
Synthesis of Chromium(II) Complexes with Chelating Bis(alkoxide) Ligand and Their Reactions with Organoazides and Diazoalkanes


Abstract
1. Introduction
2. Results and Discussion
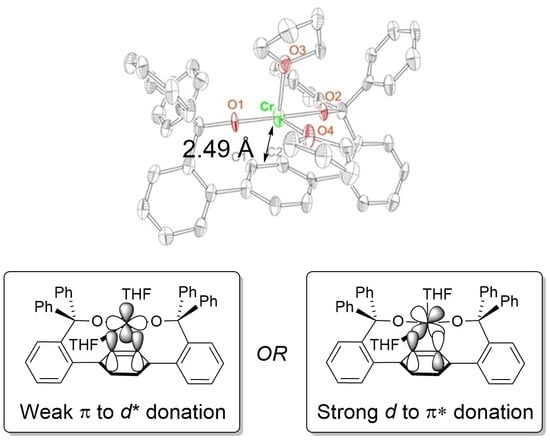
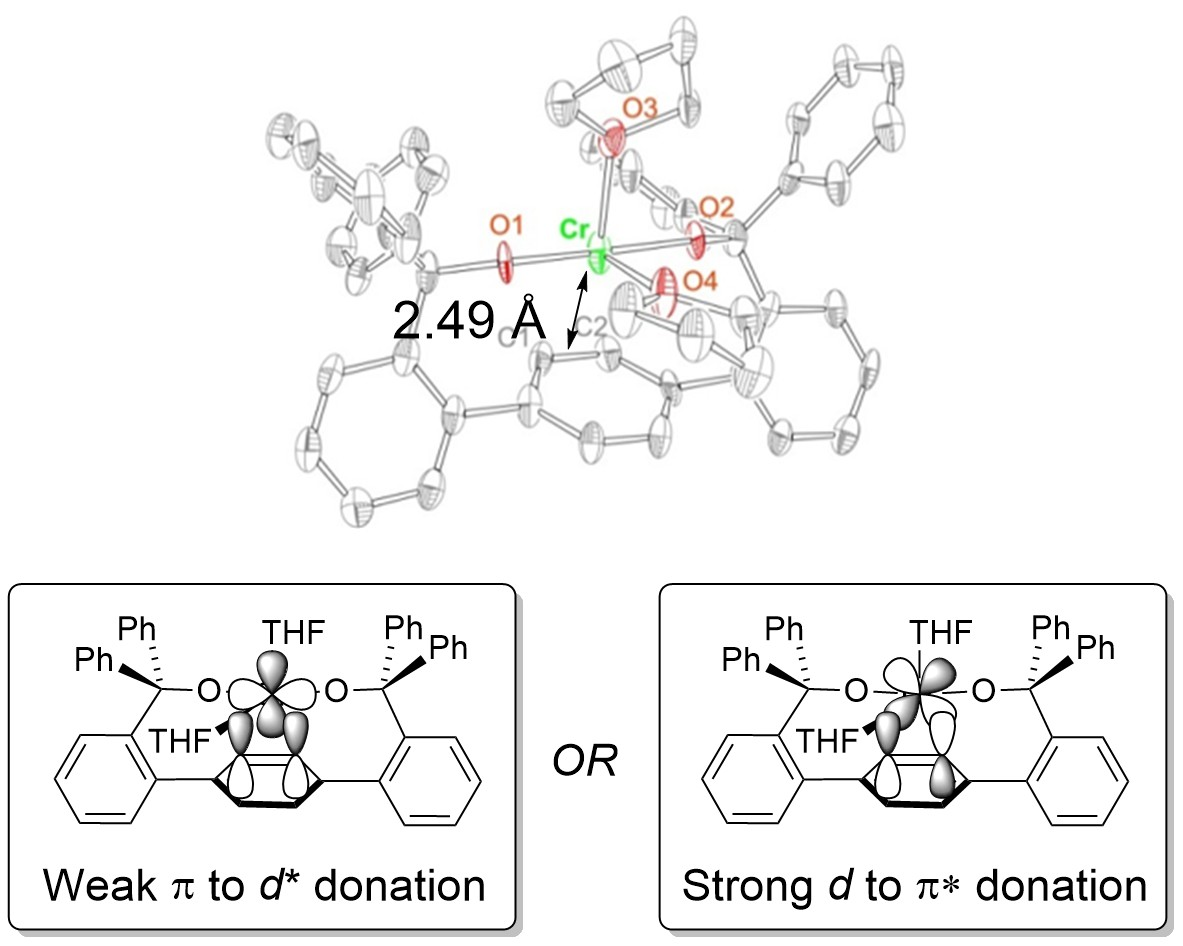
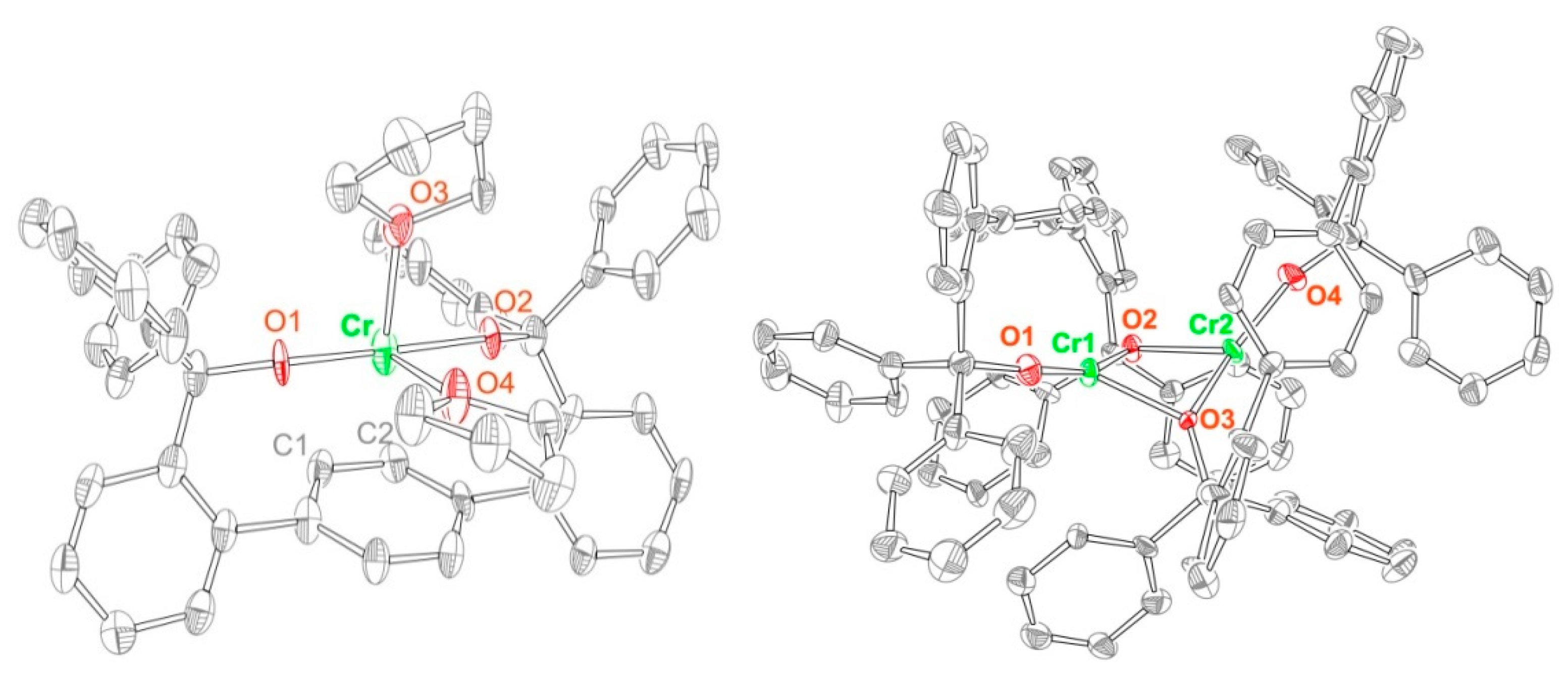
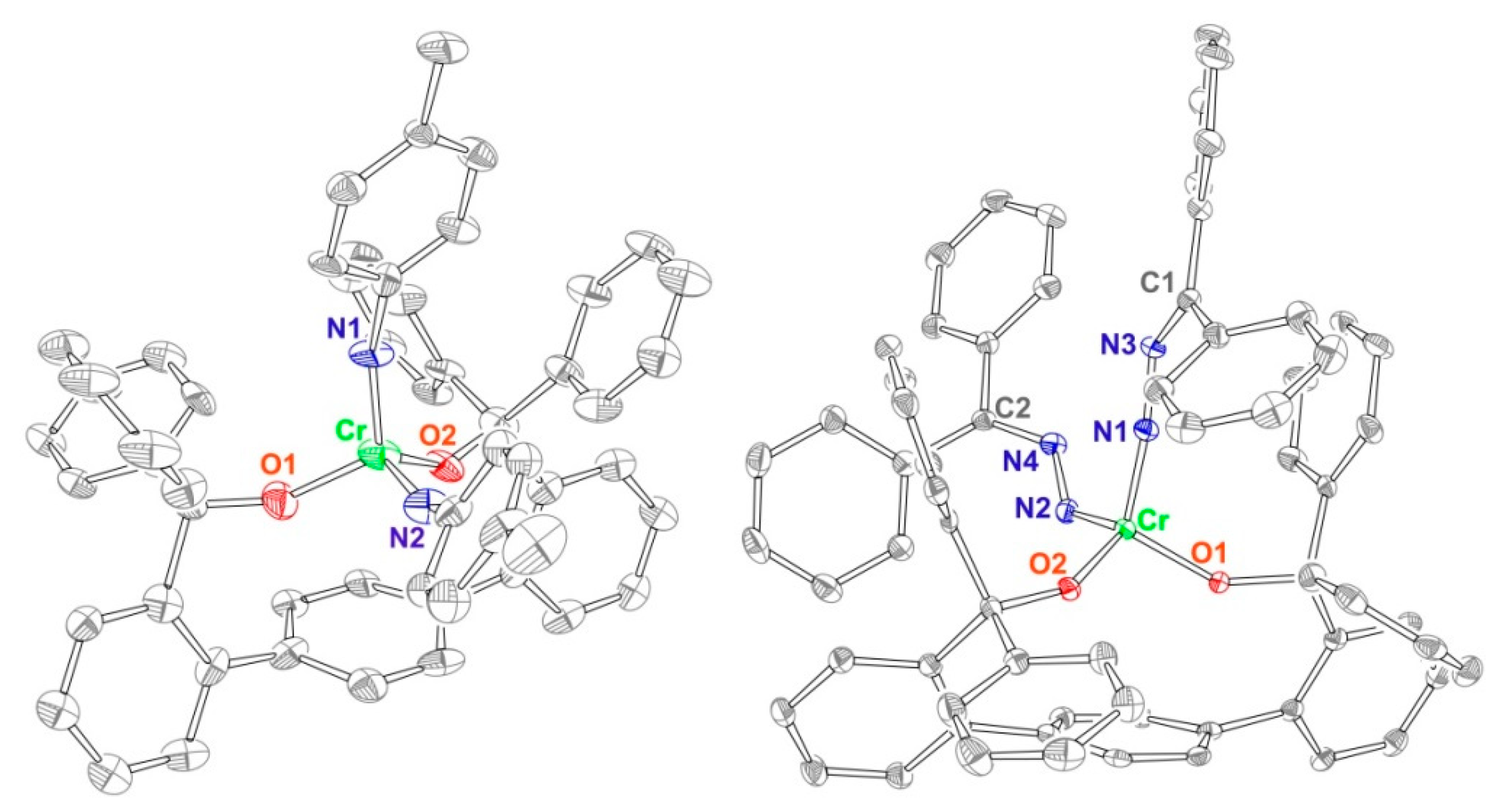
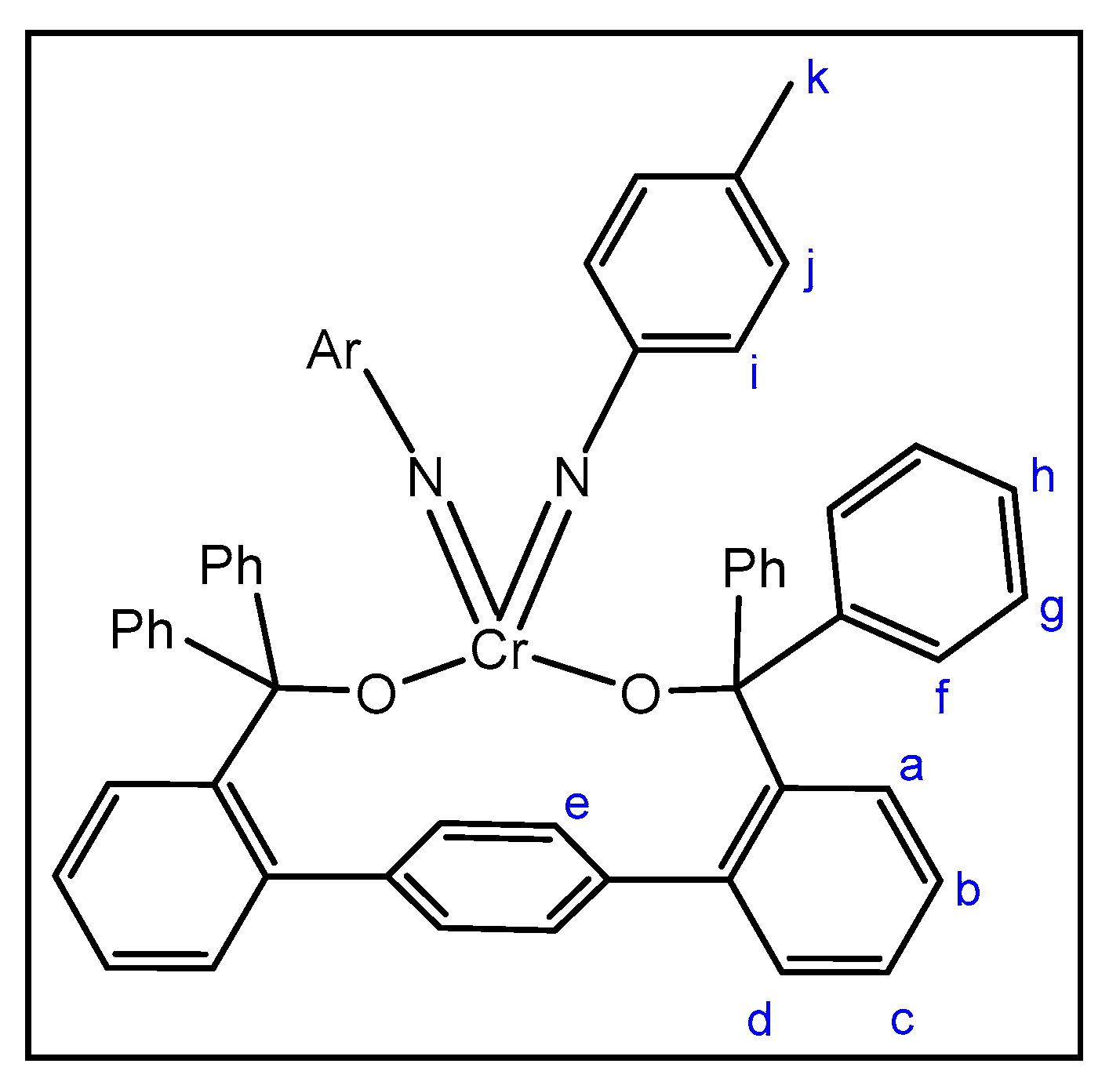
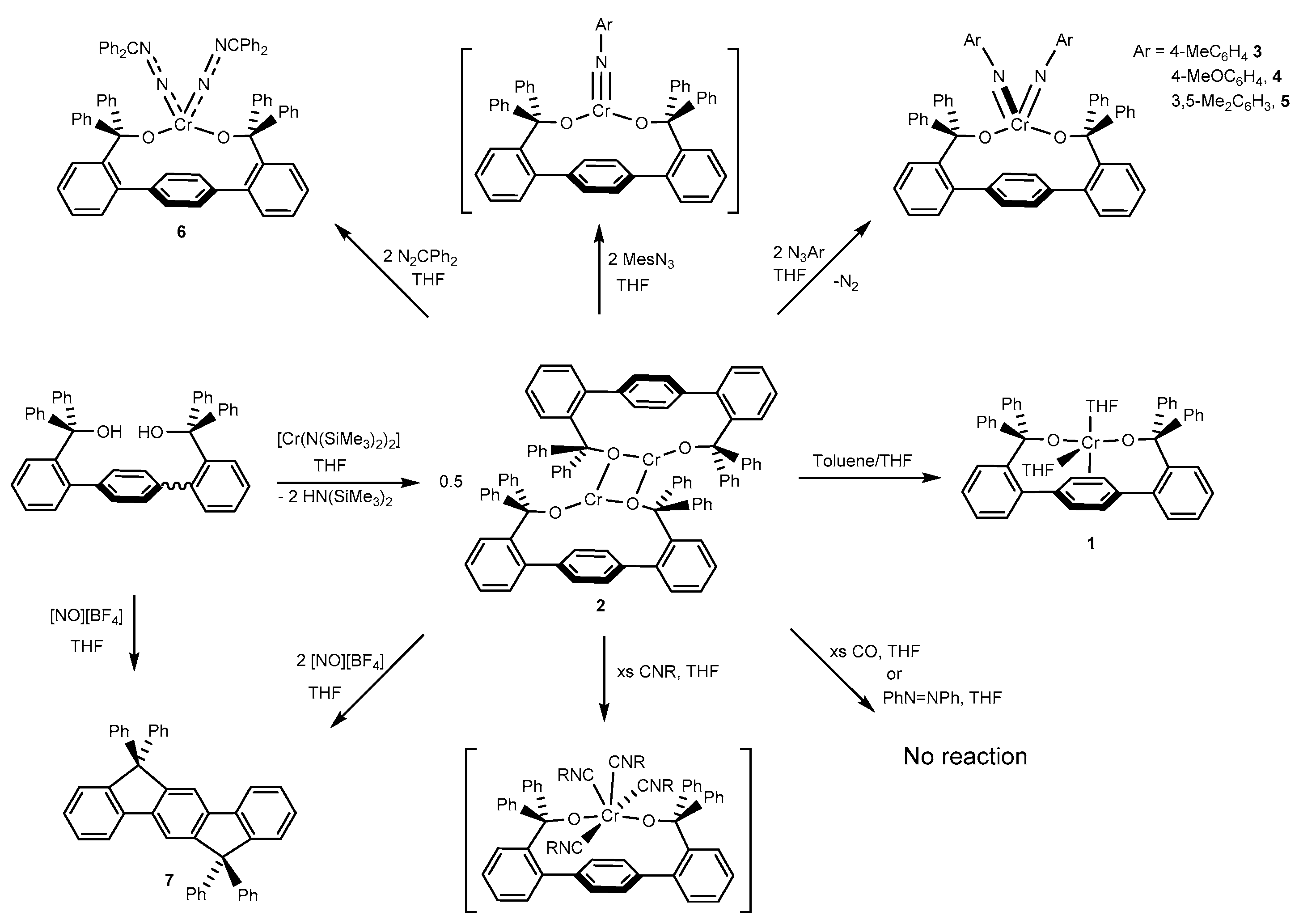
2.1. Synthesis and Structures of Cr(II) Precursors
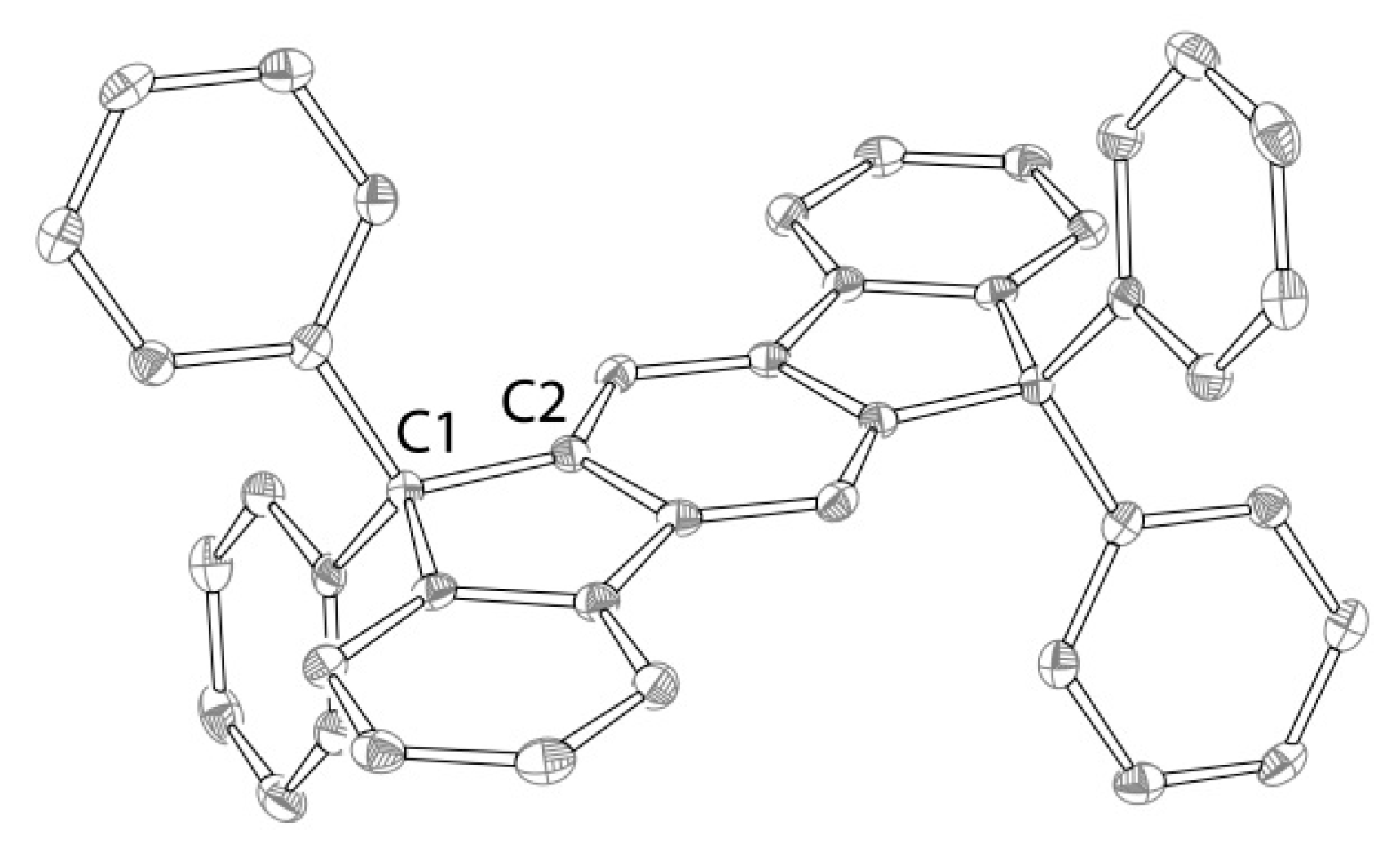
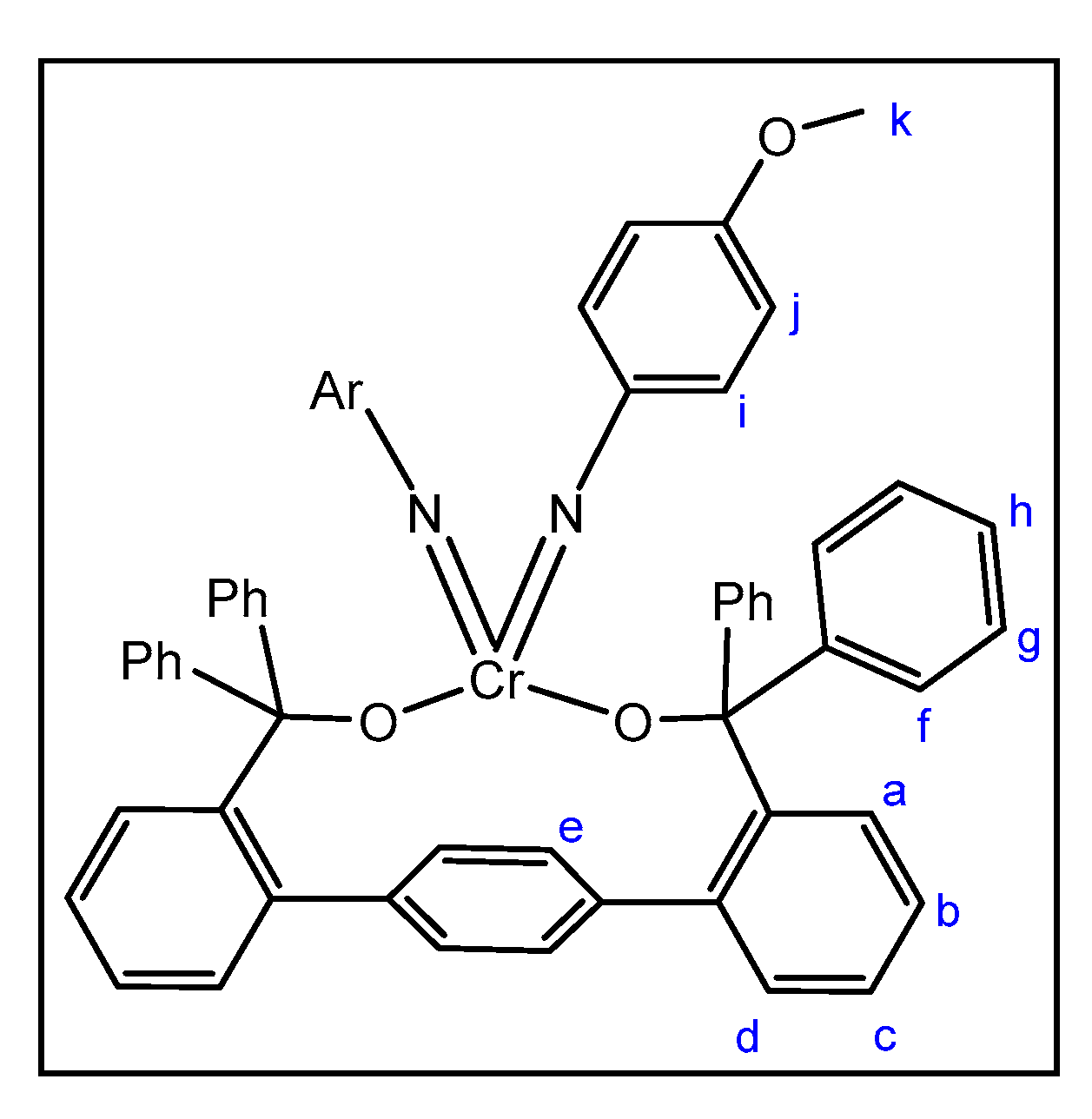
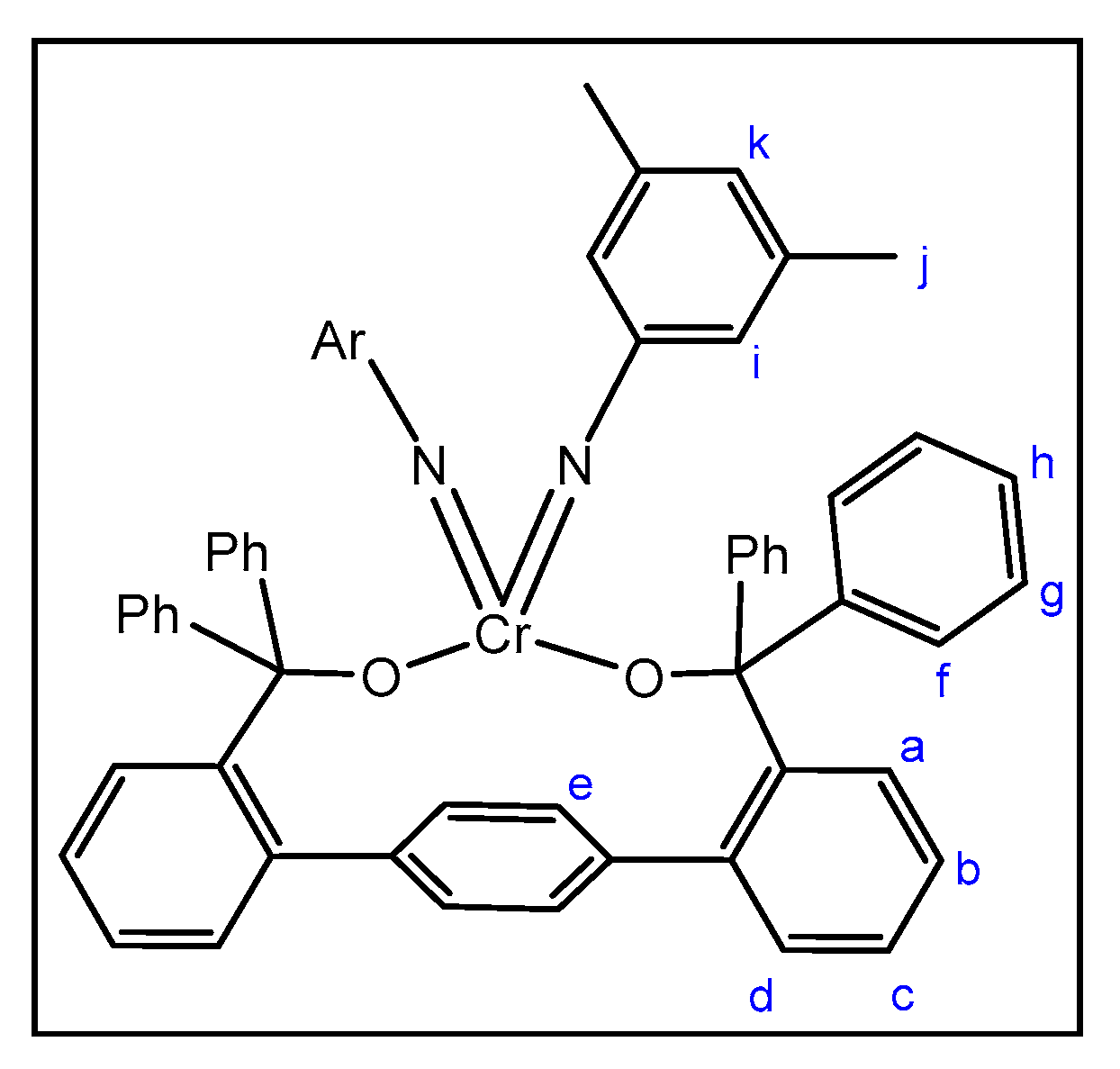
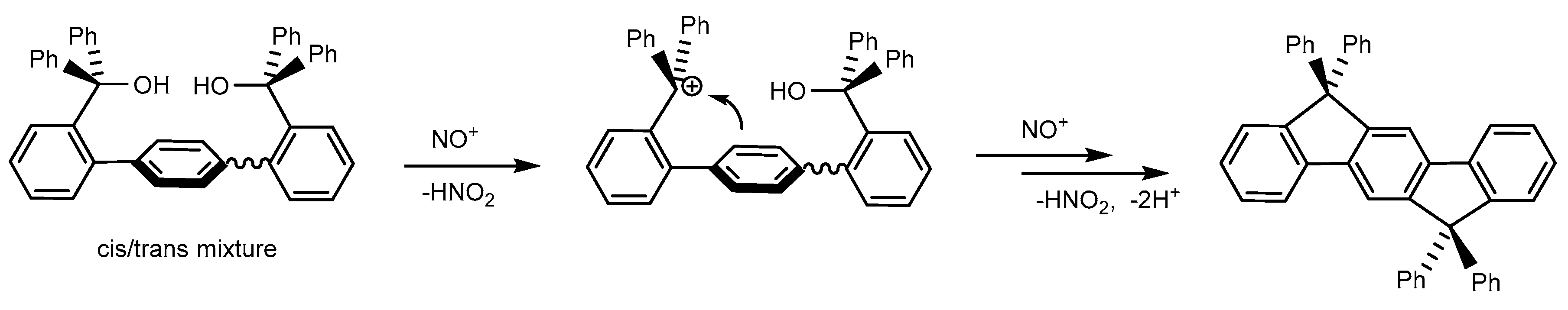
2.2. Reactions of Chromium(II) Complexes with Organoazides, Azoarenes and Diazoalkanes
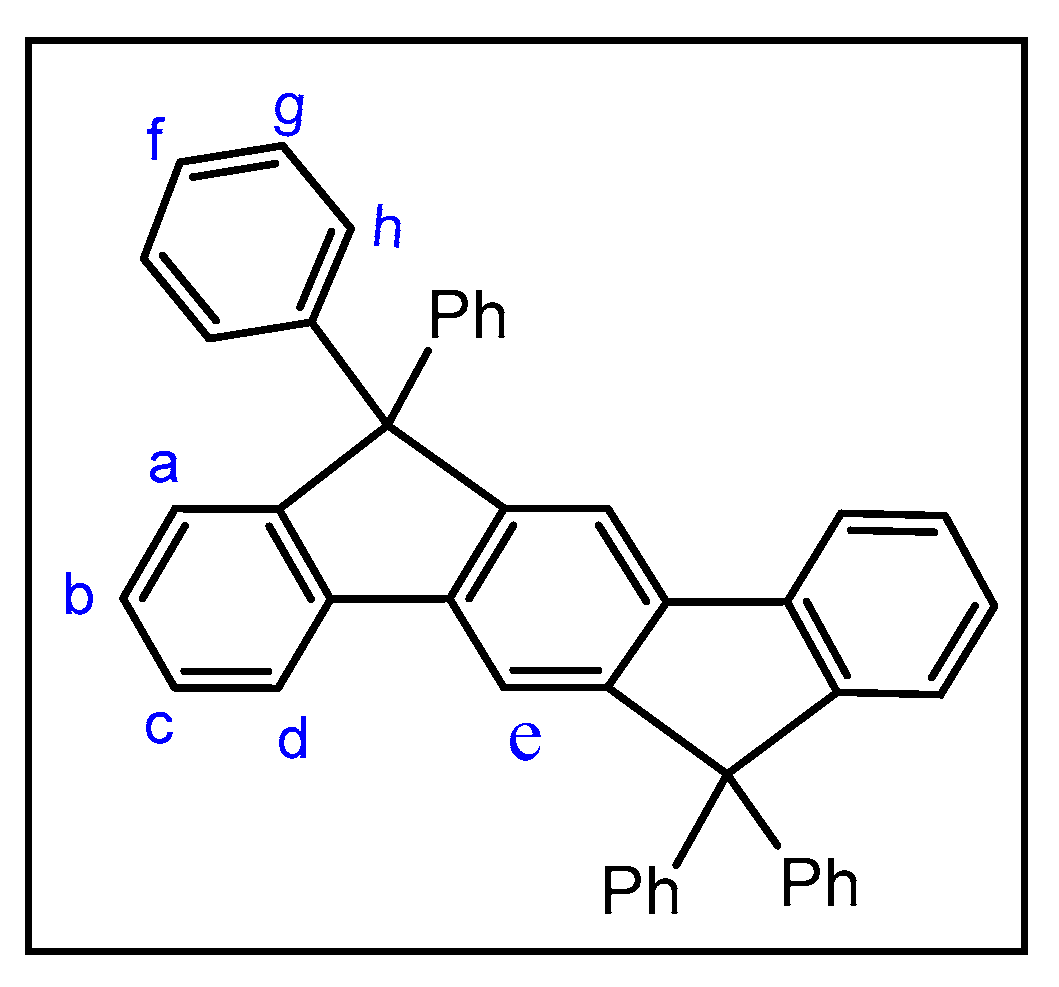
2.3. Reactivity of Cr(II) with CO, CNR and NO+
2.4. Catalytic Reactivity in the Formation of Carbodiimides
3. Materials and Methods
3.1. General
3.2. Synthesis of Cr Complexes 1–6 and Compound 7
3.3. X-ray Crystallographic Details
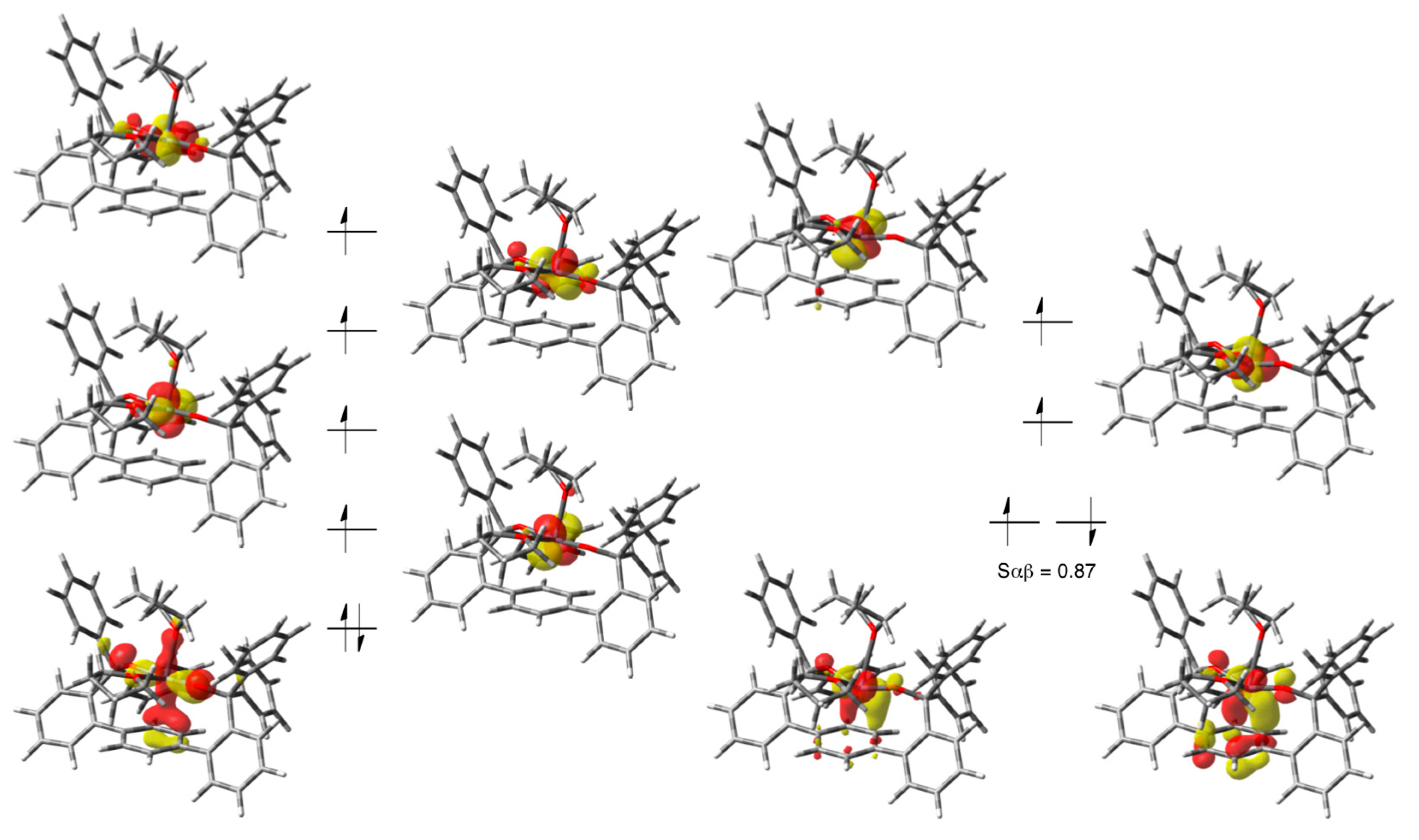
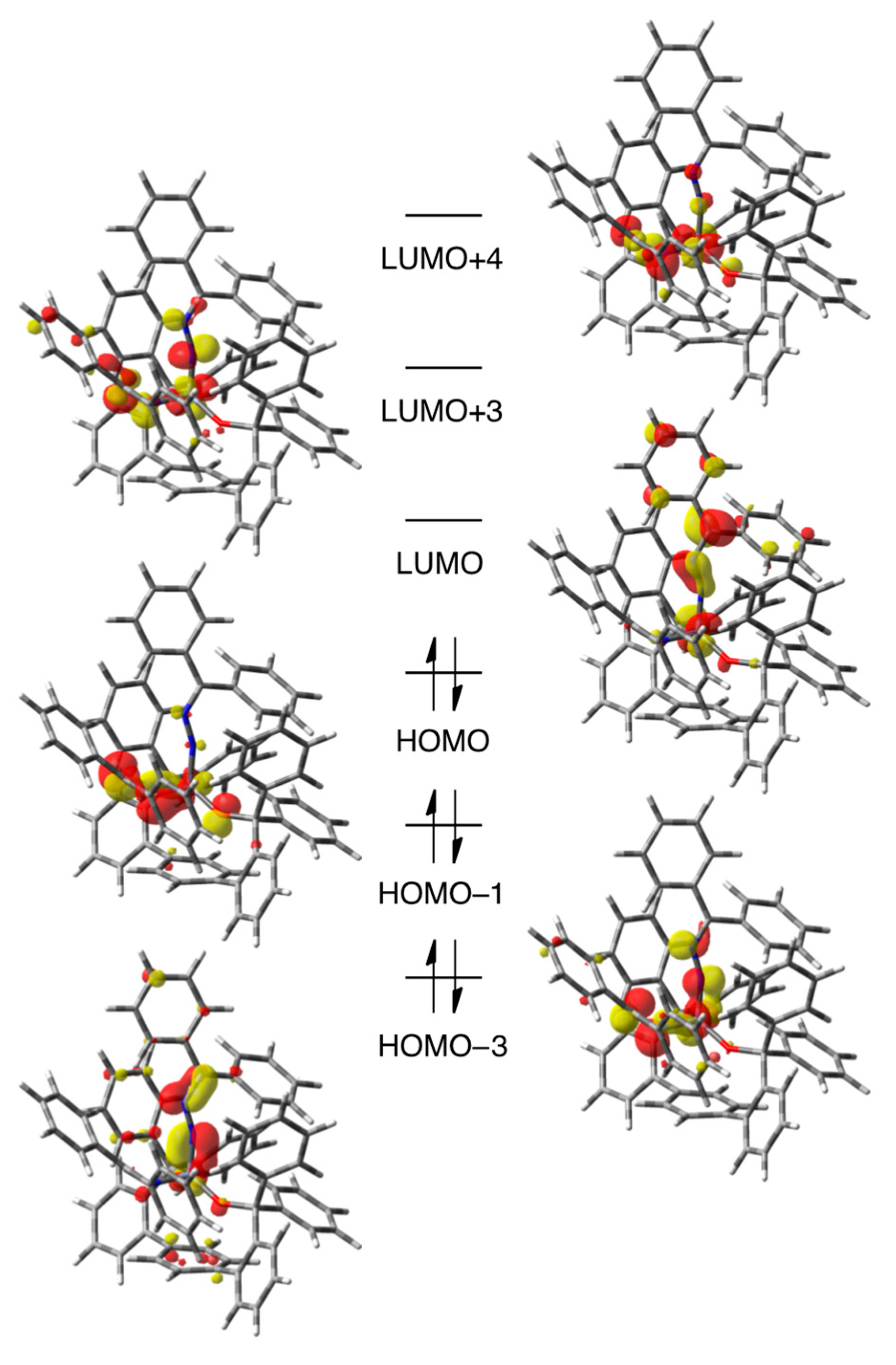
3.4. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jenkins, D.M. Atom-Economical C2 + N1 aziridination: Progress towards catalytic intermolecular reactions using alkenes and aryl azides. Synlett 2012, 23, 1267–1270. [Google Scholar] [CrossRef]
- Driver, T.G. Recent advances in transition metal-catalyzed N-Atom transfer reactions of azides. Org. Biomol. Chem. 2010, 8, 3831–3846. [Google Scholar] [CrossRef]
- Shin, K.; Kim, H.; Chang, S. Transition-Metal-Catalyzed C-N Bond forming reactions using organic azides as the nitrogen source: A journey for the mild and versatile C-H Amination. Acc. Chem. Res. 2015, 48, 1040–1052. [Google Scholar] [CrossRef]
