
NMR Study of Intercalates and Grafted Organic Derivatives of H2La2Ti3O10

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
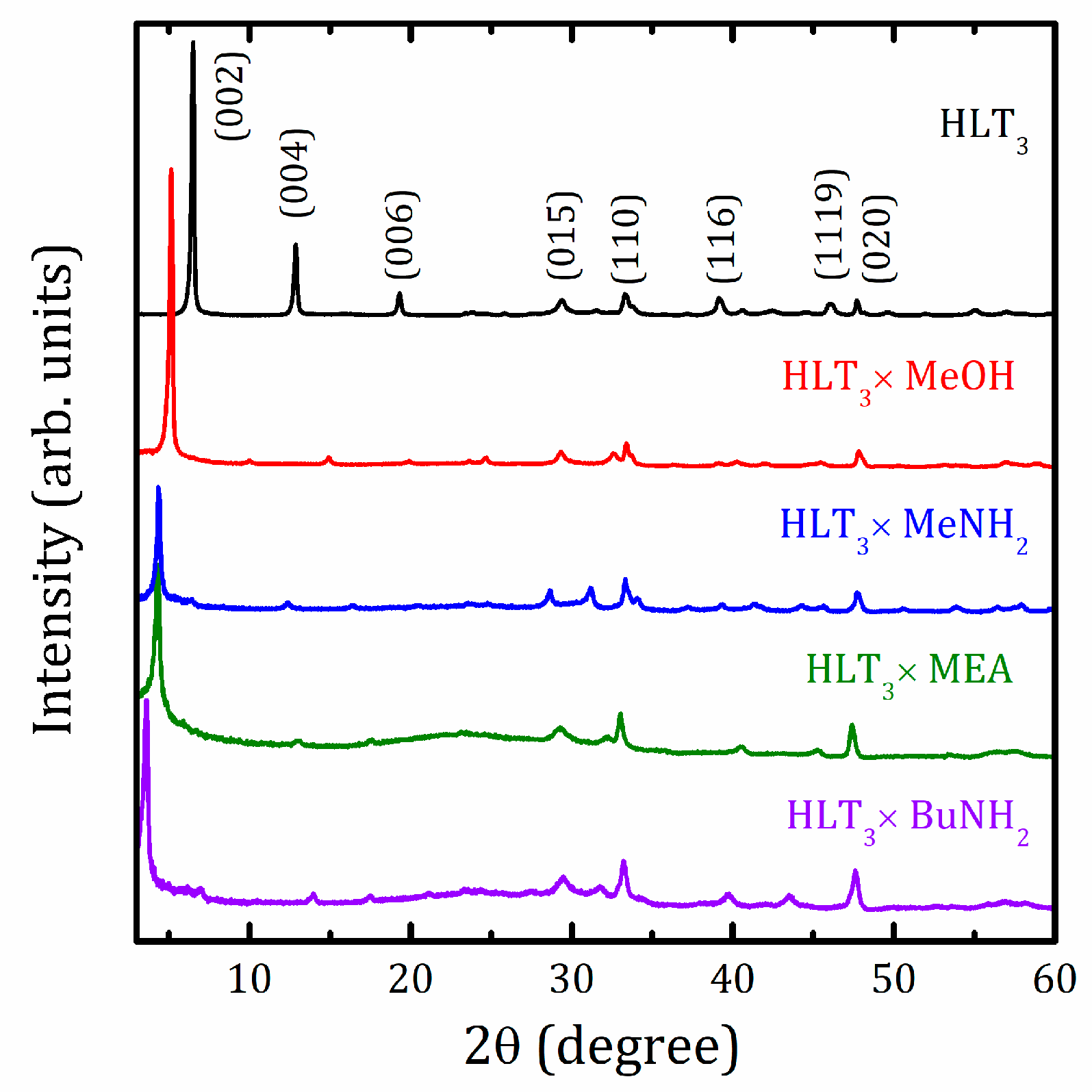
2.1. XRD Analysis
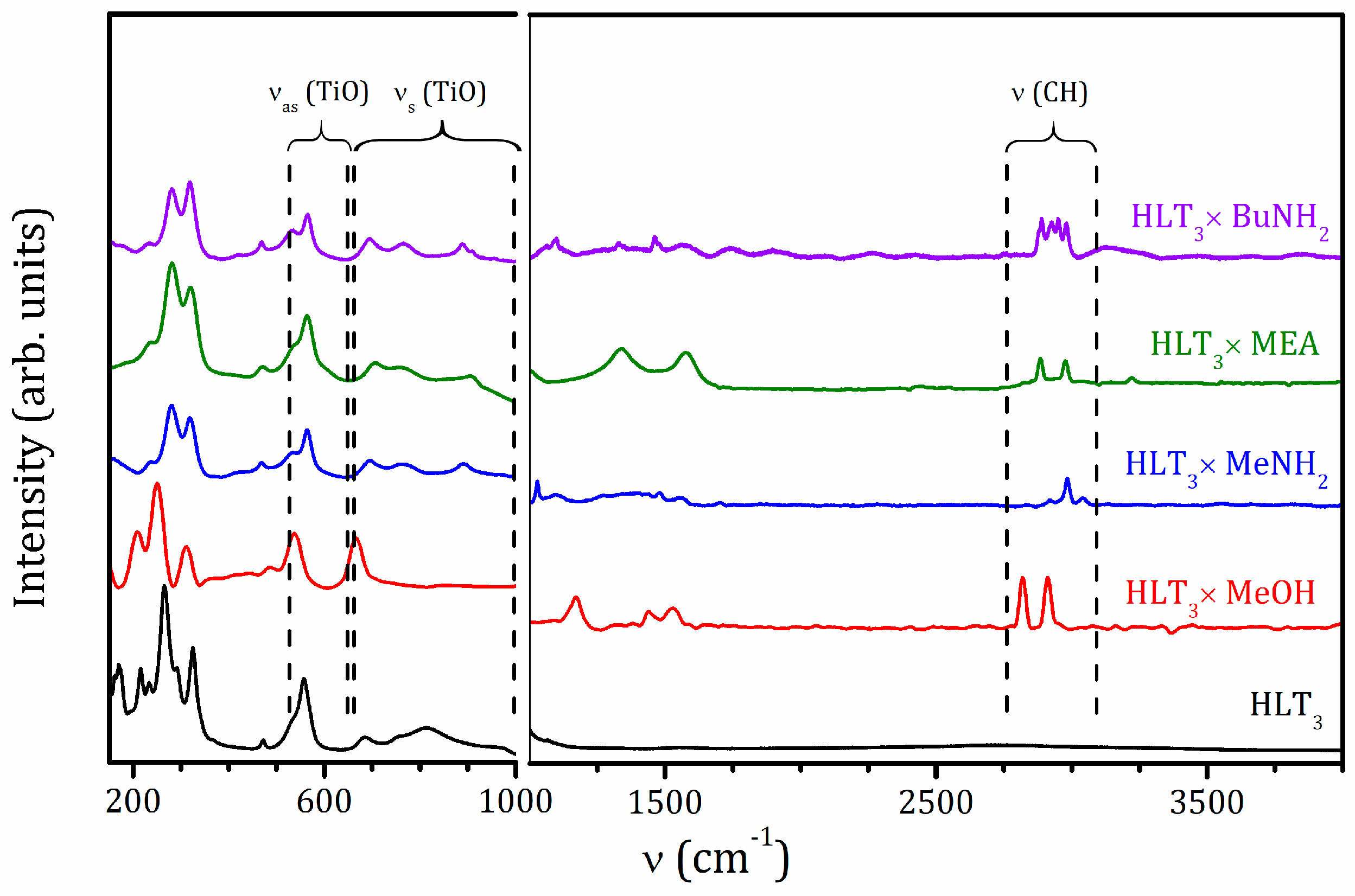
2.2. Raman and IR Spectroscopy
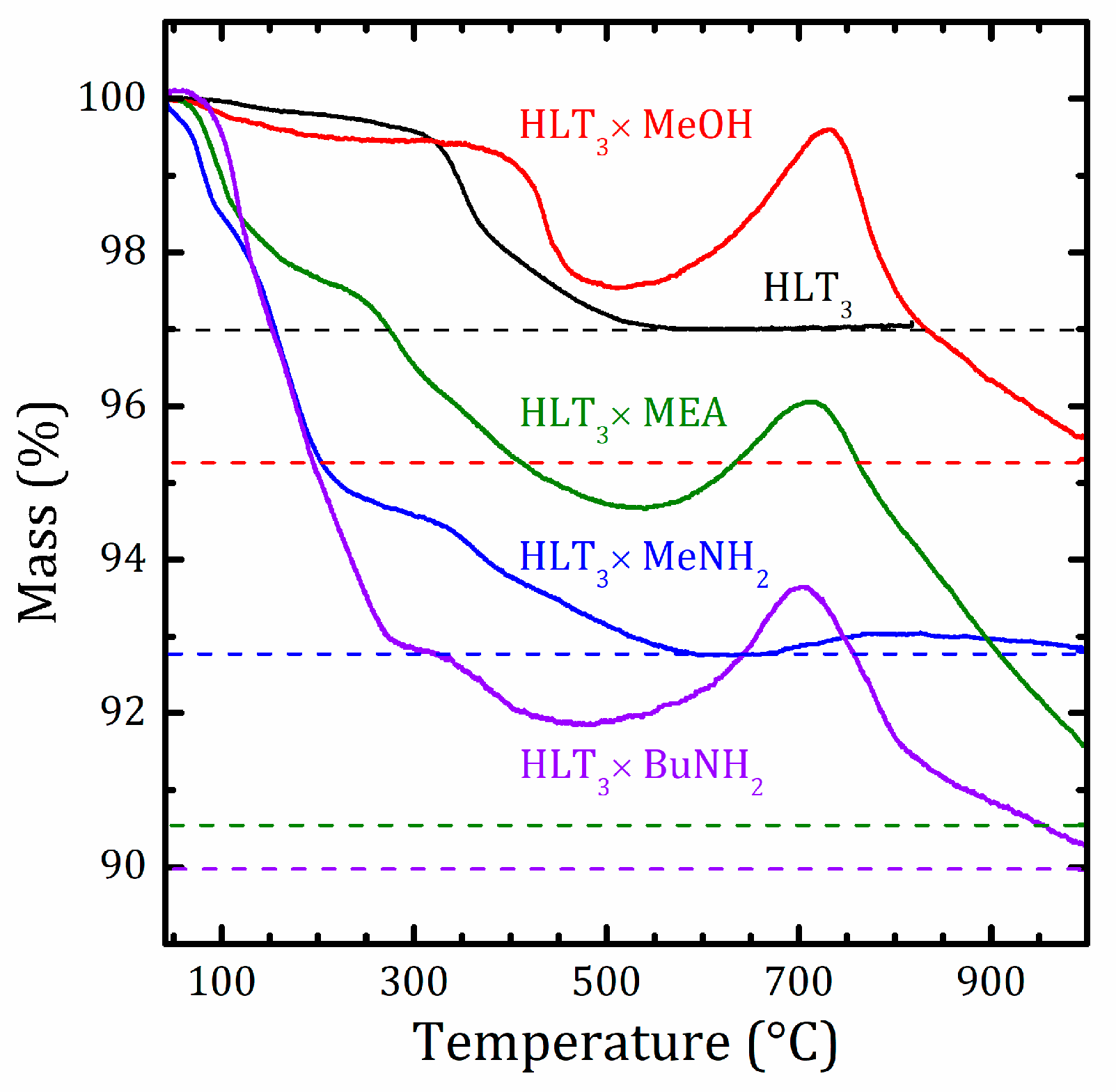
2.3. Thermal Analysis
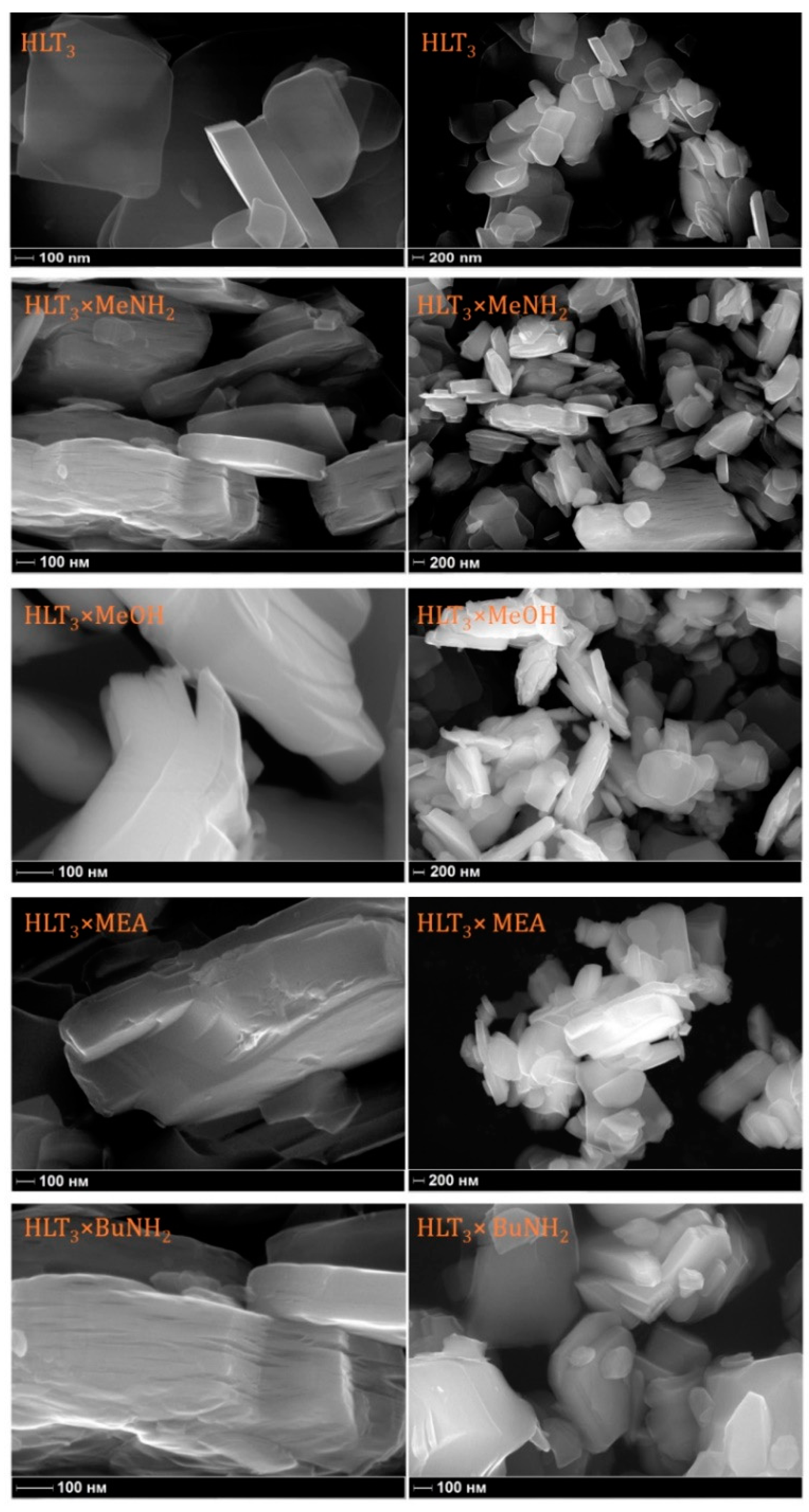
2.4. Scanning Electron Microscopy
2.5. NMR Studies
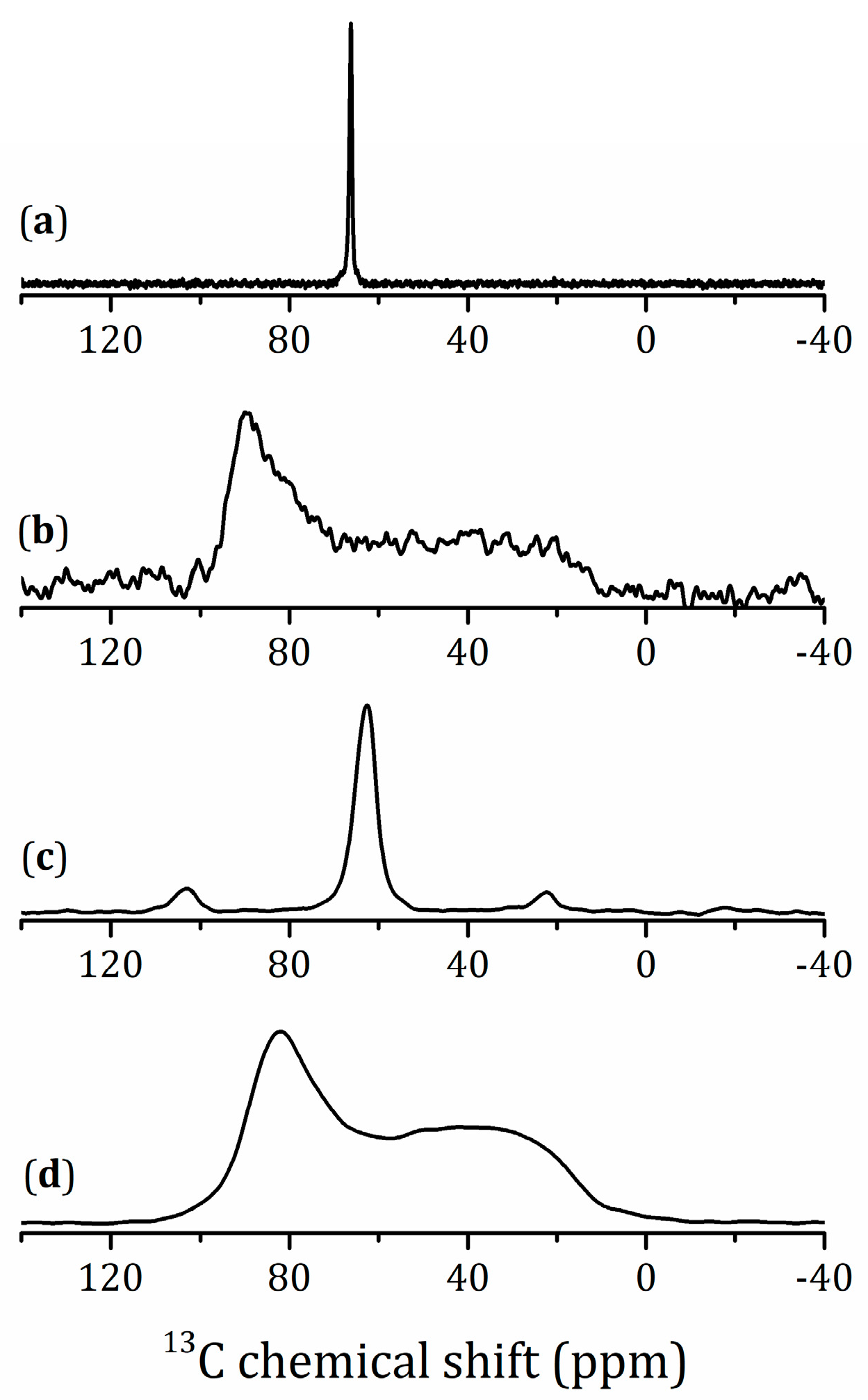
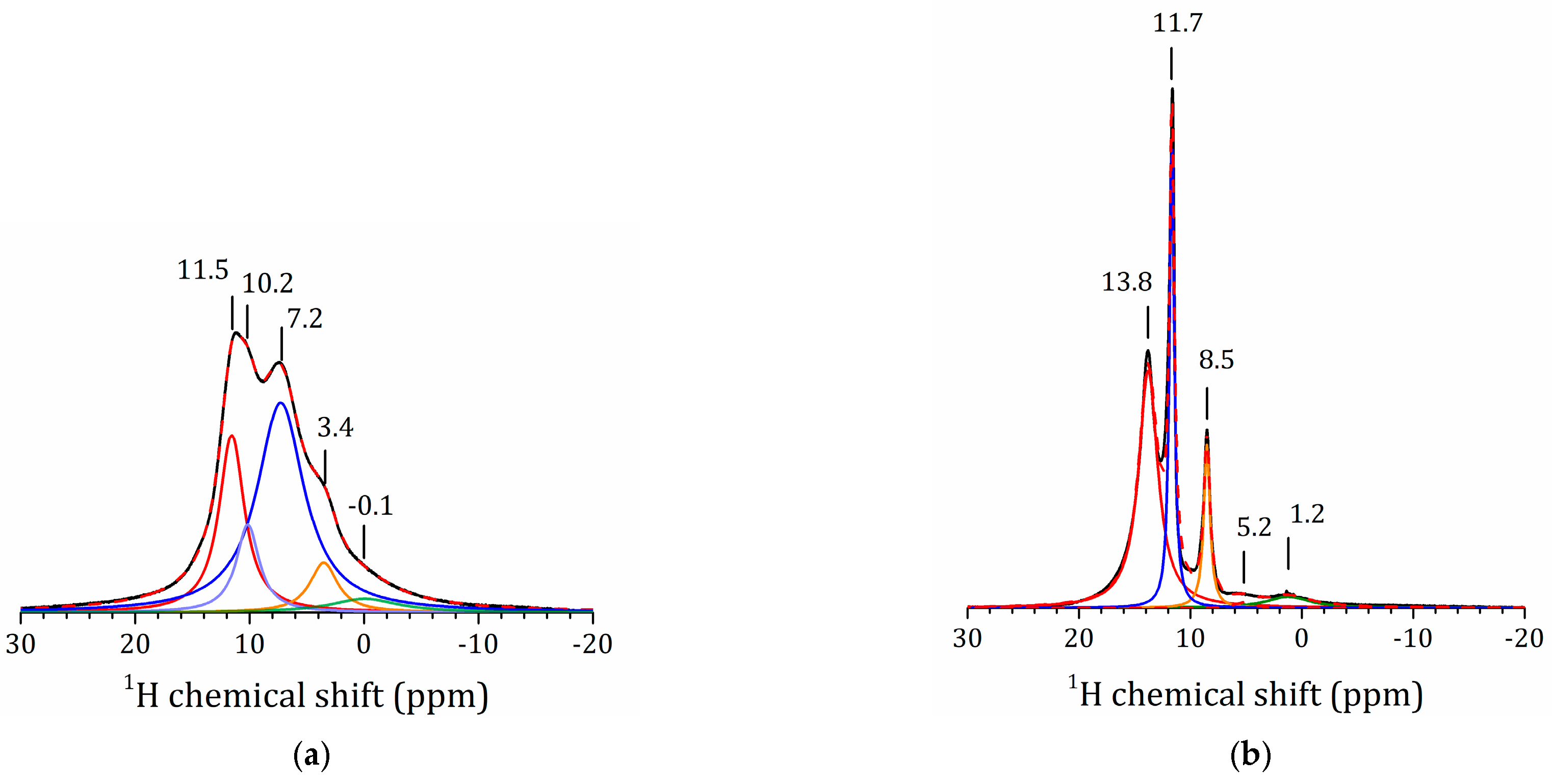
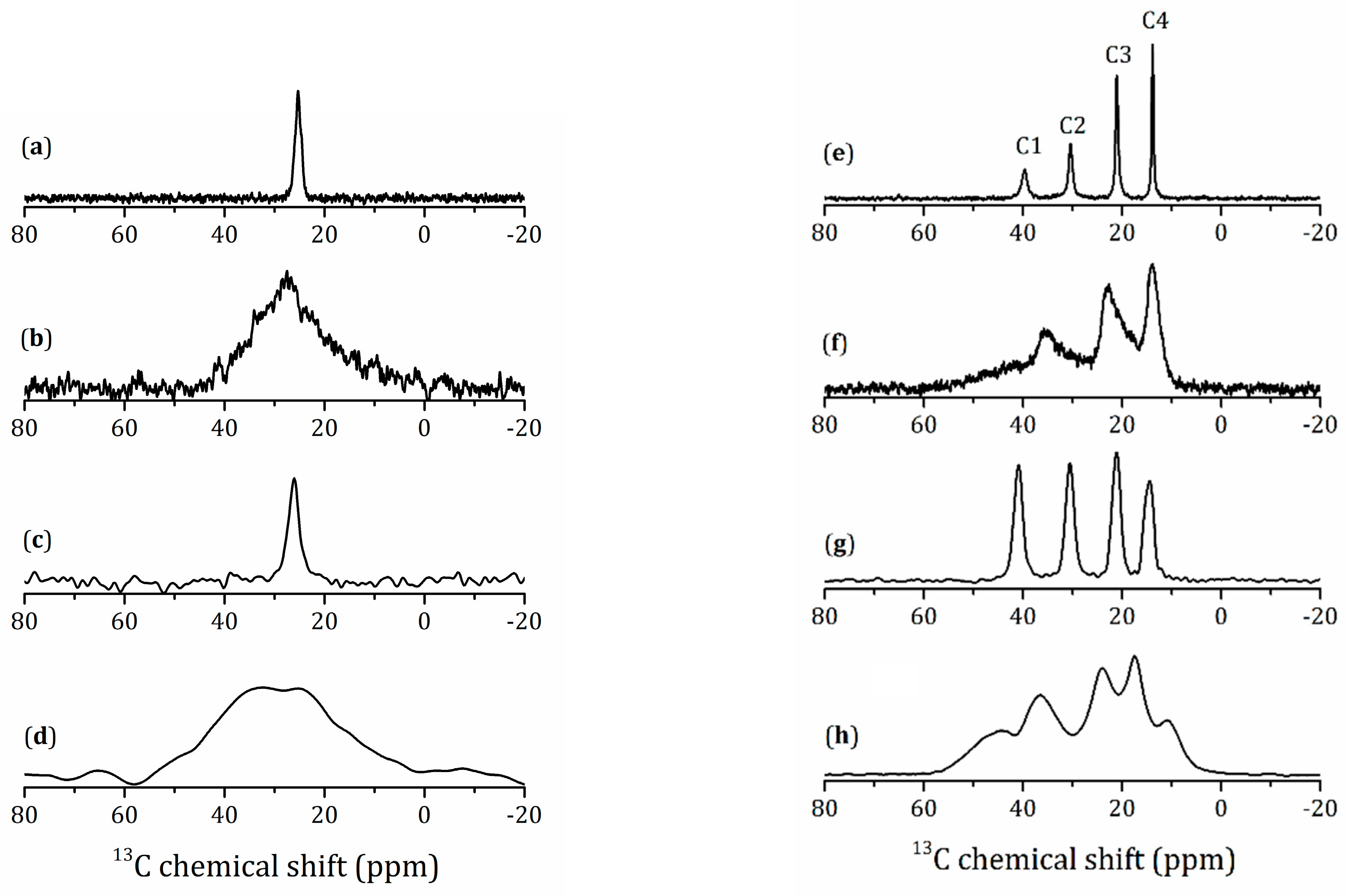
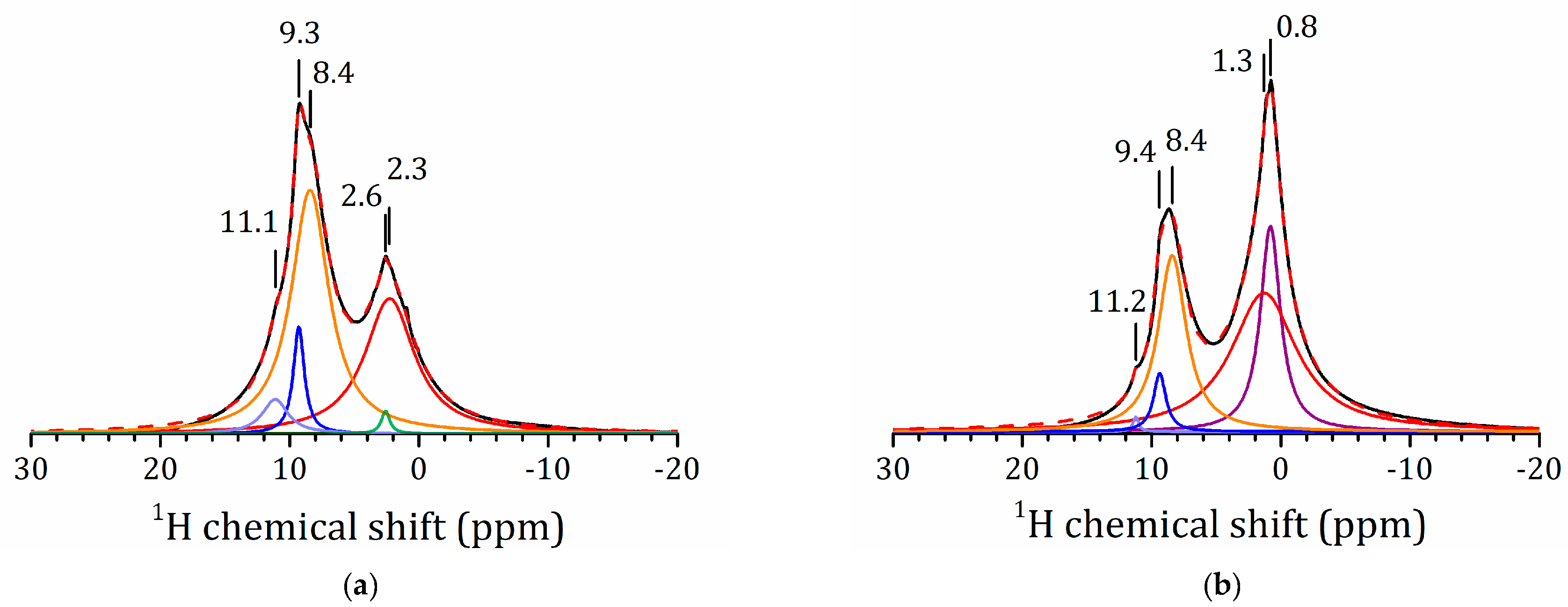
2.5.1. NMR Studies of the HLT3 × MeOH Derivative
2.5.2. NMR Studies of the HLT3 × MeNH2 and HLT3 × BuNH2 Derivatives
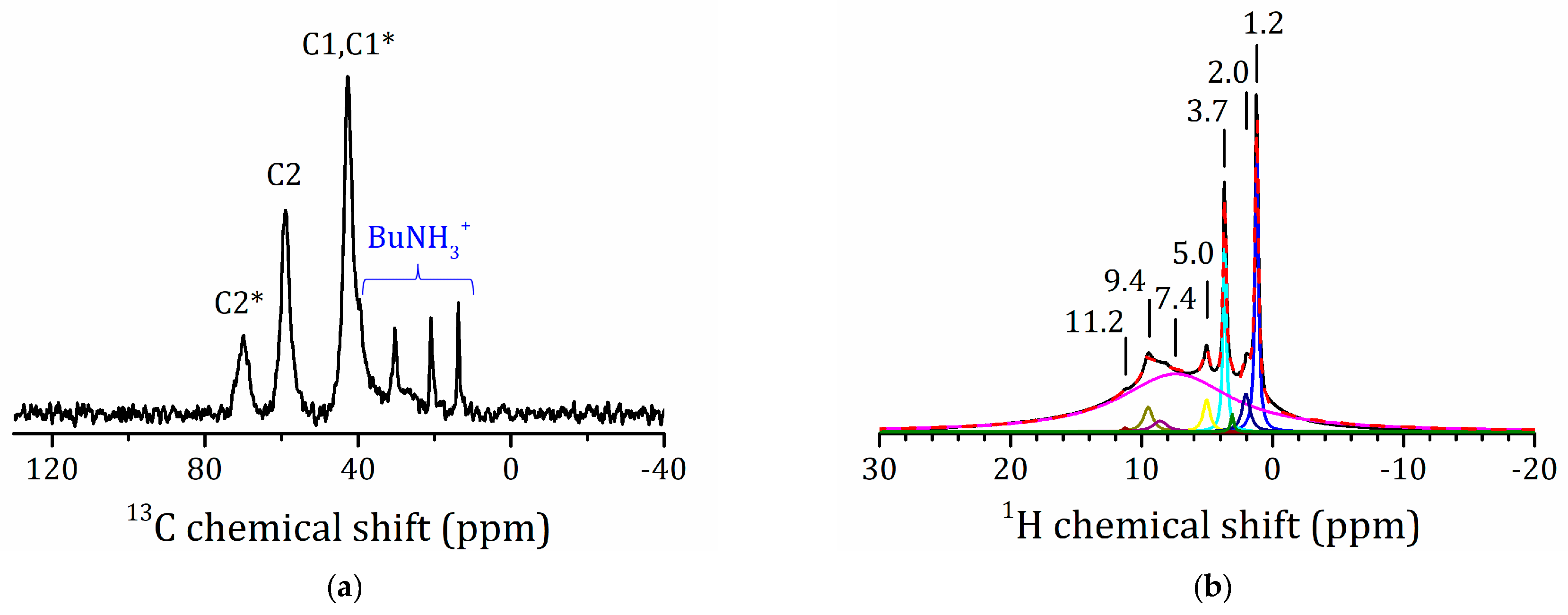
2.5.3. NMR Studies the HLT3 × MEA Derivative
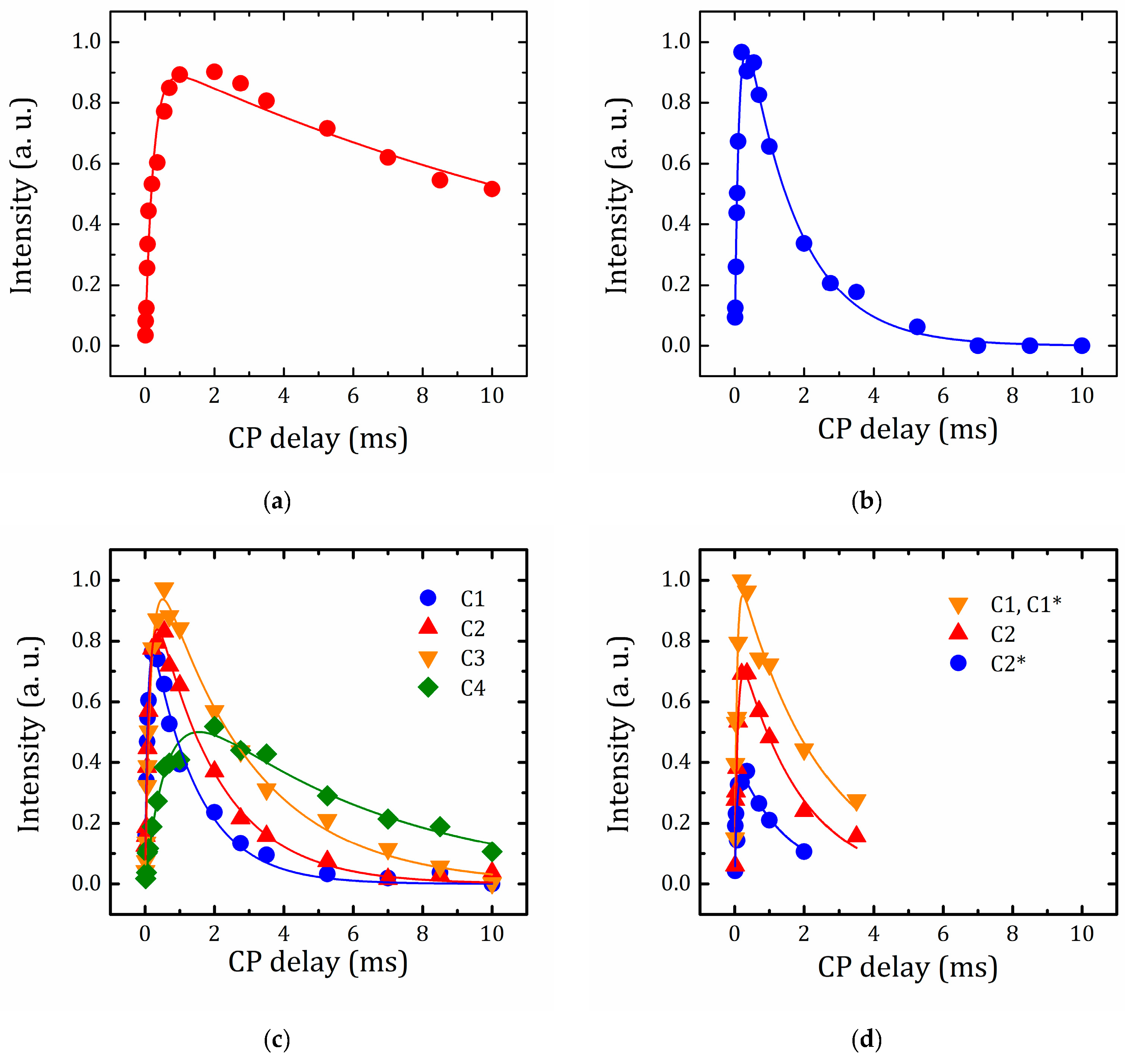
2.5.4. VCT Experiment to Study the Dynamics of Organic Molecules
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yao, K.; Wang, X.; Xu, Y.X.; Li, F.; Zhou, L. Multilayered Perovskite Materials Based on Polymeric-Ammonium Cations for Stable Large-Area Solar Cell. Chem. Mater. 2016, 28, 3131–3138. [Google Scholar] [CrossRef]
- Morell, J.; Chatterjee, S.; Klar, P.J.; Mauder, D.; Shenderovich, I.; Hoffmann, F.; Fröba, M. Synthesis and characterization of chiral benzylic ether-bridged periodic mesoporous organosilicas. Chem. Eur. J. 2008, 14, 5935–5940. [Google Scholar] [CrossRef] [PubMed]
- Lerf, A. Storylines in intercalation chemistry. Dalt. Trans. 2014, 43, 10276–10291. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Furumi, Y.; Shinohara, K.; Tanaka, A.; Hara, M.; Kondo, J.N.; Domen, K. Photocatalytic decomposition of water on spontaneously hydrated layered perovskites. Chem. Mater. 1997, 9, 1063–1064. [Google Scholar] [CrossRef]
- Rodionov, I.A.; Zvereva, I.A. Photocatalytic activity of layered perovskite-like oxides in practically valuable chemical reactions. Russ. Chem. Rev. 2016, 85, 248–279. [Google Scholar] [CrossRef]
- Schaak, R.E.; Mallouk, T.E. Perovskites by design: A toolbox of solid-state reactions. Chem. Mater. 2002, 14, 1455–1471. [Google Scholar] [CrossRef]
- Zvereva, I.A.; Silyukov, O.I.; Chislov, M.V. Ion-exchange reactions in the structure of perovskite-like layered oxides: I. Protonation of NaNdTiO4 complex oxide. Russ. J. Gen. Chem. 2011, 81, 1434–1441. [Google Scholar] [CrossRef]
- Kurnosenko, S.A.; Silyukov, O.I.; Zvereva, I.A. Preparation of Porous Particles of Layered Perovskite-Like Titanate HLaTiO4. Glas. Phys. Chem. 2020, 46, 272–276. [Google Scholar] [CrossRef]
