
Pentaborate(1-) Salts and a Tetraborate(2-) Salt Derived from C2- or C3-Linked Bis(alkylammonium) Dications: Synthesis, Characterization, and Structural (XRD) Studies

 ,
,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
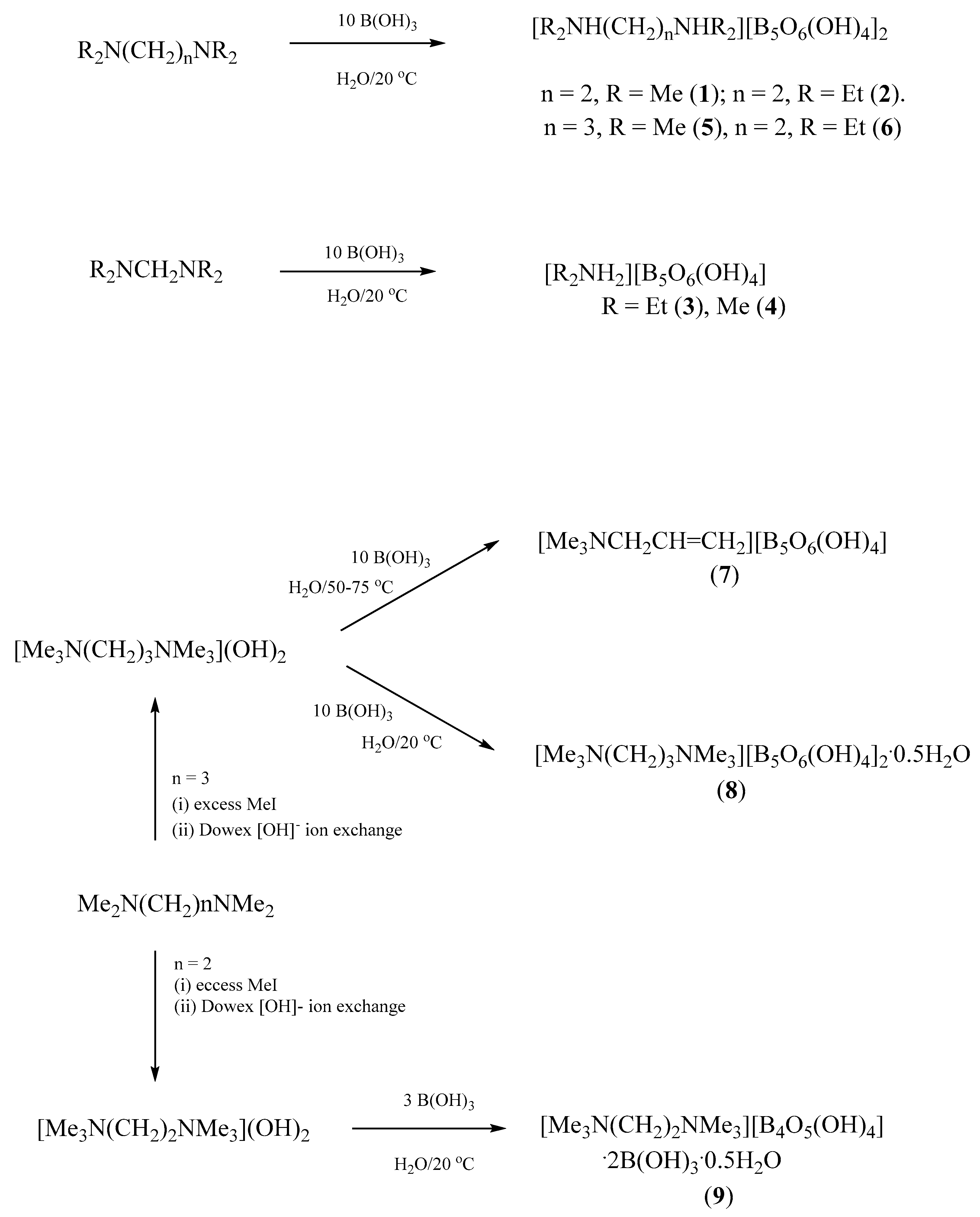
2.1. Synthesis
2.2. Thermal Analysis, Porosity Measurements and Spectroscopic Data
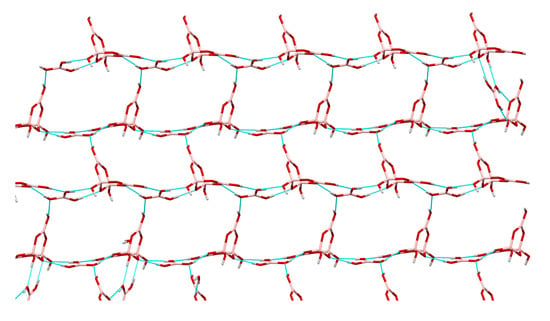
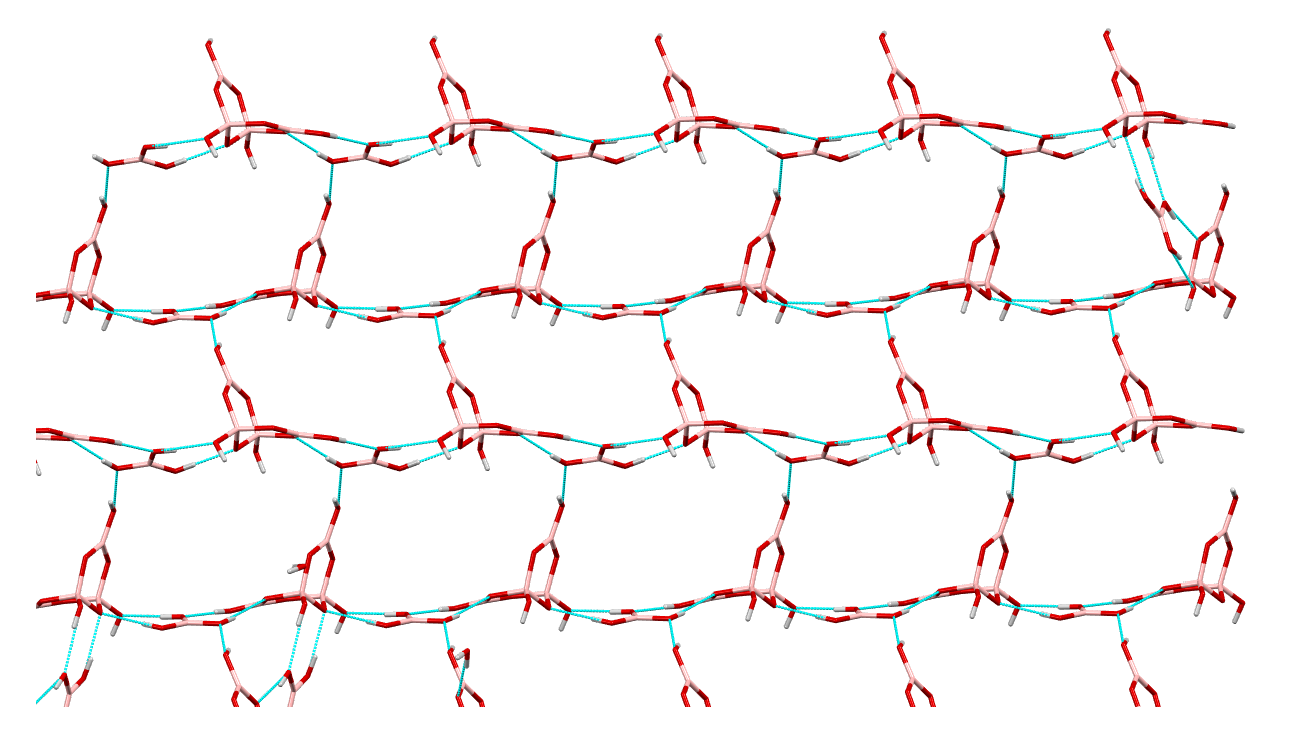
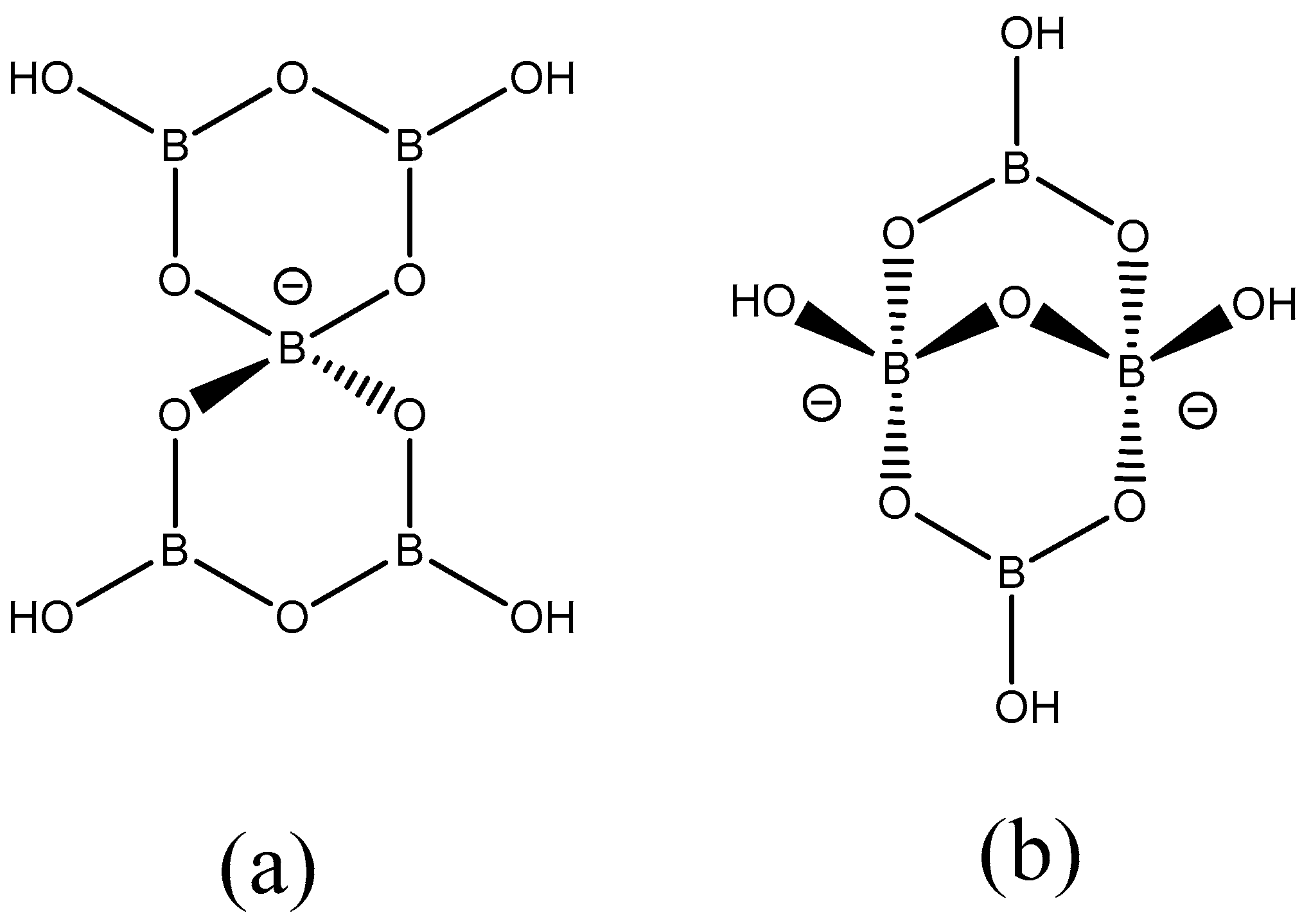
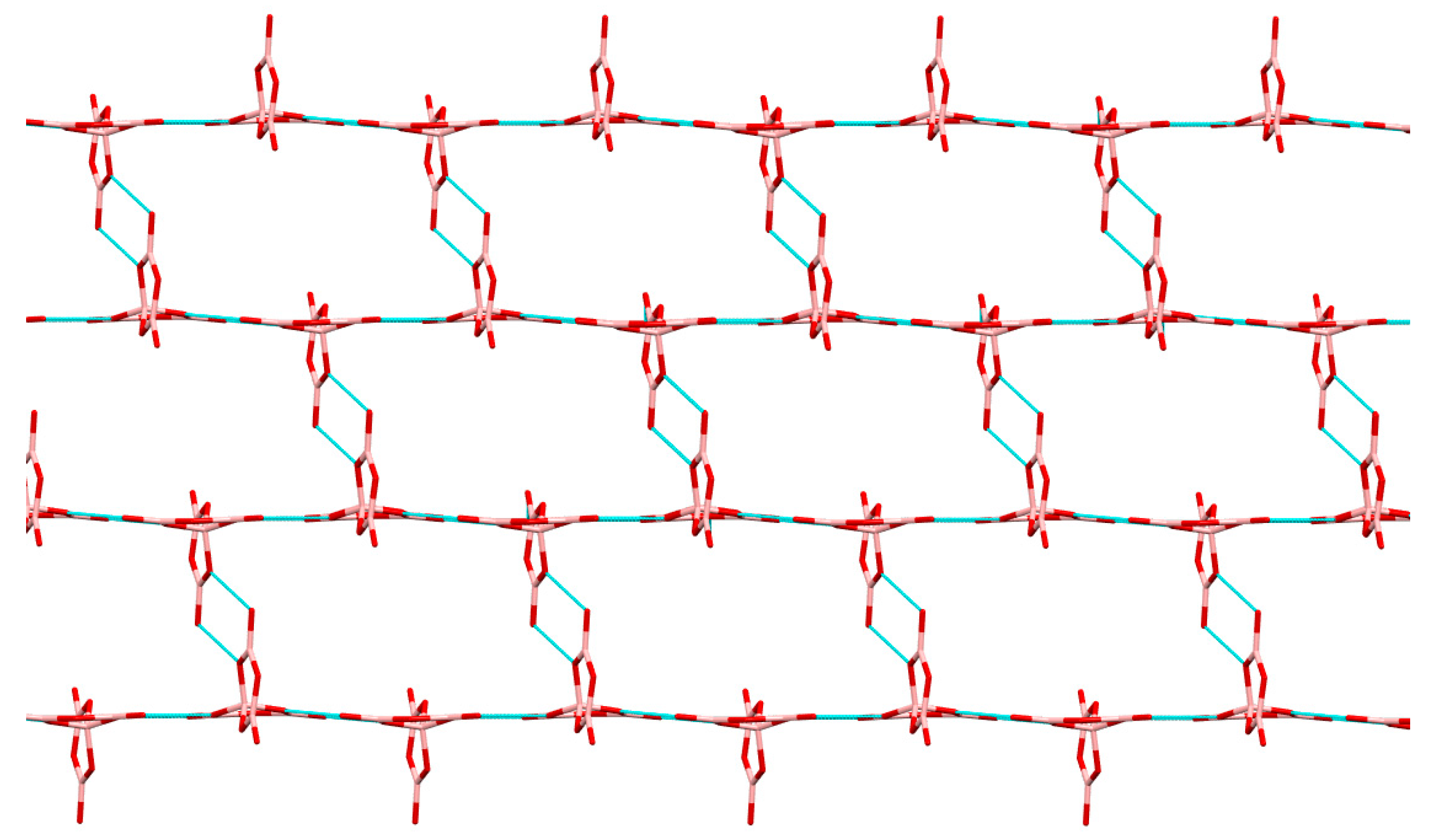
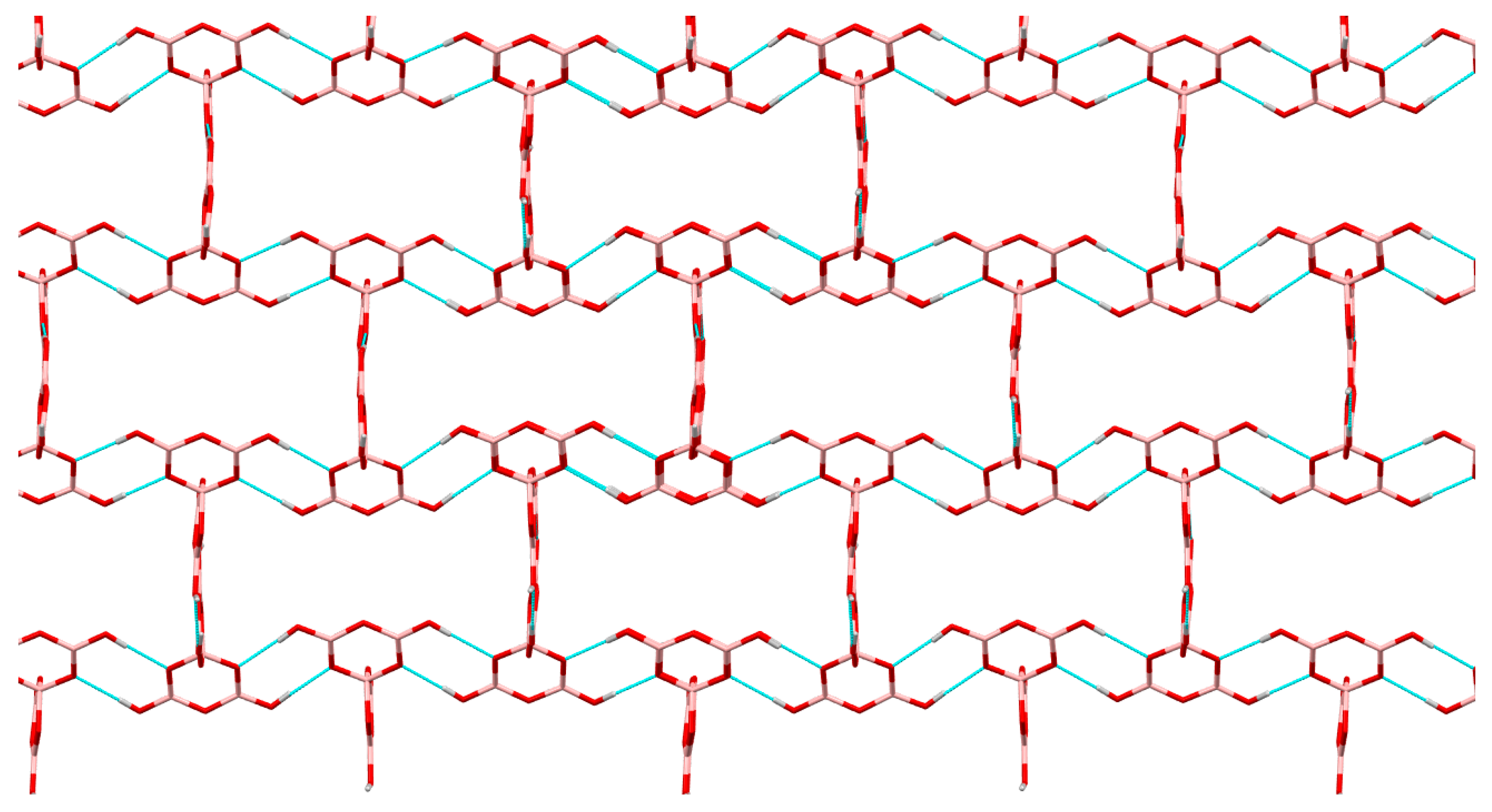
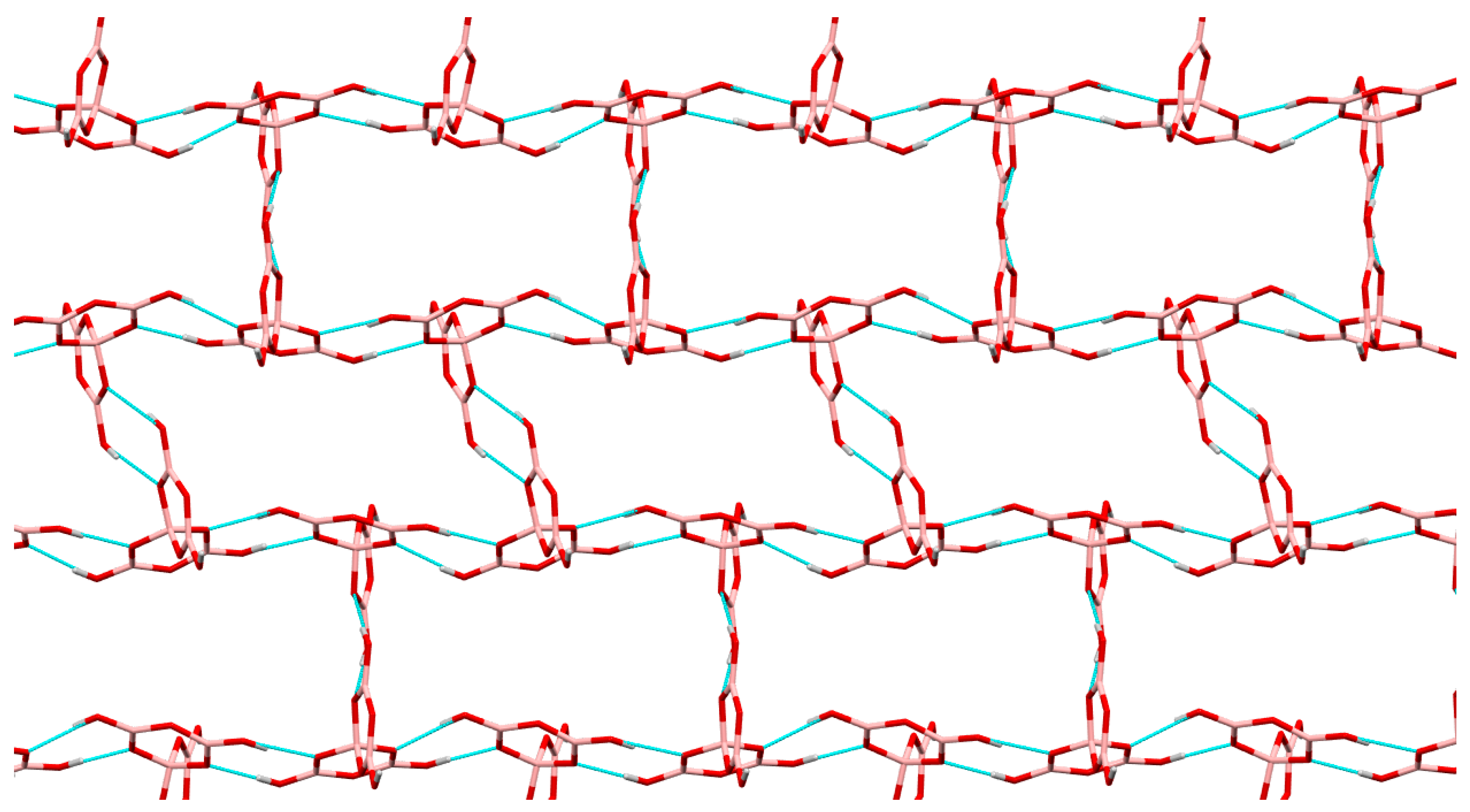
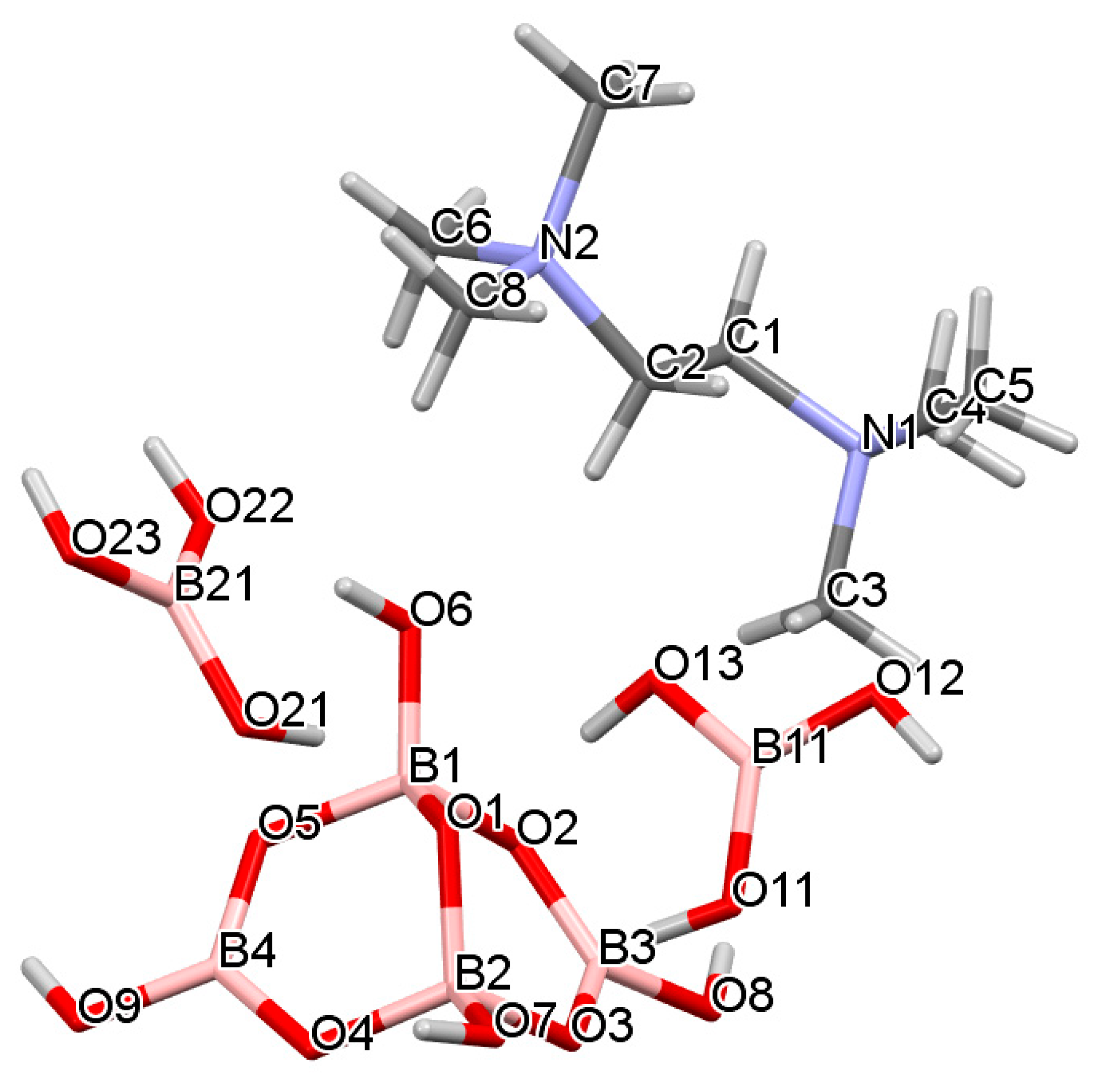
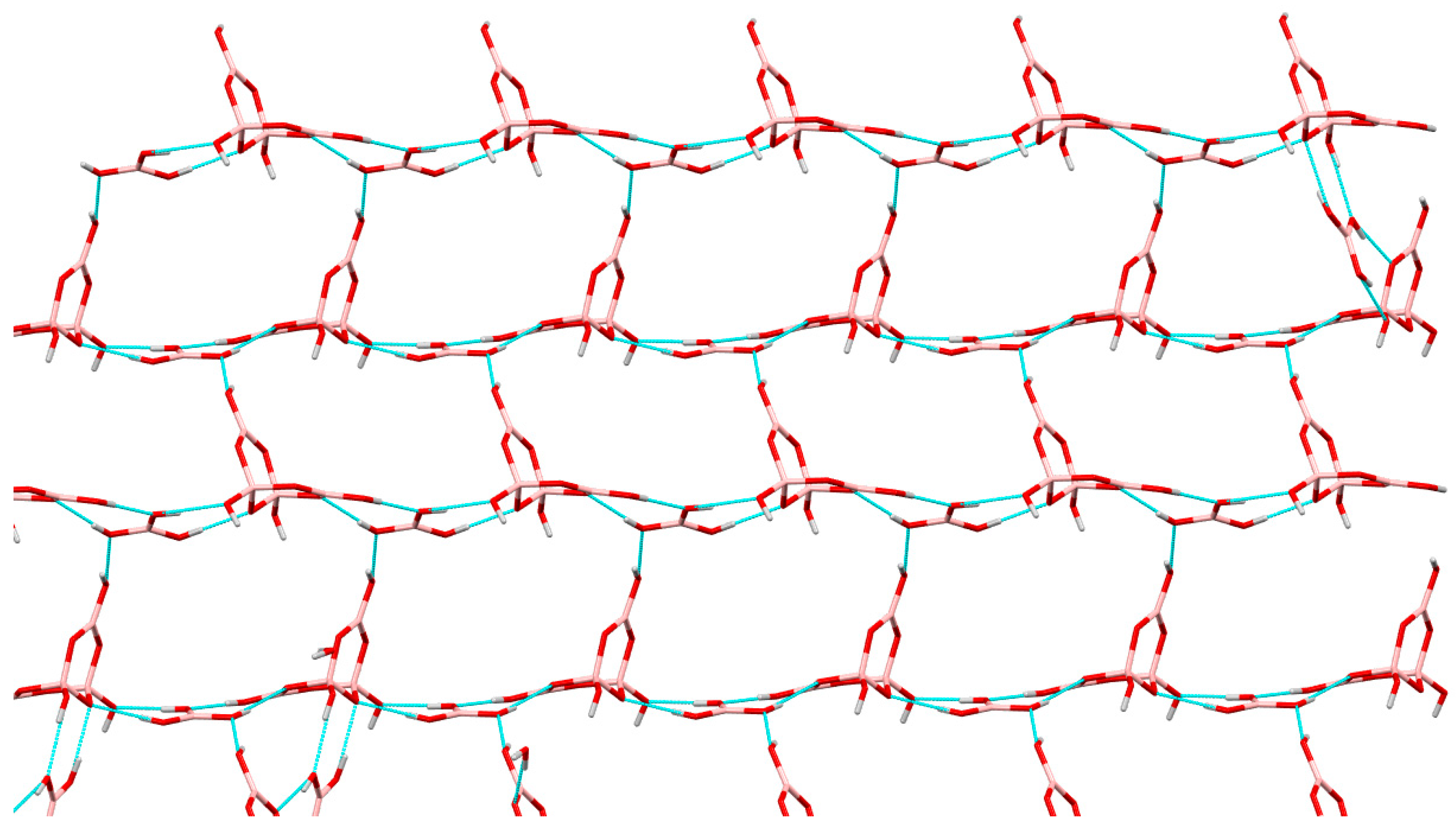
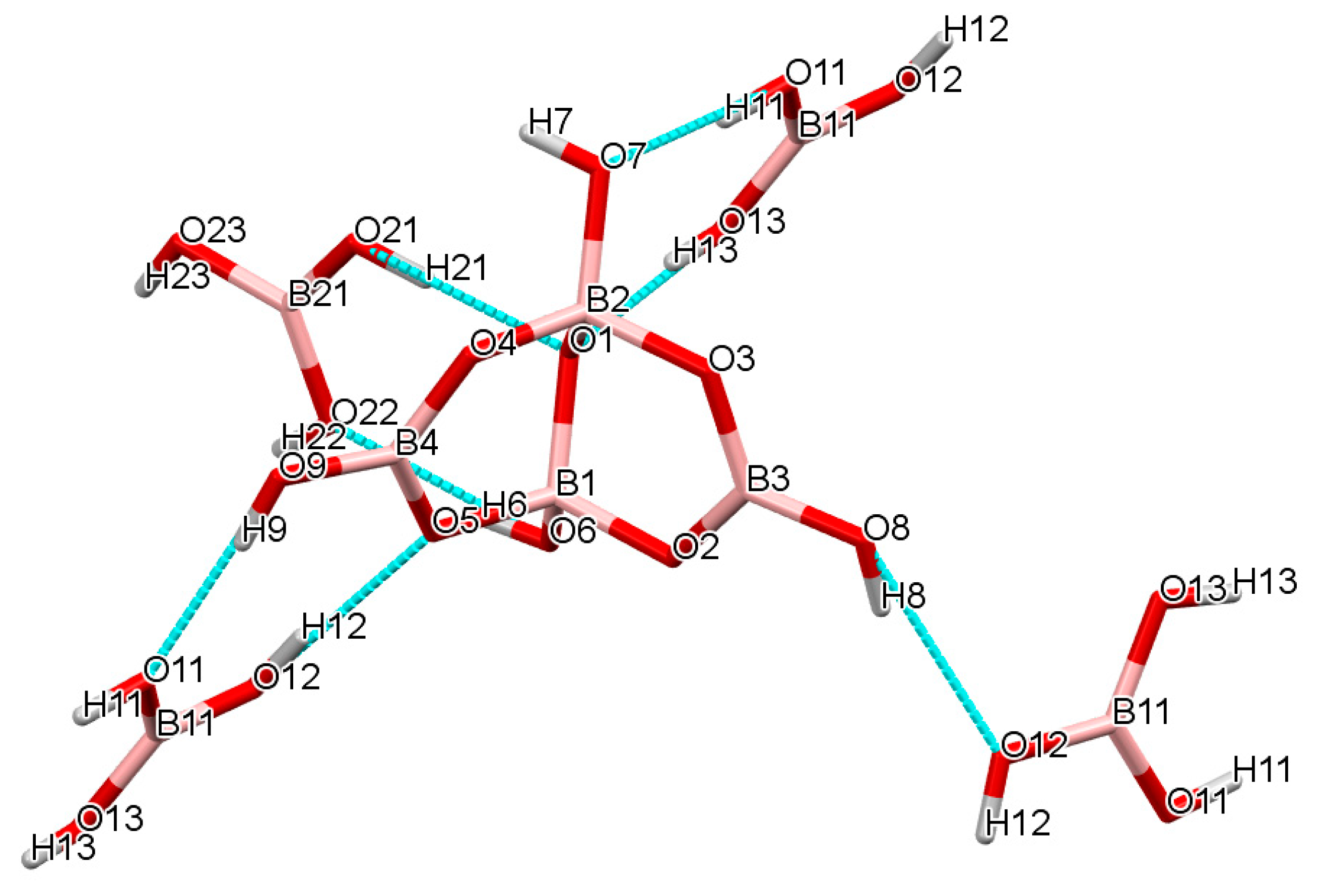
2.3. Single-Crystal XRD Studies
3. Materials and Methods
3.1. General
3.2. Synthesis, Spectroscopic, and Analytical Data for 1
3.3. Synthesis, Spectroscopic, Analytical, and Crystallographic Data for 2
3.4. Synthesis, Spectroscopic, Analytical, and Crystallographic Data for 3
3.5. Synthesis, Spectroscopic and Analytical Data for 4
3.6. Synthesis, Spectroscopic and Analytical Data for 5
3.7. Synthesis, Spectroscopic and Analytical Data for 6
3.8. Synthesis, Spectroscopic, Analytical, and Crystallographic Data for 7
3.9. Synthesis, Spectroscopic, Analytical, and Crystallographic Data for 8
3.10. Synthesis, Spectroscopic, Analytical, and Crystallographic Data for 9
3.11. X-Ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farmer, J.B. Metal borates. Adv. Inorg. Chem. Radiochem. 1982, 25, 187–237. [Google Scholar]
- Heller, G. A survey of structural types of borates and polyborates. Top. Curr. Chem. 1986, 131, 39–98. [Google Scholar]
- Schubert, D.M.; Smith, R.A.; Vis, M.Z. Studies of crystalline non-metal borates. Glass Technol. 2003, 44, 63–70. [Google Scholar]