- Wang, P.; Deng, L. Recent advances in iron-catalyzed C-H bond amination via iron imido intermediate. Chin. J. Chem. 2018, 36, 1222–1240. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, X.P. Catalytic C–H functionalization by metalloporphyrins: Recent developments and future directions. Chem. Soc. Rev. 2011, 40, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Katsuki, T. Asymmetric nitrene transfer reactions: Sulfimidation, aziridination and C–H amination using azide compounds as nitrene precursors. Chem. Rec. 2014, 14, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, P.F.; van der Vlugt, J.I.; Schneider, S.; de Bruin, B. Nitrene radical intermediates in catalytic synthesis. Chem. Eur. J. 2017, 23, 13819–13829. [Google Scholar] [CrossRef]
- Baek, Y.; Hennessy, E.T.; Betley, T.A. Direct manipulation of metal imido geometry: Key principles to enhance C–H amination efficacy. J. Am. Chem. Soc. 2019, 141, 16944–16953. [Google Scholar] [CrossRef]
- Wilding, M.J.T.; Iovan, D.A.; Betley, T.A. High-Spin iron imido complexes competent for C–H bond amination. J. Am. Chem. Soc. 2017, 139, 12043–12049. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, J.; Leng, X.; Lohmiller, T.; Schnegg, A.; Bill, E.; Ye, S.; Deng, L. A Two-Coordinate iron(II) imido complex with NHC ligation: Synthesis, characterization, and its diversified reactivity of nitrene transfer and C–H bond activation. Inorg. Chem. 2019, 58, 7634–7644. [Google Scholar] [CrossRef]
- Du, J.; Wang, L.; Xie, M.; Deng, L. A Two-Coordinate cobalt(II) imido complex with NHC ligation: Synthesis, structure, and reactivity. Angew. Chem. Int. Ed. 2015, 54, 12640–12644. [Google Scholar] [CrossRef] [PubMed]
- Cowley, R.E.; Golder, M.R.; Eckert, N.A.; Al-Afyouni, M.H.; Holland, P.L. Mechanism of catalytic nitrene transfer using iron(I)-isocyanide complexes. Organometallics 2013, 32, 5289–5298. [Google Scholar] [CrossRef]
- Bagh, B.; Broere, D.L.J.; Sinha, V.; Kuijpers, P.F.; van Leest, N.P.; de Bruin, B.; Demeshko, S.; Siegler, M.A.; van der Vlugt, J.I. Catalytic synthesis of N-heterocycles via direct C(sp3)–H amination using an air-stable iron(III) species with a redox-active ligand. J. Am. Chem. Soc. 2017, 139, 5117–5124. [Google Scholar] [CrossRef] [PubMed]
