Simultaneous Determination and Risk Assessment of Pyrrolizidine Alkaloids in Artemisia capillaris Thunb. by UPLC-MS/MS Together with Chemometrics

Abstract
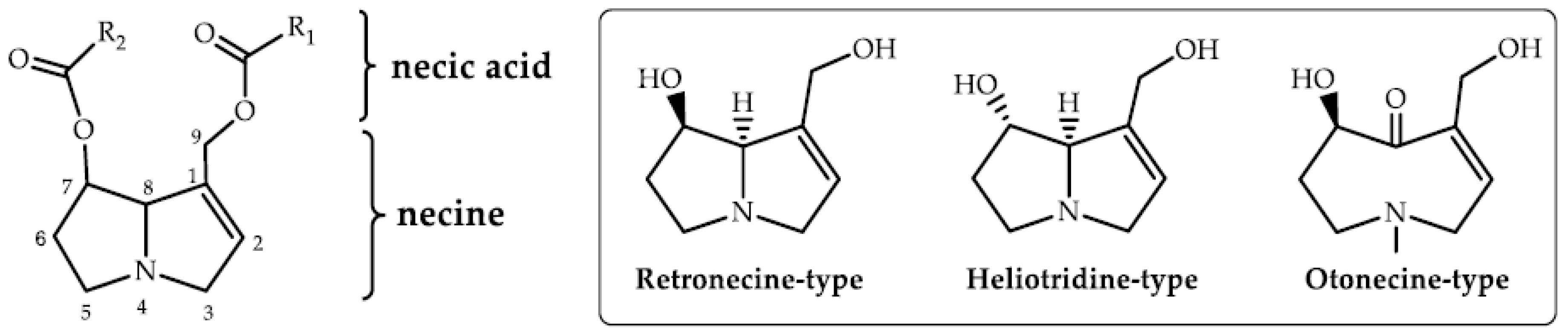
1. Introduction
2. Results and Discussion
2.1. Pretreatment Method Development