- Schubert, D.M.; Knobler, C.B. Recent studies of polyborate anions. Phys. Chem. Glasses Eur. J. Glass Sci. Technol. B 2009, 50, 71–78. [Google Scholar]
- Beckett, M.A. Recent Advances in crystalline hydrated borates with non-metal or transition-metal complex cations. Coord. Chem. Rev. 2016, 323, 2–14. [Google Scholar] [CrossRef]
- Christ, C.L.; Clark, J.R. A crystal-chemical classification of borate structures with emphasis on hydrated borates. Phys. Chem. Miner. 1977, 2, 59–87. [Google Scholar] [CrossRef]
- Burns, P.C.; Grice, J.D.; Hawthorne, F.C. Borate minerals I. Polyhedral clusters and fundamental building blocks. Can. Mineral. 1995, 33, 1131–1151. [Google Scholar]
- Grice, J.D.; Burns, P.C.; Hawthorne, F.C. Borate minerals II. A hierarchy of structures based upon the borate fundamental building block. Can. Mineral. 1999, 37, 731–762. [Google Scholar]
- Becker, P. A contribution to borate crystal chemistry: Rules for the occurrence of polyborate anion types. Z. Kristallogr. 2001, 216, 523–533. [Google Scholar] [CrossRef]
- Belokoneva, E.L. Borate crystal chemistry in terms of the extended OD theory: Topology and symmetry analysis. Crystallogr. Rev. 2005, 11, 151–198. [Google Scholar] [CrossRef]
- Topnikova, A.P.; Belokoneva, E.L. The structure and classification of complex borates. Russ. Chem. Rev. 2019, 88, 204–228. [Google Scholar] [CrossRef]
- Schubert, D.M. Borates in industrial use. Struct. Bond. 2003, 105, 1–40. [Google Scholar]
- Schubert, D.M. Boron oxide, boric acid, and borates. In Kirk-Othmer Encyclopedia of Chemical Technology, 5th ed.; J. Wiley Sons: Hoboken, NY, USA, 2011; pp. 1–68. [Google Scholar]
- Schubert, D.M. hydrated zinc borates and their industrial use. Molecules 2019, 24, 2419. [Google Scholar] [CrossRef] [PubMed]