- Vreeken, V.; Baij, L.; de Bruin, B.; Siegler, M.A.; van der Vlugt, J.I. N-Atom transfer via thermal or photolytic activation of a Co-azido complex with a PNP pincer ligand. Dalton Trans. 2017, 46, 7145–7149. [Google Scholar] [CrossRef] [PubMed]
- Hakey, B.M.; Darmon, J.M.; Akhmedov, N.G.; Petersen, J.L.; Milsmann, C. Reactivity of pyridine dipyrrolide iron(II) complexes with organic azides: C–H amination and iron tetrazene formation. Inorg. Chem. 2019, 58, 11028–11042. [Google Scholar] [CrossRef]
- Goswami, M.; de Bruin, B. Porphyrin Co(III)-nitrene radical mediated pathway for synthesis of o-aminoazobenzenes. Molecules 2018, 23, 1052. [Google Scholar] [CrossRef]
- Mankad, N.P.; Müller, P.; Peters, J.C. Catalytic N−N coupling of aryl azides to yield azoarenes via trigonal bipyramid iron−nitrene intermediates. J. Am. Chem. Soc. 2010, 132, 4083–4085. [Google Scholar] [CrossRef]
- Lang, K.; Torker, S.; Wojtas, L.; Zhang, X.P. Asymmetric induction and enantiodivergence in catalytic radical C–H Amination via enantiodifferentiative H-Atom abstraction and stereoretentive radical substitution. J. Am. Chem. Soc. 2019, 141, 12388–12396. [Google Scholar] [CrossRef]
- Jacobs, B.P.; Wolczanski, P.T.; Jiang, Q.; Cundari, T.R.; MacMillan, S.N. Rare examples of Fe(IV) alkyl-imide migratory insertions: Impact of Fe-C covalency in (Me2IPr)Fe(=NAd)R2 (R = neoPe, 1-nor). J. Am. Chem. Soc. 2017, 139, 12145–12148. [Google Scholar] [CrossRef]
- Powers, I.G.; Andjaba, J.M.; Luo, X.; Mei, J.; Uyeda, C. catalytic azoarene synthesis from aryl azides enabled by a dinuclear Ni complex. J. Am. Chem. Soc. 2018, 140, 4110–4118. [Google Scholar] [CrossRef]
- Spasyuk, D.M.; Carpenter, S.H.; Kefalidis, C.E.; Piers, W.E.; Neidig, M.L.; Maron, L. Facile hydrogen atom transfer to Iron(III) imido radical complexes supported by a dianionic pentadentate ligand. Chem. Sci. 2016, 7, 5939–5944. [Google Scholar] [CrossRef] [PubMed]