- Silyukov, O.I.; Kurnosenko, S.A.; Zvereva, I.A. Intercalation of Methylamine into the Protonated Forms of Layered Perovskite-Like Oxides HLnTiO4 (Ln = La and Nd). Glas. Phys. Chem. 2018, 44, 428–432. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Lushpinskaya, I.P.; Kurnosenko, S.A.; Silyukov, O.I.; Zvereva, I.A. Identification of intercalates and grafted organic derivatives of H2La2Ti3O10 by multinuclear NMR. Russ. J. Gen. Chem. 2020, 90, 760–761. [Google Scholar] [CrossRef]
- Lee, W.-J.; Yeo, H.J.; Kim, D.-Y.; Paek, S.-M.; Kim, Y.-I. Exfoliation of Dion-Jacobson Layered Perovskite into Macromolecular Nanoplatelet. Bull. Korean Chem. Soc. 2013, 34, 2041–2043. [Google Scholar] [CrossRef]
- Wang, T.H.; Henderson, C.N.; Draskovic, T.I.; Mallouk, T.E. Synthesis, exfoliation, and electronic/protonic conductivity of the dion-jacobson phase layer perovskite HLa2TiTa2O10. Chem. Mater. 2014, 26, 898–906. [Google Scholar] [CrossRef]
- Han, Y.-S.; Park, I.; Choy, J.-H. Exfoliation of layered perovskite, KCa2Nb3O10, into colloidal nanosheets by a novel chemical process. J. Mater. Chem. 2001, 11, 1277–1282. [Google Scholar] [CrossRef]
- Hojamberdiev, M.; Bekheet, M.F.; Zahedi, E.; Wagata, H.; Kamei, Y.; Yubuta, K.; Gurlo, A.; Matsushita, N.; Domen, K.; Teshima, K. New Dion-Jacobson Phase Three-Layer Perovskite CsBa2Ta3O10 and Its Conversion to Nitrided Ba2Ta3O10 Nanosheets via a Nitridation-Protonation-Intercalation-Exfoliation Route for Water Splitting. Cryst. Growth Des. 2016, 16, 2302–2308. [Google Scholar] [CrossRef]
- Machida, M.; Miyazaki, K.; Matsushima, S.; Arai, M. Photocatalytic properties of layered perovskite tantalates, MLnTa2O7 (M = Cs, Rb, Na, and H; Ln = La, Pr, Nd, and Sm). J. Mater. Chem. 2003, 13, 1433. [Google Scholar] [CrossRef]
- Choi, J.; Zhang, X.; Wiley, J.B. Building alkali-metal-halide layers within a perovskite host by sequential intercalation: (A2Cl)LaNb2O7 (A = Rb, Cs). Inorg. Chem. 2009, 48, 4811–4816. [Google Scholar] [CrossRef]
- Kawashima, K.; Hojamberdiev, M.; Wagata, H.; Yubuta, K.; Domen, K.; Teshima, K. Protonated Oxide, Nitrided, and Reoxidized K2La2Ti3O10 Crystals: Visible-Light-Induced Photocatalytic Water Oxidation and Fabrication of Their Nanosheets. ACS Sustain. Chem. Eng. 2017, 5, 232–240. [Google Scholar] [CrossRef]
- Rodionov, I.A.; Silyukov, O.I.; Utkina, T.D.; Chislov, M.V.; Sokolova, Y.P.; Zvereva, I.A. Photocatalytic properties and hydration of perovskite-type layered titanates A2Ln2Ti3O10 (A = Li, Na, K; Ln = La, Nd). Russ. J. Gen. Chem. 2012, 82, 1191–1196. [Google Scholar] [CrossRef]
- Silyukov, O.I.; Abdulaeva, L.D.; Burovikhina, A.A.; Rodionov, I.A.; Zvereva, I.A. Phase transformations during HLnTiO4 (Ln=La, Nd) thermolysis and photocatalytic activity of obtained compounds. J. Solid State Chem. 2015, 226, 101–106. [Google Scholar] [CrossRef]