- Becker, P. Borate materials in nonlinear optics. Adv. Mater. 1998, 10, 979–992. [Google Scholar] [CrossRef]
- Flores, H.R.; Mattenella, L.E.; Kwok, L.H. Slow release boron micronutients from pelletized borates of the northwest of Argentinia. Miner. Eng. 2006, 19, 364–367. [Google Scholar] [CrossRef]
- Laane, H.-M. The effects of foliar sprays with different silicon compounds. Plants 2018, 7, 45. [Google Scholar] [CrossRef]
- Hoebbel, D.; Garzo, G.; Englehardt, G.; Vargha, A. On the Constitution and Distribution of Silicate Anions in Aqueous Tetramethylammonium Silicate Solutions. Z. Anorg. Allg. Chem. 1992, 494, 31–42. [Google Scholar] [CrossRef]
- Visi, M.Z.; Knobler, C.B.; Owen, J.J.; Khan, M.I.; Schubert, D.M. Structures of self-assembled nonmetal borates derived from α,ω-diaminoalkanes. Cryst. Growth Des. 2006, 6, 538–545. [Google Scholar] [CrossRef]
- Bergum, F.; Twamley, B.; Baker, R.J. The solid state structure of [TMEDAH]2[B5O6(OH)4]2. J. Chem. Cryststallgr. 2019. [Google Scholar] [CrossRef]
- Baber, R.A.; Charmont, P.H.; Norman, N.C.; Orpen, A.G.; Rossi, J. Dimethylammonium tetrahydropentaborate. Acta Cryst. 2004, E60, o1086–o1088. [Google Scholar] [CrossRef]
- Abel, E.W.; Goldsworthy, D.H.; Heard, P.J.; Kite, K. Formation of dimethylamine complexes of trimethylplatinum(IV) from reactions with N,N,N’,N’-trtramethyldiaminomethane. Polyhedron 1995, 14, 515–519. [Google Scholar] [CrossRef]
- Sawatsky, E.; Drakopoulos, A.; Rolz, M.; Sotriffer, C.; Engels, B.; Decker, M. Experimental and theoretical investigations into stability of cyclic aminals. Bielstein J. Org. Chem. 2016, 12, 2280–2292. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.K.; Gonzalez, P.E.; Craig, A.L.; Chakrabarty, S.; Metta-Magana, A.; Pannell, K.H. Siloxymethylamines as aminomethylation reagents for amines leading to labile diaminomethanes that can be trapped as their [Mo(CO)4] complexes. Chem. Eur. J. 2016, 22, 7363–7366. [Google Scholar] [CrossRef] [PubMed]