- Nugent, W.A.; Mayer, J.M. Metal-Ligand Multiple Bonds: The Chemistry of Transition Metal Complexes Containing Oxo, Nitrido, Imido, Alkylidene, or Alkylidyne Ligands, 1st ed.; Wiley-Interscience: New York, NY, USA, 1988. [Google Scholar]
- Danopoulos, A.A.; Hussain-Bates, B.; Hursthouse, M.B.; Leung, W.H.; Wilkinson, G. t-Butylimido complexes of chromium(V). X-ray crystal structure of t-butylimidotrichlorobis(ethyldiphenylphosphine)chromium(V). J. Chem. Soc. Chem. Commun. 1990, 23, 1678–1679. [Google Scholar] [CrossRef]
- Danopoulos, A.A.; Hankin, D.M.; Wilkinson, G.; Cafferkey, S.M.; Sweet, T.K.N.; Hursthouse, M.B. Amido, imido and carbene complexes of chromium. Polyhedron 1997, 16, 3879–3892. [Google Scholar] [CrossRef]
- Edwards, N.Y.; Eikey, R.A.; Loring, M.I.; Abu-Omar, M.M. High-Valent imido complexes of manganese and chromium corroles. Inorg. Chem. 2005, 44, 3700–3708. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Yap, G.P.A.; Theopold, K.H. Structure and reactivity of chromium(VI) alkylidenes. J. Am. Chem. Soc. 2018, 140, 7088–7091. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.-H. Synthesis and reactivity of organoimido complexes of chromium. Eur. J. Inorg. Chem. 2003, 583–593. [Google Scholar] [CrossRef]
- Beaumier, E.P.; Billow, B.S.; Singh, A.K.; Biros, S.M.; Odom, A.L. A complex with nitrogen single, double, and triple bonds to the same chromium atom: Synthesis, structure, and reactivity. Chem. Sci. 2016, 7, 2532–2539. [Google Scholar] [CrossRef]
- Keller, C.L.; Kern, J.L.; Terry, B.D.; Roy, S.; Jenkins, D.M. Catalytic aziridination with alcoholic substrates via a chromium tetracarbene catalyst. Chem. Commun. 2018, 54, 1429–1432. [Google Scholar] [CrossRef]
- Elpitiya, G.R.; Malbrecht, B.J.; Jenkins, D.M. A Chromium(II) tetracarbene complex allows unprecedented oxidative group transfer. Inorg. Chem. 2017, 56, 14101–14110. [Google Scholar] [CrossRef]
- Yousif, M.; Tjapkes, D.J.; Lord, R.L.; Groysman, S. Catalytic formation of asymmetric carbodiimides at mononuclear Chromium (II/IV) bis(alkoxide) complexes. Organometallics 2015, 34, 5119–5128. [Google Scholar] [CrossRef]