- Rodionov, I.A.; Silyukov, O.I.; Zvereva, I.A. Study of photocatalytic activity of layered oxides: NaNdTiO4, LiNdTiO4, and HNdTiO4 titanates. Russ. J. Gen. Chem. 2012, 82, 635–638. [Google Scholar] [CrossRef]
- Rodionov, I.A.; Maksimova, E.A.; Pozhidaev, A.Y.; Kurnosenko, S.A.; Silyukov, O.I.; Zvereva, I.A. Layered Titanate H2Nd2Ti3O10 Intercalated with n-Butylamine: A New Highly Efficient Hybrid Photocatalyst for Hydrogen Production from Aqueous Solutions of Alcohols. Front. Chem. 2019, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Voytovich, V.V.; Kurnosenko, S.A.; Silyukov, O.I.; Rodionov, I.A.; Minich, I.A.; Zvereva, I.A. Study of n-alkylamine Intercalated Layered Perovskite-Like Niobates HCa2Nb3O10 as Photocatalysts for Hydrogen Production From an Aqueous Solution of Methanol. Front. Chem. 2020, 8, 300. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Kim, C.H.; Mallouk, T.E. Dielectric Properties of the Lamellar Niobates and Titanoniobates AM2Nb3O10 and ATiNbO5 (A = H, K, M = Ca, Pb), and Their Condensation Products Ca4Nb6O19 and Ti2Nb2O9. Chem. Mater. 1999, 11, 1519–1525. [Google Scholar] [CrossRef]
- Takayanagi, S.; Ogawa, S. Superconducting properties of Layered Perovskite KCa2Nb3O10 and KLaNb2O7. Solid State Ion. 1997, 103, 215–217. [Google Scholar]
- Toda, K.; Honma, T.; Sato, M. Unusual concentration quenching of europium luminescence in new layered perovskite compound, RbLa1−xEuxTa2O7. J. Lumin. 1997, 71, 71–75. [Google Scholar] [CrossRef]
- Sugahara, Y. Chemical processes employing inorganic layered compounds for inorganic and inorganic-organic hybrid materials. J. Ceram. Soc. Jpn. 2014, 122, 523–529. [Google Scholar] [CrossRef]
- Ranmohotti, K.G.S.; Josepha, E.; Choi, J.; Zhang, J.; Wiley, J.B. Topochemical manipulation of perovskites: Low-temperature reaction strategies for directing structure and properties. Adv. Mater. 2011, 23, 442–460. [Google Scholar] [CrossRef]
- Tahara, S.; Ichikawa, T.; Kajiwara, G.; Sugahara, Y. Reactivity of the Ruddlesden-Popper Phase H2La2Ti3O10 with Organic Compounds: Intercalation and Grafting Reactions. Chem. Mater. 2007, 19, 2352–2358. [Google Scholar] [CrossRef]
- Tahara, S.; Sugahara, Y. Interlayer Surface Modification of the Protonated Triple-Layered Perovskite HCa2Nb3O10·xH2O with n-Alcohols. Langmuir 2003, 19, 9473–9478. [Google Scholar] [CrossRef]
- Tahara, S.; Takeda, Y.; Sugahara, Y. Preparation of Organic-Inorganic Hybrids Possessing Nanosheets with Perovskite-Related Structures via Exfoliation during a Sol-Gel Process. Chem. Mater. 2005, 17, 6198–6204. [Google Scholar] [CrossRef]
- Wang, C.; Tang, K.; Wang, D.; Liu, Z.; Wang, L.; Zhu, Y.; Qian, Y. A new carbon intercalated compound of Dion–Jacobson phase HLaNb2O7. J. Mater. Chem. 2012, 22, 11086. [Google Scholar] [CrossRef]