- Godin, G.; Levrand, B.; Trachsel, A.; Lehn, J.-M.; Herrmann, A. Reversible formation of aminals: A new strategy to control the release of bioactive volatiles from dynamic mixtures. Chem. Commun. 2010, 46, 3125–3127. [Google Scholar] [CrossRef]
- Hofmann, A.W. Beiträge zur Kenntniss der flüchtigen organischen Basen. Ann. Chem. Pharm. (Eur. J. Org. Chem.) 1851, 78, 253–286. [Google Scholar] [CrossRef]
- Cope, A.C.; LeBel, N.A.; Moore, P.T.; Moore, W.R. Mechanism of the Hofmann elimination reaction: Evidence that an ylide intermediate is not involved in simple compounds. J. Am. Chem. Soc. 1961, 83, 3861–3865. [Google Scholar] [CrossRef]
- Beckett, M.A.; Coles, S.J.; Horton, P.N.; Rixon, T.A. Synthesis and XRD study of a C2-linked bis(quaternary ammonium) pentaborate: [Me3NCH2CH2NMe3][B5O6(OH)4]2. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194. [Google Scholar] [CrossRef]
- Wiebcke, M.; Freyhardt, C.C.; Felsche, J.; Engelhardt, G. Clathrates with three-dimensional host structures of hydrogen bonded pentaborate [B5O6(OH)4]− ions: Pentaborates with the cations NMe4+, NEt4+, NPhMe3+ and pipH+ (pipH+ = piperidinium). Z. Naturforsch. 1993, 48b, 978–985. [Google Scholar] [CrossRef]
- Brauner, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Beckett, M.A.; Horton, P.N.; Hursthouse, M.B.; Knox, D.A.; Timmis, J.L. Structural (XRD) and thermal (DSC, TGA) and BET analysis of materials derived from non-metal cation pentaborate salts. Dalton Trans. 2010, 39, 3944–3951. [Google Scholar] [CrossRef]