- Heins, S.P.; Morris, W.D.; Wolczanski, P.T.; Lobkovsky, E.B.; Cundari, T.R. Nitrene insertion into C-C and C-H bonds of diamide diimine ligands ligated to chromium and iron. Angew. Chem. Int. Ed. 2015, 54, 14407–14411. [Google Scholar] [CrossRef] [PubMed]
- Bellow, J.A.; Yousif, M.; Groysman, S. Discrete complexes of 3d metals with monodentate bulky alkoxide ligands and their reactivity in bond activation and bond formation reactions. Comments Inorg. Chem. 2015, 36, 92–122. [Google Scholar] [CrossRef]
- Grass, A.; Wannipurage, D.; Lord, R.L.; Groysman, S. Group-transfer chemistry at transition metal centers in bulky alkoxide ligand environments. Coord. Chem. Rev. 2019, 400, 1–16. [Google Scholar] [CrossRef]
- Kurup, S.S.; Wannipurage, D.; Lord, R.L.; Groysman, S. An iron complex with a new chelating bis(alkoxide) ligand leads to an active nitrene dimerization catalyst for a variety of para- and meta-substituted azide precursors. Chem. Commun. 2019, 55, 10780–10783. [Google Scholar] [CrossRef] [PubMed]
- Yousif, M.; Wannipurage, D.; Huizenga, C.D.; Washnock-Schmid, E.; Peraino, N.J.; Ozarowski, A.; Stoian, S.A.; Lord, R.L.; Groysman, S. Catalytic nitrene homocoupling by an iron(II) bis(alkoxide) complex: Bulking up the alkoxide enables a wider range of substrates and provides insight into the reaction mechanism. Inorg. Chem. 2018, 57, 9425–9438. [Google Scholar] [CrossRef] [PubMed]
- Bellow, J.A.; Yousif, M.; Cabelof, A.C.; Lord, R.L.; Groysman, S. Reactivity modes of an iron bis(alkoxide) complex with aryl azides: Catalytic nitrene coupling vs formation of iron(III) imido dimers. Organometallics 2015, 34, 2917–2923. [Google Scholar] [CrossRef]
- Yousif, M.; Cabelof, A.C.; Martin, P.D.; Lord, R.L.; Groysman, S. Synthesis of a mononuclear, non-square-planar chromium(ii) bis(alkoxide) complex and its reactivity toward organic carbonyls and CO2. Dalton Trans. 2016, 45, 9794–9804. [Google Scholar] [CrossRef]