- Kimura, N.; Kato, Y.; Suzuki, R.; Shimada, A.; Tahara, S.; Nakato, T.; Matsukawa, K.; Mutin, P.H.; Sugahara, Y. Single- and Double-Layered Organically Modified Nanosheets by Selective Interlayer Grafting and Exfoliation of Layered Potassium Hexaniobate. Langmuir 2014, 30, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Corriu, R.J.P.; Leclercq, D. Recent Developments of Molecular Chemistry for Sol-Gel Processes. Angew. Chem. 1996, 35, 1420–1436. [Google Scholar] [CrossRef]
- Sanchez, C.; Soler-Illia, G.J.D.A.A.; Ribot, F.; Lalot, T.; Mayer, C.R.; Cabuil, V. Designed hybrid organic-inorganic nanocomposites from functional nanobuilding blocks. Chem. Mater. 2001, 13, 3061–3083. [Google Scholar] [CrossRef]
- Faustini, M.; Nicole, L.; Ruiz-Hitzky, E.; Sanchez, C. History of organic–inorganic hybrid materials: Prehistory, art, science, and advanced applications. Adv. Funct. Mater. 2018, 28, 1–30. [Google Scholar] [CrossRef]
- Bin, D.; Huo, W.; Yuan, Y.; Huang, J.; Liu, Y.; Zhang, Y.; Dong, F.; Wang, Y.; Xia, Y. Organic-inorganic-induced polymer intercalation into layered composites for aqueous zinc-ion battery. Chem 2020, 6, 968–984. [Google Scholar] [CrossRef]
- Wang, Y.; Nikolopoulou, M.; Delahaye, E.; Leuvrey, C.; Leroux, F.; Rabu, P.; Rogez, G. Microwave-assisted functionalization of the Aurivillius phase Bi2SrTa2O9: Diol grafting and amine insertion vs. alcohol grafting. Chem. Sci. 2018, 9, 7104–7114. [Google Scholar] [CrossRef]
- Wang, Y.; Delahaye, E.; Leuvrey, C.; Leroux, F.; Rabu, P.; Rogez, G. Post-synthesis modification of the aurivillius phase Bi2SrTa2O9 via in situ microwave-assisted “click reaction”. Inorg. Chem. 2016, 55, 9790–9797. [Google Scholar] [CrossRef]
- Idota, N.; Fukuda, S.; Tsukahara, T.; Sugahara, Y. Preparation of thermoresponsive nanosheets exhibiting phase transitions in water via surface modification of layered perovskite nanosheets with poly(N-isopropylacrylamide) (PNIPAAm). Chem. Lett. 2015, 44, 203–205. [Google Scholar] [CrossRef]
- Sato, S.; Shintani, K.; Idota, N.; Nishino, T.; Sugahara, Y. Effect of the graft density of cellulose diacetate-modified layered perovskite nanosheets on mechanical properties of the transparent organic-inorganic hybrids bearing covalent bonds at the interface. Cellulose 2017, 24, 5463–5473. [Google Scholar] [CrossRef]
- Asai, Y.; Ariake, Y.; Saito, H.; Idota, N.; Matsukawa, K.; Nishino, T.; Sugahara, Y. Layered perovskite nanosheets bearing fluoroalkoxy groups: Their preparation and application in epoxy-based hybrids. RSC Adv. 2014, 4, 26932–26939. [Google Scholar] [CrossRef]
- Byeon, S.; Kileung, P.; Park, K. Structure and Ionic Conductivity of NaLnTiO4, Comparison with Those of Na2Ln2Ti3O10 (Ln = La, Nd, Sm, and Gd). J. Solid State Chem. 1996, 121, 430–436. [Google Scholar] [CrossRef]