- Beckett, M.A.; Horton, P.N.; Hursthouse, M.B.; Timmis, J.L.; Varma, K.S. Templated heptaborate and pentaborate salts of cyclo-alkylammonium cations: Structural and thermal properties. Dalton Trans. 2012, 41, 4396–4403. [Google Scholar] [CrossRef] [PubMed]
- Beckett, M.A.; Coles, S.J.; Horton, P.N.; Jones, C.L. Polyborate anions partnered with large non-metal cations: Triborate(1-), pentaborate(1-) and heptaborate(2-) salts. Eur. J. Inorg. Chem. 2017, 4510–4518. [Google Scholar] [CrossRef]
- Beckett, M.A.; Coles, S.J.; Davies, R.A.; Horton, P.N.; Jones, C.L. Pentaborate(1−) salts templated by substituted pyrrolidinium cations: Synthesis, structural characterization, and modelling of solid-state H-bond interactions by DFT calculations. Dalton Trans. 2015, 44, 7032–7040. [Google Scholar] [CrossRef] [PubMed]
- Salentine, G. High-field 11B NMR of alkali borate. Aqueous polyborate equilibria. Inorg. Chem. 1983, 22, 3920–3924. [Google Scholar] [CrossRef]
- Anderson, J.L.; Eyring, E.M.; Whittaker, M.P. Temperature jump rate studies of polyborate formation in aqueous boric acid. J. Phys. Chem. 1964, 68, 1128–1132. [Google Scholar] [CrossRef]
- Beckett, M.A.; Bland, C.C.; Horton, P.N.; Hursthouse, M.B.; Varma, K.S. Supramolecular structures containing “isolated” pentaborate anions and non-metal cations: Crystal structures of [Me3NCH2CH2OH][B5O6(OH)4] and [4-Mepy,4-MepyH][B5O6(OH)4]. J. Organomet. Chem. 2007, 692, 2832–2838. [Google Scholar] [CrossRef]
- Li, J.; Xia, S.; Gao, S. FT-IR and Raman spectroscopic study of hydrated borates. Spectrochim. Acta 1995, 51A, 519–532. [Google Scholar]
- Beckett, M.A.; Horton, P.N.; Coles, S.J.; Kose, D.A.; Kreuziger, A.-M. Structurral and thermal studies of non-metal cation pentaborate salts with cations derived from 1,5-diazobicyclo[4.3.0]non-5-ene, 1,8-diazobicyclo[5.4.0]undec-7-ene and 1,8-bis(dimethylamino)naphthalene. Polyhedron 2012, 38, 157–161. [Google Scholar] [CrossRef]