- Murray, B.D.; Hope, H.; Power, P.P. An Unusual C-C Bond cleavage in a bulky metal alkoxide: Syntheses and X-ray crystal structures of three-coordinate Mn(II) and Cr(II) complexes containing the di-tert-butylmethoxide ligand. J. Am. Chem. Soc. 1985, 107, 169–173. [Google Scholar] [CrossRef]
- Sydora, O.L.; Kuiper, D.S.; Wolczanski, P.T.; Lobkovsky, E.B.; Dinescu, A.; Cundari, T.R. The butterfly dimer [(tBu3SiO)Cr]2(μ-OSitBu3)2 and its oxidative cleavage to (tBu3SiO)2Cr(N−NCPh2)2 and (tBu3SiO)2CrN(2,6-Ph2-C6H3). Inorg. Chem. 2006, 45, 2008–2021. [Google Scholar] [CrossRef]
- Ohki, Y.; Takikawa, Y.; Hatanaka, T.; Tatsumi, K. Reductive N−N Bond cleavage of diphenylhydrazine and azobenzene induced by coordinatively unsaturated Cp*Fe{N(SiMe3)2}. Organometallics 2006, 25, 3111–3113. [Google Scholar] [CrossRef]
- Bellows, S.M.; Arnet, N.A.; Gurubasavaraj, P.M.; Brennessel, W.W.; Bill, E.; Cundari, T.R.; Holland, P.L. The Mechanism of N-N double bond cleavage by an Iron(II)-hydride complex. J. Am. Chem. Soc. 2016, 138, 12112–12123. [Google Scholar] [CrossRef] [PubMed]
- Davis-Gilbert, Z.W.; Wen, X.; Goodpaster, J.D.; Tonks, I.A. Mechanism of Ti-catalyzed oxidative nitrene transfer in [2 + 2 + 1] pyrrole synthesis from alkynes and azobenzene. J. Am. Chem. Soc. 2018, 140, 7267–7281. [Google Scholar] [CrossRef]
- Kawakita, K.; Beaumier, E.P.; Kakiuchi, Y.; Tsurugi, H.; Tonks, I.A.; Mashima, K. Bis(imido)vanadium(V)-catalyzed [2 + 2 + 1] coupling of alkynes and azobenzenes giving multisubstituted pyrroles. J. Am. Chem. Soc. 2019, 141, 4194–4198. [Google Scholar] [CrossRef] [PubMed]
- Copéret, C.; Comas-Vives, A.; Conley, M.P.; Estes, D.P.; Fedorov, A.; Mougel, V.; Nagae, H.; Núñez-Zarur, F.; Zhizhko, P.A. surface organometallic and coordination chemistry toward single-site heterogeneous catalysts: Strategies, methods, structures, and activities. Chem. Rev. 2016, 116, 323–421. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Yap, G.P.A.; Theopold, K.H. Synthesis, characterization, and reactivity of chromium(VI) alkylidenes. Organometallics 2019, 38, 4593–4600. [Google Scholar] [CrossRef]
