 12.77 | 12.67 | 9.63 | 9.8 | 8.68 | 8.87 | 8.23 | 7.67 | 7.57 |
| Unidentified 679.5133 C40H71O8 or C41H67N4O4 | 6.46 | 7.89 | 7.47 | 8.01 | 8.48 | 6.94 | 6.24 | 5.93 | 4.94 | 4.6 |
| SUM % | 64.15 | 55.83 | 52.52 | 57.04 | 57.16 | 53.26 | 55.01 | 53.95 | 53.96 | 59.85 |
| Isolated Compounds | Fraction Number | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Acetylfawcettiine | 12.1 | - | - | - | - | 1.2 | 4.9 |
| Lycopodine N-oxide | 12.3 | 8.4 | 6.7 | 3.7 | 2.2 | - | - |
| Lycopodine | 9.7 | 7.3 | 6.5 | 6.2 | 2.2 | 4.3 | 12.6 |
| Dihydrolycopodine | 4.3 | 4.6 | 5.3 | 6.4 | 5.0 | 5.1 | 7.3 |
| Flabelline | - | 0.8 | 1.4 | - | - | - | - |
| Fawcettimine | - | 1.8 | 3.6 | 2.6 | 2.7 | 2.2 | 1.5 |
| 8β-Hydroxylycoposerramine K | - | 2.4 | 3.6 | 2.8 | 1.5 | 1.9 | - |
| Lycoposerramine K | 2.9 | - | - | - | - | - | - |
| 4,6α-Dihydroxylycopodine or epimer | - | - | - | - | - | 1.1 | 1.4 |
| α-Lofoline or epimer | 2.1 | 3.1 | 2.4 | 2.3 | 1.1 | - | 1.8 |
| Deacetylfawcettiine | - | - | 1.8 | 2.0 | 3.7 | 4.7 | 2.5 |
| Serratezomine E | - | 8.1 | 6.2 | - | 2.3 | - | - |
| Japonicumin B or lycoclavanin | 0.3 | 4.2 | 4.1 | - | 4.2 | - | 2.6 |
| 16-Oxolyclanitin | 1.2 | 5.3 | 1.9 | - | - | 2.1 | |
| Unidentified 474.3794 C27H48N5O2 or C26H52NO6 | - | - | - | - | - | - | 7.2 |
| Unidentified 371.2290 C17H31N4O5 or C19H33NO6 | 9.9 | 8.0 | 9.0 | 10.6 | 4.0 | 9.8 | - |
| Unidentified 415.2532 C19H35N4O6 | 10.5 | 8.4 | 9.6 | 11.3 | 4.2 | 10.1 | 5.5 |
| Unidentified 679.5133 C40H71O8 or C41H67N4O4 | 7.39 | 6.7 | 6.87 | 7.12 | 4.75 | 8.17 | 3.91 |
| % SUM | 71.43 | 64.95 | 72.29 | 56.76 | 37.64 | 48.59 | 53.24 |
| Isolated Compounds | Fraction Number | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
| Lycopodine | 0.8 | - | 28.5 | 31.8 | 27.6 | 30.0 | 33.5 | 39.1 | 36.8 | 35.4 | 37.2 |
| Dihydrolycopodine | - | - | 8.2 | 12.3 | 12.7 | 8.5 | 6.2 | 6.0 | 7.6 | 11.3 | 7.0 |
| Lycopodine N-oxide | - | - | - | 8.5 | 11.8 | 13.0 | 10.1 | 8.8 | 11.0 | 13.2 | 13.2 |
| 8β-Hydroxylycoposerramine K | - | 46.3 | 22.8 | 13.3 | 8.9 | 7.3 | 3.7 | 4.8 | 3.2 | 5.5 | 4.3 |
| Acetylfawcettiine | - | - | - | - | - | - | 1.1 | 0.9 | - | 0.8 | 1.1 |
| 8-β,11-α Dihydroxylycopodine or epimer | - | - | 3.9 | - | - | 1.0 | - | 1.2 | 0.9 | 0.7 | 1.0 |
| Lycodine | - | - | - | 1.2 | 1.1 | 0.8 | - | - | 1.1 | 0.9 | 0.6 |
| α-Lofoline or epimer | - | - | - | 1.1 | 1.1 | 1.0 | 1.1 | 1.1 | 2.0 | 1.0 | 1.2 |
| β-Obscurine | - | - | - | 1.2 | 2.1 | 1.2 | - | - | - | - | 1.1 |
| Deacetylfawcettiine | - | 1.1 | 4.6 | 3.2 | 3.5 | 4.6 | 2.2 | 1.7 | 3.6 | 1.9 | 3.4 |
| Serratezomine E | - | - | - | 1.5 | 2.9 | 1.5 | 1.0 | 1.3 | 1.2 | 3.2 | 3.6 |
| N-Methyl lycodine | - | - | - | - | - | - | - | - | 0.5 | 0.6 | 0.8 |
| Unidentified 236.1489 C10H22NO5 | - | 20.1 | 11.7 | 10.3 | 9.8 | 10.0 | 9.3 | 9.9 | 11.1 | 12.5 | 11.5 |
| SUM % | 0.8 | 67.5 | 79.8 | 84.4 | 81.4 | 78.7 | 68.0 | 74.7 | 79.0 | 87.0 | 85.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dymek, A.; Widelski, J.; Wojtanowski, K.K.; Vivcharenko, V.; Przekora, A.; Mroczek, T. Fractionation of Lycopodiaceae Alkaloids and Evaluation of Their Anticholinesterase and Cytotoxic Activities. Molecules 2021, 26, 6379. https://doi.org/10.3390/molecules26216379
Dymek A, Widelski J, Wojtanowski KK, Vivcharenko V, Przekora A, Mroczek T. Fractionation of Lycopodiaceae Alkaloids and Evaluation of Their Anticholinesterase and Cytotoxic Activities. Molecules. 2021; 26(21):6379. https://doi.org/10.3390/molecules26216379
Chicago/Turabian StyleDymek, Aleksandra, Jarosław Widelski, Krzysztof Kamil Wojtanowski, Vladyslav Vivcharenko, Agata Przekora, and Tomasz Mroczek. 2021. "Fractionation of Lycopodiaceae Alkaloids and Evaluation of Their Anticholinesterase and Cytotoxic Activities" Molecules 26, no. 21: 6379. https://doi.org/10.3390/molecules26216379
APA StyleDymek, A., Widelski, J., Wojtanowski, K. K., Vivcharenko, V., Przekora, A., & Mroczek, T. (2021). Fractionation of Lycopodiaceae Alkaloids and Evaluation of Their Anticholinesterase and Cytotoxic Activities. Molecules, 26(21), 6379. https://doi.org/10.3390/molecules26216379