- Richard, M.; Brohan, L.; Tournoux, M. Synthesis, Characterization, and Acid Exchange of the Layered Perovskites A2Nd2Ti3O10 (A—Na, K). J. Solid State Chem. 1994, 112, 345–354. [Google Scholar] [CrossRef]
- Rodionov, I.A.; Sokolova, I.P.; Silyukov, O.I.; Burovikhina, A.A.; Fateev, S.A.; Zvereva, I.A. Protonation and Photocatalytic Activity of the Rb2La2Ti3O10 Layered Oxide in the Reaction of Hydrogen Production. Int. J. Photoenergy 2017, 2017, 1–8. [Google Scholar] [CrossRef]
- Tong, Z.; Zhang, G.; Takagi, S.; Shimada, T.; Tachibana, H.; Inoue, H. Preparation and Characterization of a Transparent Thin Film of the Layered Perovskite, K2La2Ti3O10, Intercalated with an Ionic Porphyrin. Chem. Lett. 2005, 34, 632–633. [Google Scholar] [CrossRef]
- Akbarian-Tefaghi, S.; Wiley, J.B. Microwave-assisted routes for rapid and efficient modification of layered perovskites. Dalt. Trans. 2018, 47, 2917–2924. [Google Scholar] [CrossRef]
- Chizhik, V.I.; Chernyshev, Y.S.; Donets, A.V.; Frolov, V.V.; Komolkin, A.V.; Shelyapina, M.G. Magnetic Resonance and Its Applications; Springer International Publishing: Cham, Switzerland, 2014; ISBN 9783319052984. [Google Scholar]
- Kharkov, B.B.; Corkery, R.W.; Dvinskikh, S.V. Phase transitions and chain dynamics of surfactants intercalated into the galleries of naturally occurring clay mineral magadiite. Langmuir 2014, 30, 7859–7866. [Google Scholar] [CrossRef]
- Krylova, E.A.; Shelyapina, M.G.; Nowak, P.; Harańczyk, H.; Chislov, M.; Zvereva, I.A.; Privalov, A.F.; Becker, M.; Vogel, M.; Petranovskii, V. Mobility of water molecules in sodium- and copper-exchanged mordenites: Thermal analysis and 1H NMR. Microporous Mesoporous Mater. 2018, 265, 132–142. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Nefedov, D.Y.; Kostromin, A.V.; Silyukov, O.I.; Zvereva, I.A. Proton mobility in Ruddlesden–Popper phase H2La2Ti3O10 studied by 1H-NMR. Ceram. Int. 2019, 45, 5788–5795. [Google Scholar] [CrossRef]
- Kharkov, B.B.; Dvinskikh, S. Chain dynamics of surfactants in mesoporous silica. Phys. Chem. Chem. Phys. 2013, 42, 18620–18626. [Google Scholar] [CrossRef]
- Khimyak, Y.Z.; Klinowski, J. Solid-state NMR studies of the organic template in mesostructured aluminophosphates. Phys. Chem. Chem. Phys. 2001, 3, 616–626. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Yocupicio-Gaxiola, R.I.; Zhelezniak, I.V.; Chislov, M.V.; Antúnez-García, J.; Murrieta-Rico, F.N.; Galván, D.H.; Petranovskii, V.; Fuentes-Moyado, S. Local structures of two-dimensional zeolites—Mordenite and ZSM-5—Probed by multinuclear NMR. Molecules 2020, 25, 4678. [Google Scholar] [CrossRef] [PubMed]