- Grass, A.; Dewey, N.S.; Lord, R.L.; Groysman, S. Ketenimine formation catalyzed by a high valent cobalt carbene in bulky alkoxide ligand environment. Organometallics 2019, 38, 962–972. [Google Scholar] [CrossRef]
- Grass, A.; Stoian, S.A.; Lord, R.L.; Groysman, S. Transition metal-mediated reductive coupling of diazoesters. Chem. Commun. 2019, 55, 8458–8461. [Google Scholar] [CrossRef]
- Hempe, M.; Reggelin, M. Molecular Packing and Morphological Stability of Dihydro-indeno[1,2-b]fluorenes in the Context of Their substitution Pattern. RSC Adv. 2017, 7, 47183–47189. [Google Scholar] [CrossRef]
- Poriel, C.; Liang, J.-J.; Rault-Berthelot, J.; Barrière, F.; Cocherel, N.; Slawin, A.M.Z.; Horhant, D.; Virboul, M.; Alcaraz, G.; Audebrand, N.; et al. Dispirofluorene–Indenofluorene derivatives as new building blocks for blue organic electroluminescent devices and electroactive polymers. Chem. Eur. J. 2007, 13, 10055–10069. [Google Scholar] [CrossRef]
- Bradley, D.C.; Hursthouse, M.B.; Newing, C.W.; Welch, A. Square planar and tetrahedral chromium(II) complexes; crystal structure determinations. J. Chem. Soc. Chem. Commun. 1972, 567–568. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence, and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Bootsma, A.N.; Wheeler, S. Popular integration grids can result in large errors in dft-computed free energies. ChemRxiv Preprint 2019. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
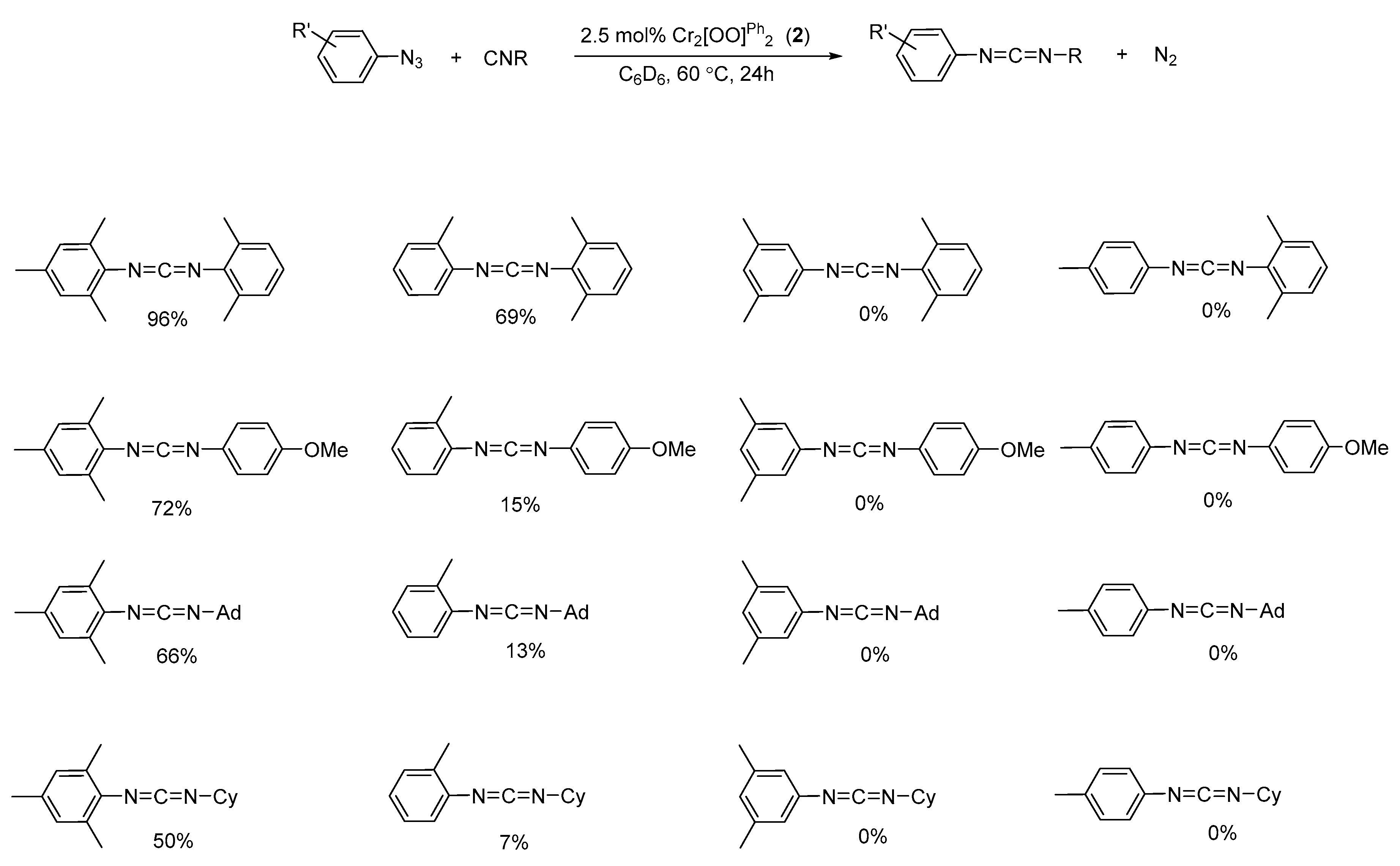{kind=link}
| Complex | 1 | 2 | 3 | 6 | 7 |
|---|---|---|---|---|---|
| formula | C52H46CrO4 × 2.5C7H8 | 2C88H64Cr2O4 | C58H46CrN2O2 | C70H52CrN4O2 × 0.5C6H12 × C4H8O | C22H15 × CH2Cl2 |
| fw, g/mol | 1020.25 | 2578.80 | 854.97 | 1147.34 | 364.27 |
| crystal system | Triclinic | Triclinic | Monoclinic | Triclinic | Triclinic |
| space group | P-1 | P-1 | P21/c | P-1 | P-1 |
| a (Å) | 9.946 (11) | 13.9851 (4) | 17.38 (15) | 10.1831 (6) | 8.7464 (5) |