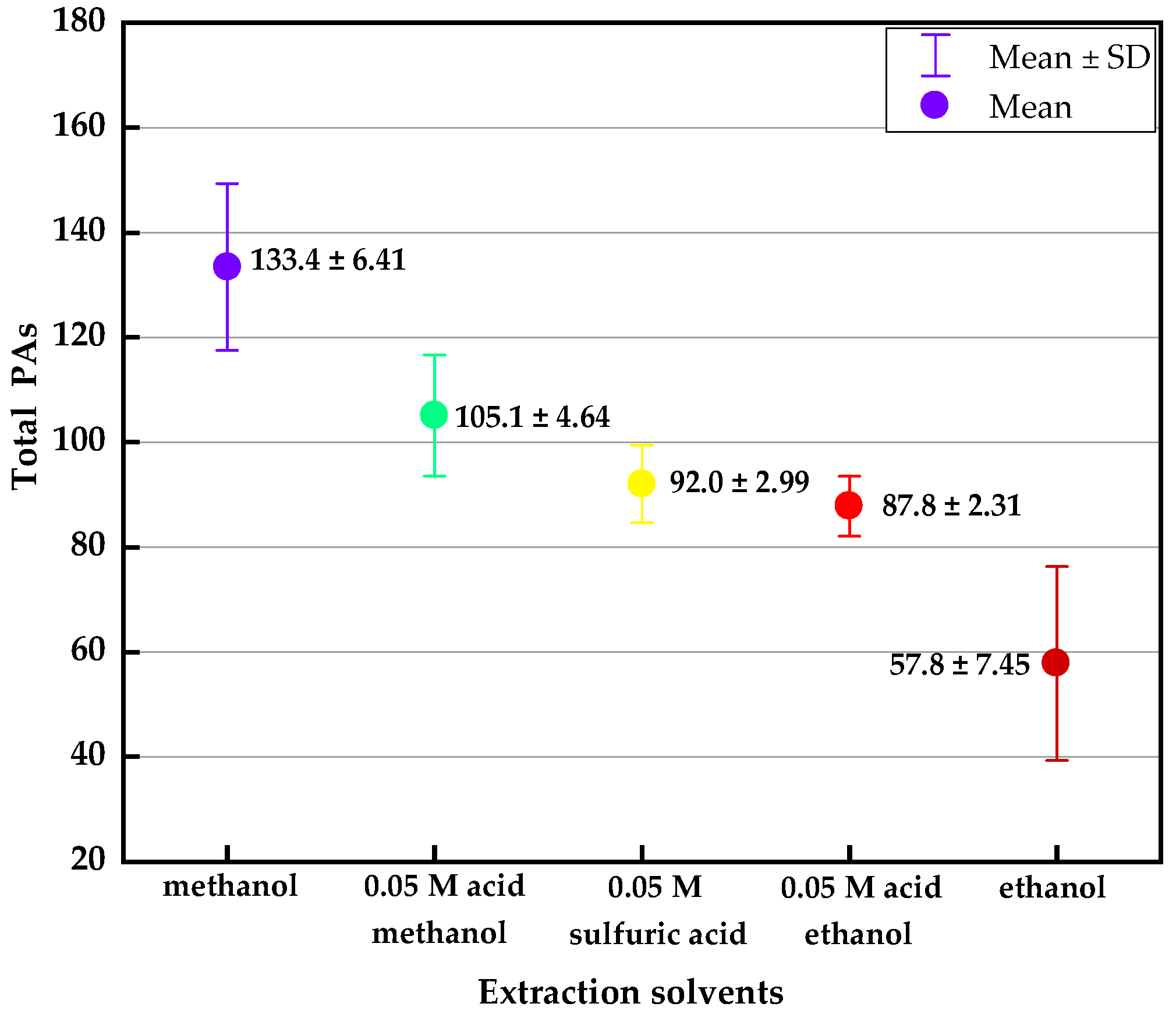
2.1.1. Extraction Optimization
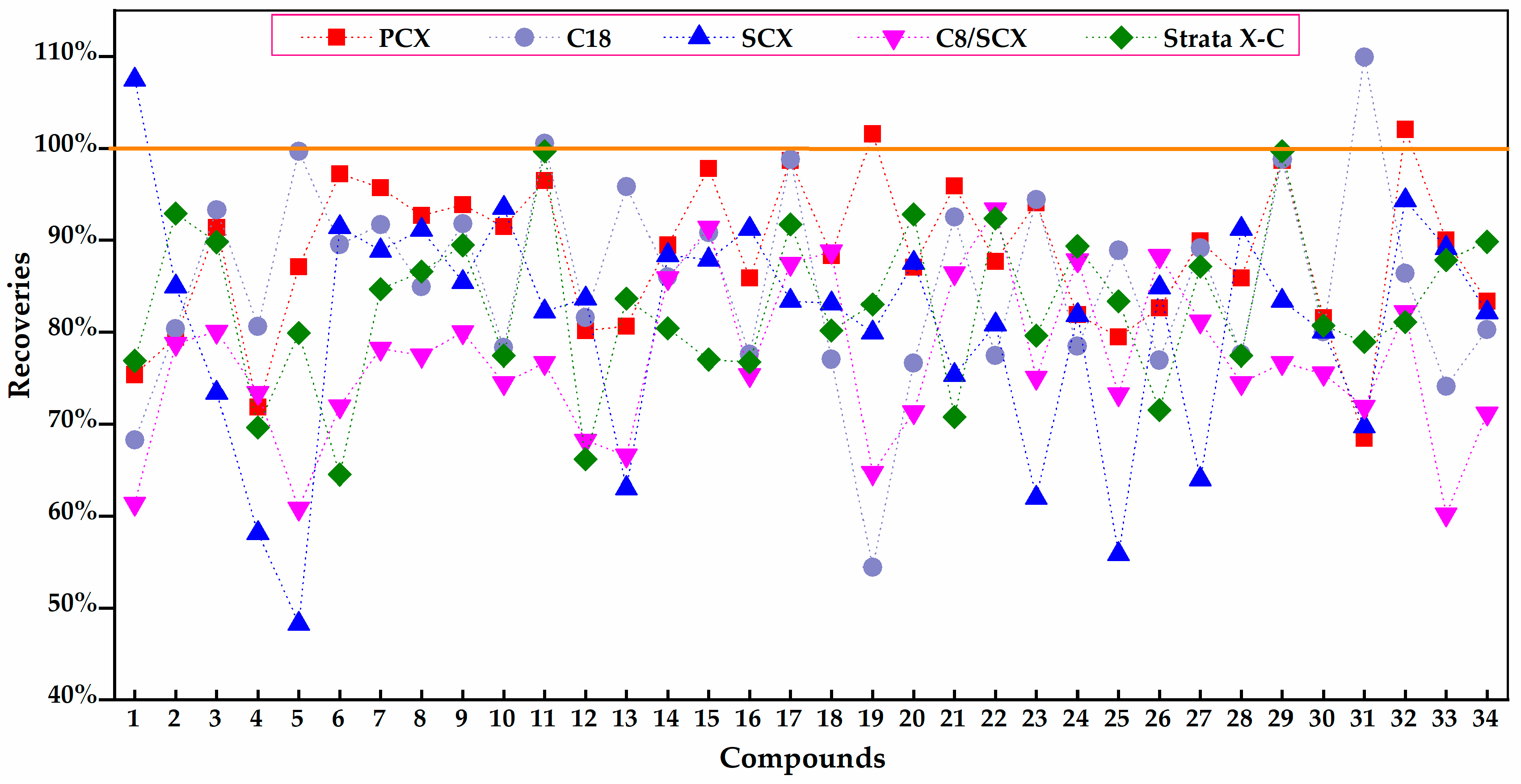
2.1.2. Purification Optimization