- Kurnosenko, S.A.; Silyukov, O.I.; Mazur, A.S.; Zvereva, I.A. Synthesis and thermal stability of new inorganic-organic perovskite-like hybrids based on layered titanates HLnTiO4 (Ln = La, Nd). Ceram. Int. 2019, 46, 5058–5068. [Google Scholar] [CrossRef]
- Byeon, S.; Nam, H. Neutron Diffraction and FT-Raman Study of Ion-Exchangeable Layered Titanates and Niobates. Chem. Mater. 2000, 12, 1771–1778. [Google Scholar] [CrossRef]
- Nozaki, R.; Kondo, J.N.; Hirose, C.; Domen, K.; Wada, A. Vibrational Study of Layered Perovskites M2La2Ti3O10 (M = Li, Na, K, Rb): Raman Spectra and Normal Mode Analysis. J. Phys. Chem. B 2001, 105, 7950–7953. [Google Scholar] [CrossRef]
- Torres-Barthelemy, V.; Pérez-Hernández, N.; Shenderovich, I.G.; Tolstoy, P.M.; Denisov, G.S.; Limbach, H.-H. NMR-detected host-guest proton exchange as a tool to explore surface/volume ratios and fluid filling of internal and external spaces of porous solids containing surface OH groups. J. Phys. Chem. C 2020, 124, 22082–22095. [Google Scholar] [CrossRef]
- Kong, S.; Borissova, A.O.; Lesnichin, S.B.; Hartl, M.; Daemen, L.L.; Eckert, J.; Antipin, M.Y.; Shenderovich, I.G. Geometry and spectral properties of the protonated homodimer of pyridine in the liquid and solid states. A combined NMR, X-ray diffraction and inelastic neutron scattering study. J. Phys. Chem. A 2011, 115, 8041–8048. [Google Scholar] [CrossRef]
- Grünberg, B.; Emmler, T.; Gedat, E.; Shenderovich, I.; Findenegg, G.H.; Limbach, H.; Buntkowsky, G. Hydrogen bonding of water confined in mesoporous silica MCM-41 and SBA-15 studied by 1H solid-state NMR. Chem. Eur. J. 2004, 10, 5689–5696. [Google Scholar] [CrossRef]
- Shenderovich, I.G. For whom a puddle is the sea? Adsorption of organic guests on hydrated MCM-41 silica. Langmuir 2020, 36, 11383–11392. [Google Scholar] [CrossRef]
- Lorente, P.; Shenderovich, I.G.; Golubev, N.S.; Denisov, G.S.; Buntkowsky, G.; Limbach, H.H. 1H/15N NMR chemical shielding, dipolar 15N,2H coupling and hydrogen bond geometry correlations in a novel series of hydrogen-bonded acid-base complexes of collidine with carboxylic acids. Magn. Reson. Chem. 2001, 39, 18–29. [Google Scholar] [CrossRef]
- Cattaneo, A.S.A.S.; Ferrara, C.; Marculescu, A.M.A.M.; Giannici, F.; Martorana, A.; Mustarelli, P.; Tealdi, C. Solid-state NMR characterization of the structure and thermal stability of hybrid organic-inorganic compounds based on a HLaNb2O7 Dion-Jacobson layered perovskite. Phys. Chem. Chem. Phys. 2016, 18, 21903–21912. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Crocker, M.; Herold, R.H.M.; Wilson, A.E.; Mackay, M.; Emeis, C.A.; Hoogendoorn, A.M. 1H NMR spectroscopy of titania: Chemical shift assignments for hydroxy groups in crystalline and amorphous forms of TiO2. J. Chem. Soc. Faraday Trans. 1996, 92, 2791–2798. [Google Scholar] [CrossRef]