| b (Å) | 14.298 (16)) | 19.4602 (6) | 13.03 (11) | 13.0107 (7) | 8.7610 (5) |
| c (Å) | 19.19 (2) | 30.4287 (10) | 28.58 (19) | 23.0961 (13) | 12.4072 (7) |
| α (deg) | 83.38 (3) | 81.683 (2) | 90.00 | 100.964 (3) | 76.995 (2) |
| β (deg) | 82.55 (2) | 77.609 (2) | 127.4 (4) | 101.580 (3) | 78.928 (2) |
| γ (deg) | 82.44 (2) | 74.125 (2) | 90.00 | 99.999 (3) | 70.505 (2) |
| V (Å3) | 2668 (5) | 7747.2 (4) | 5142 (71) | 2870.7 (3) | 866.13 (9) |
| Z | 2 | 4 | 4 | 2 | 2 |
| dcalcd, g/cm3 | 1.270 | 1.1055 | 1.104 | 1.327 | 1.397 |
| μ, mm−1 | 0.266 | 2.673 | 0.263 | 0.257 | 0.377 |
| T (K) | 100(2) | 172.99 | 100 (2) | 100 (2) | 100 (2) |
| 2θ, deg | 49.00 | 128.14 | 47.44 | 52.96 | 55.02 |
| R1a [(I > 2σ)] | 0.0981 | 0.0827 | 0.0727 | 0.0730 | 0.0388 |
| wR2b [(I > 2σ)] | 0.2070 | 0.1875 | 0.1512 | 0.1710 | 0.0803 |
| GOFc (F2) | 0.973 | 0.7573 | 0.881 | 1.037 | 1.041 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurup, S.S.; Staples, R.J.; Lord, R.L.; Groysman, S. Synthesis of Chromium(II) Complexes with Chelating Bis(alkoxide) Ligand and Their Reactions with Organoazides and Diazoalkanes. Molecules 2020, 25, 273. https://doi.org/10.3390/molecules25020273
Kurup SS, Staples RJ, Lord RL, Groysman S. Synthesis of Chromium(II) Complexes with Chelating Bis(alkoxide) Ligand and Their Reactions with Organoazides and Diazoalkanes. Molecules. 2020; 25(2):273. https://doi.org/10.3390/molecules25020273
Chicago/Turabian StyleKurup, Sudheer S., Richard J. Staples, Richard L. Lord, and Stanislav Groysman. 2020. "Synthesis of Chromium(II) Complexes with Chelating Bis(alkoxide) Ligand and Their Reactions with Organoazides and Diazoalkanes" Molecules 25, no. 2: 273. https://doi.org/10.3390/molecules25020273
APA StyleKurup, S. S., Staples, R. J., Lord, R. L., & Groysman, S. (2020). Synthesis of Chromium(II) Complexes with Chelating Bis(alkoxide) Ligand and Their Reactions with Organoazides and Diazoalkanes. Molecules, 25(2), 273. https://doi.org/10.3390/molecules25020273