- Beckett, M.A.; Horton, P.N.; Colers, S.J.; Martin, D.W. Syntheis and structural chatertization of an unprecedented non-metal cation polyborate salt containing two different ‘isolated’ polyborate anions: [H2en][B4O6(OH)4[B7O9(OH)5].2H2O (en = NH2CH2CH2NH2). Inorg. Chem. 2011, 50, 12215–12218. [Google Scholar] [CrossRef]
- Beckett, M.A.; Coles, S.J.; Light, M.E.L.; Fischer, B.M.; Stiefvater-Thomas, K.S. Varma, Synthesis and X-ray characterization of the orgaonotriboroxinate salts [Me3NCH2CH2OH][Ph4B3O3] and [NEt3H][Ph3B3O3(OH)], and the X-ray structure of the triarylboroxine, (4-MeOC6H4)3B3O3. Polyhedron 2006, 25, 1011–1016. [Google Scholar] [CrossRef]
- Sola, J.; Lafuente, M.; Atcher, J.; Alfonso, I. Constitutional self-selection from dynamic combinatorial libraries in aqueous solution through supramolecular interactions. Chem. Commun. 2014, 50, 4564–4566. [Google Scholar] [CrossRef] [PubMed]
- Corbett, P.T.; Leclaire, J.; Vial, L.; West, K.R.; Wietor, J.-L.; Sanders, J.K.M.; Otto, S. Dynamic combinatorial chemistry. Chem. Rev. 2006, 106, 3652–3711. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D.; Gavezzotti, A. Supramolecular synthons: Validation and ranking of intermolecular interaction energies. Cryst. Growth Des. 2012, 12, 5873–5877. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic chemistry. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Levy, H.A.; Lisensky, G.C. Crystal Structures of sodium sulfate decahydrate (Glauber’s salt) and sodium tetraborate decahydrate (borax). Redetermination by neutron diffraction. Acta Crystallogr. Sect. B 1978, 34, 3502–3510. [Google Scholar] [CrossRef]