- Yang, L.; Feng, N.; Wang, Q.; Chu, Y.; Xu, J.; Deng, F. Surface water loading on titanium dioxide modulates photocatalytic water splitting. Cell Rep. Phys. Sci. 2020, 1, 100013. [Google Scholar] [CrossRef]
- Jeantelot, G.; Ould-Chikh, S.; Sofack-Kreutzer, J.; Abou-Hamad, E.; Anjum, D.H.; Lopatin, S.; Harb, M.; Cavallo, L.; Basset, J.M. Morphology control of anatase TiO2 for well-defined surface chemistry. Phys. Chem. Chem. Phys. 2018, 20, 14362–14373. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Bodensteiner, M.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G. P=O Moiety as an ambidextrous hydrogen bond acceptor. J. Phys. Chem. C 2018, 122, 1711–1720. [Google Scholar] [CrossRef]
- Gutiérrez-Ortega, J.A.; Gómez-Salazar, S.; Shenderovich, I.G.; Manríquez-González, R. Efficiency and lead uptake mechanism of a phosphonate functionalized mesoporous silica through P/Pb association ratio. Mater. Chem. Phys. 2020, 239, 122037. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Rozhkova, Y.A.; Zukal, A.; Čejka, J.; Shenderovich, I.G. Mutable Lewis and Brønsted acidity of aluminated SBA-15 as revealed by NMR of adsorbed pyridine-15N. Langmuir 2011, 27, 12115–12123. [Google Scholar] [CrossRef]
- Witanowski, M.; Stefaniak, L.; Szymański, S.; Januszewski, H. External neat nitromethane scale for nitrogen chemical shifts. J. Magn. Reson. 1977, 28, 217–226. [Google Scholar] [CrossRef]
- Bishop, G.R.; Valente, E.J.; Whitehead, T.L.; Brown, K.L.; Hicks, R.P.; Davidson, V.L. Direct detection by 15N NMR of the tryptophan tryptophylquinone aminoquinol reaction intermediate of methylamine dehydrogenase. J. Am. Chem. Soc. 1996, 118, 12868–12869. [Google Scholar] [CrossRef]
- Banert, K.; Lehmann, J.; Quast, H.; Meichsner, G.; Regnat, D.; Seiferling, B. 15N-NMR spectra, tautomerism and diastereomerism of 4,5-dihydro-1H-1,2,3-triazoles. J. Chem. Soc. Perkin Trans. 2002, 2, 126–134. [Google Scholar] [CrossRef]
- Duthaler, R.O.; Roberts, J.D. Steric and electronic effects on 15N chemical shifts of saturated aliphatic amines and their hydrochlorides. J. Am. Chem. Soc. 1978, 100, 3889–3895. [Google Scholar] [CrossRef]
- Liu, H.; Li, M.; Luo, X.; Liang, Z.; Idem, R.; Tontiwachwuthikul, P. Investigation mechanism of DEA as an activator on aqueous MEA solution for postcombustion CO2 capture. AIChE J. 2018, 64, 2515–2525. [Google Scholar] [CrossRef]
- Birnie, D.P.; Bendzko, N.J. 1H- and 13C-NMR observation of the reaction of acetic acid with titanium isopropoxide. Mater. Chem. Phys. 1999, 59, 26–35. [Google Scholar] [CrossRef]
- Voelkel, R. High-resolution solid-state 13C-NMR spectroscopy of polymers. Angew. Chem. Int. Ed. Engl. 1988, 27, 1468–1483. [Google Scholar] [CrossRef]
- Rodionov, I.A.; Fateev, S.A.; Zvereva, I.A. Synthesis of a New Layered Rb2Nd2Ti3O10 Oxide, Its Hydration and Protonation. Glas. Phys. Chem. 2017, 43, 593–596. [Google Scholar] [CrossRef]
- Silyukov, O.; Chislov, M.; Burovikhina, A.; Utkina, T.; Zvereva, I. Thermogravimetry study of ion exchange and hydration in layered oxide materials. J. Therm. Anal. Calorim. 2012, 110, 187–192. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors upon request. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | a (Å) | c (Å) | d (Å) |
|---|---|---|---|
| HLT3 | 3.809 | 27.55 | 13.78 |
| HLT3 × MeNH2 | 3.830 | 39.94 | 18.97 |
| HLT3 × BuNH2 | 3.828 | 24.23 | 24.23 |
| HLT3 × MeOH | 3.800 | 35.40 | 17.70 |
| HLT3 × MEA | 3.832 | 38.97 | 19.49 |
| Sample | Composition | Total Mass Loss in TG (%) | Td (°C) |
|---|---|---|---|
| HLT3 | H2La2Ti3O10(H2O)0.05 | 3.01 | 280 |
| HLT3 × MeNH2 | H2La2Ti3O10(MeNH2)0.75(H2O)0.20 | 7.29 | 75 |
| HLT3 × BuNH2 | H2La2Ti3O10(BuNH2)0.60(H2O)0.20 | 10.3 | 75 |
| HLT3 × MeOH | H1.3La2Ti3O9.3(MeO)0.75(H2O)0.05 | 4.75 | 300 |
| HLT3 × MEA | H2La2Ti3O10(MEA)0.65(H2O)0.10 | 9.47 | 250 |
| Compound | Carbon Site | δiso (ppm) | TCH (μs) | T1ρ(H) (ms) |
|---|---|---|---|---|
| HLT3 × MeOH | C | 66.2 ± 0.1 | 233 ± 29 | 17.1 ± 2.8 |
| HLT3 × MeNH2 | C | 25.2 ± 0.1 | 131 ± 10 | 1.5 ± 0.1 |
| HLT3 × BuNH2 | C1 | 39.7 ± 0.1 | 79 ± 6 | 1.3 ± 0.1 |
| C2 | 30.4 ± 0.1 | 129 ± 9 | 1.8 ± 0.1 | |
| C3 | 21.1 ± 0.1 | 182 ± 13 | 2.3 ± 0.2 | |
| C4 | 13.8 ± 0.1 | 700 ± 93 | 5.8 ± 0.6 | |
| HLT3 × MEA | C1,C1* | 42.4 ± 0.1 | 64 ± 9 | 2.3 ± 0.4 |
| C2 | 58.7 ± 0.1 | 87 ± 11 | 1.8 ± 0.3 | |
| C2* | 69.4 ± 0.1 | 67 ± 14 | 1.5 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shelyapina, M.G.; Silyukov, O.I.; Lushpinskaia, I.P.; Kurnosenko, S.A.; Mazur, A.S.; Shenderovich, I.G.; Zvereva, I.A. NMR Study of Intercalates and Grafted Organic Derivatives of H2La2Ti3O10. Molecules 2020, 25, 5229. https://doi.org/10.3390/molecules25225229
Shelyapina MG, Silyukov OI, Lushpinskaia IP, Kurnosenko SA, Mazur AS, Shenderovich IG, Zvereva IA. NMR Study of Intercalates and Grafted Organic Derivatives of H2La2Ti3O10. Molecules. 2020; 25(22):5229. https://doi.org/10.3390/molecules25225229
Chicago/Turabian StyleShelyapina, Marina G., Oleg I. Silyukov, Irina P. Lushpinskaia, Sergey A. Kurnosenko, Anton S. Mazur, Ilya G. Shenderovich, and Irina A. Zvereva. 2020. "NMR Study of Intercalates and Grafted Organic Derivatives of H2La2Ti3O10" Molecules 25, no. 22: 5229. https://doi.org/10.3390/molecules25225229
APA StyleShelyapina, M. G., Silyukov, O. I., Lushpinskaia, I. P., Kurnosenko, S. A., Mazur, A. S., Shenderovich, I. G., & Zvereva, I. A. (2020). NMR Study of Intercalates and Grafted Organic Derivatives of H2La2Ti3O10. Molecules, 25(22), 5229. https://doi.org/10.3390/molecules25225229