- Gainsford, G.J.; Kemmitt, T.; Higham, C. Redetermination of the borax structure from laboratory X-ray data at 145 K. Acta Cryst. 2008, E64, i24–i25. [Google Scholar] [CrossRef]
- Pan, C.Y.; Wang, G.-M.; Zheng, S.-T.; Yang, G.Y. Cyclohexane-1,4-diammonium tetrahydroxotetraborate 2.5 hydrate. Acta Crystallogr. 2007, E63, o1207–o1209. [Google Scholar] [CrossRef]
- Janda, R.; Heller, G. Die kristallstruktur von synthetischem ammoniumtetraboratdihydrat, (NH4)2 [B4O5(OH)4]·2H2O. Z. Kristallogr. 1981, 154, 1–9. [Google Scholar]
- Weakley, T.J.R. Guanidinium tetraborate(2-) dihydrate, (CH6N3)2[B4O5(OH)4].2H2O. Acta Crystallogr. Sect. C 1985, 41, 377–379. [Google Scholar] [CrossRef]
- Wang, G.M.; Sun, Y.Q.; Yang, G.Y. Synthesis and crystal structures of two new organically templated borates. J. Solid State Chem. 2004, 177, 4648–4654. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, J.; Liu, Z. Synthesis, Crystal Structure and Thermal Behavior of Co(en)3[B4O5(OH)4]Cl·3H2O and [Ni(en)3][B5O6(OH)4]2·2H2O. Chin. J. Chem. 2009, 27, 494–500. [Google Scholar] [CrossRef]
- Lin, D.; You, X.; Zhu, L. Synthesis, crystal structure and vibrational spectroscopy of NH4[Co(NH3)5(H2O)][B4O5(OH)4].6H2O. Chin. J. Chem. 2011, 29, 468–472. [Google Scholar] [CrossRef]
- Lin, D.; You, X.; Zhu, L. A new organic-inorganic hybrid copper pentaborate with free boric acid. Chin. J. Chem. 2011, 29, 463–467. [Google Scholar] [CrossRef]
- Altahan, M.A.; Beckett, M.A.; Coles, S.J.; Horton, P.N. Synthesis and characterization of polyborates templated by cationic copper(II) complexes: Structural (XRD), spectroscopic, thermal (TGA/DSC) and magnetic properties. Polyhedron 2017, 135, 247–257. [Google Scholar] [CrossRef]
- Freyhardt, C.C.; Wiebcke, M.; Felsche, J.; Englehardt, G. N(nPr4)[B5O6(OH)4][B(OH)3]2 and N(nBu4)[B5O6(OH)4][B(OH)3]2: Clathrates with a diamondoid arrangement of hydrogen bonded pentaborate anions. J. Inclusion Phenom. Mol. Recogn. Chem. 1994, 18, 161–175. [Google Scholar] [CrossRef]
- Yang, Y.; Fu, D.S.; Li, G.F.; Zhang, Y. Synthesis, crystal structure and variable temperature luminescent property of the organically template pentaborate [C10N2H9][B5O6(OH)4]·H3BO3·H2O. Z. Anorg. Allg. Chem. 2013, 639, 722–727. [Google Scholar] [CrossRef]
- Sheldrick, G.M. ShelXT-intergrated space-group and crystal structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beckett, M.A.; Meena, B.I.; Rixon, T.A.; Coles, S.J.; Horton, P.N. Pentaborate(1-) Salts and a Tetraborate(2-) Salt Derived from C2- or C3-Linked Bis(alkylammonium) Dications: Synthesis, Characterization, and Structural (XRD) Studies. Molecules 2020, 25, 53. https://doi.org/10.3390/molecules25010053
Beckett MA, Meena BI, Rixon TA, Coles SJ, Horton PN. Pentaborate(1-) Salts and a Tetraborate(2-) Salt Derived from C2- or C3-Linked Bis(alkylammonium) Dications: Synthesis, Characterization, and Structural (XRD) Studies. Molecules. 2020; 25(1):53. https://doi.org/10.3390/molecules25010053
Chicago/Turabian StyleBeckett, Michael A., Bashdar I. Meena, Thomas A. Rixon, Simon J. Coles, and Peter N. Horton. 2020. "Pentaborate(1-) Salts and a Tetraborate(2-) Salt Derived from C2- or C3-Linked Bis(alkylammonium) Dications: Synthesis, Characterization, and Structural (XRD) Studies" Molecules 25, no. 1: 53. https://doi.org/10.3390/molecules25010053
APA StyleBeckett, M. A., Meena, B. I., Rixon, T. A., Coles, S. J., & Horton, P. N. (2020). Pentaborate(1-) Salts and a Tetraborate(2-) Salt Derived from C2- or C3-Linked Bis(alkylammonium) Dications: Synthesis, Characterization, and Structural (XRD) Studies. Molecules, 25(1), 53. https://doi.org/10.3390/molecules25010053