2.1.3. Redissolution Optimization
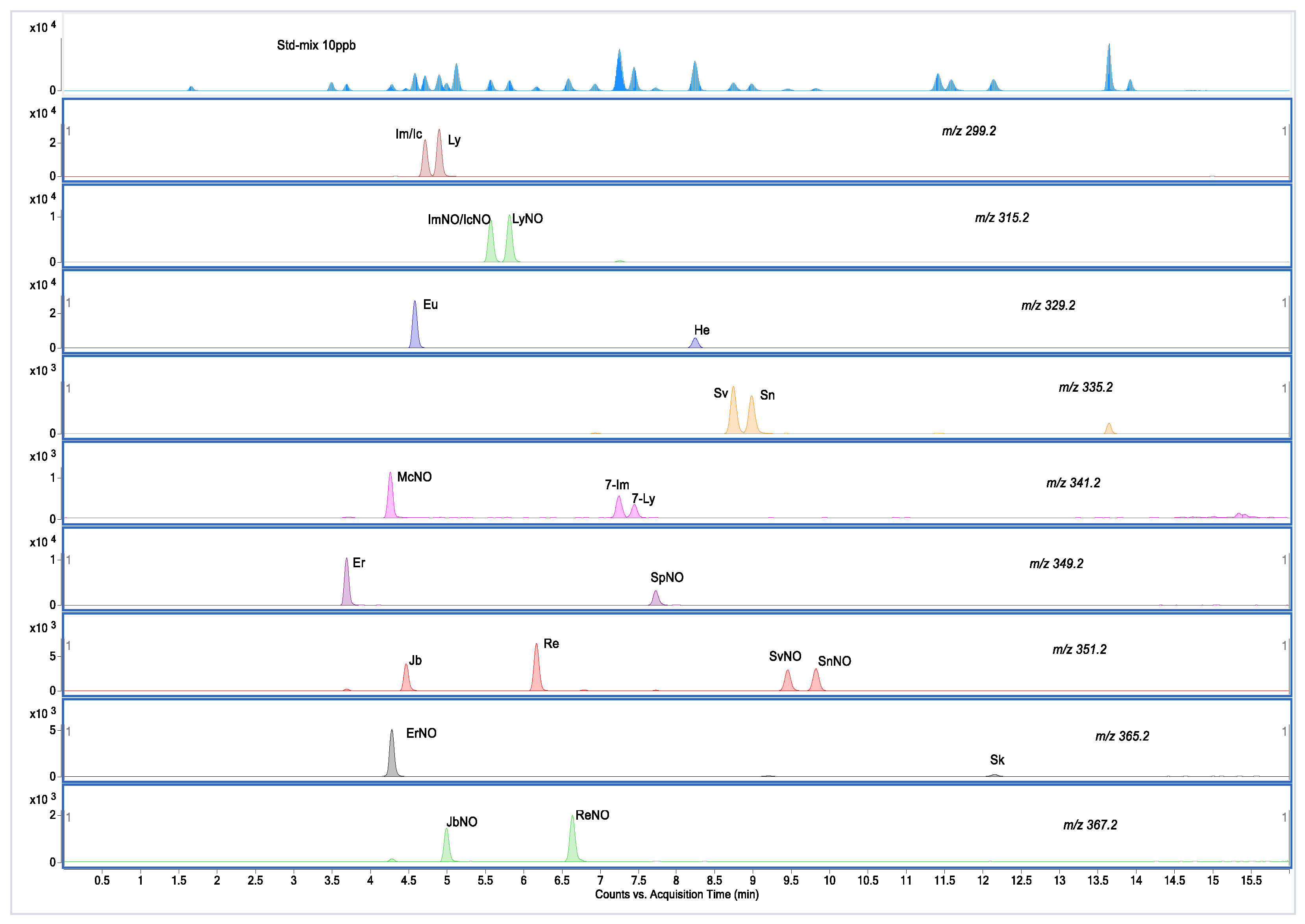
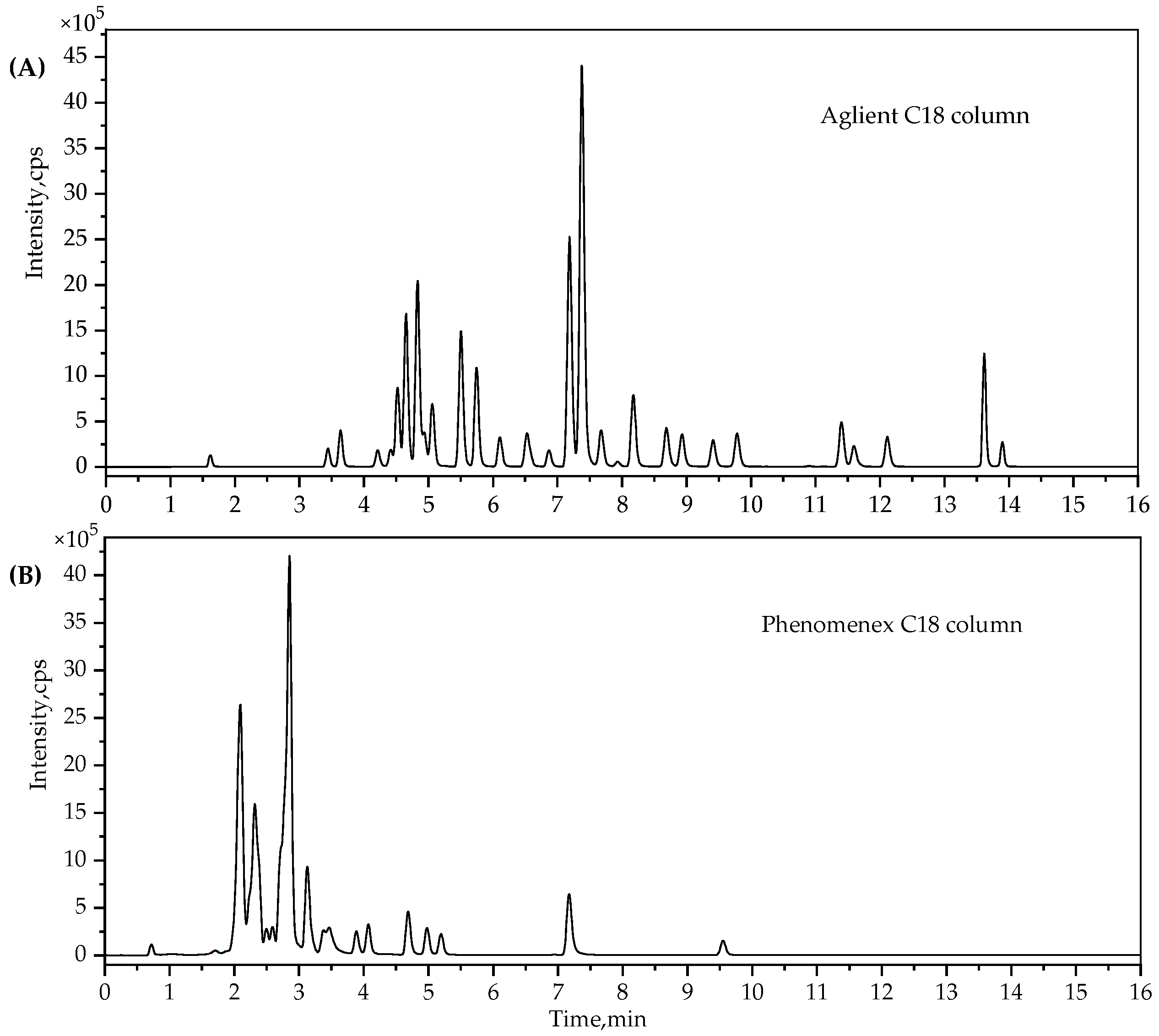
2.2. UPLC-MS/MS Method Development
2.3. Method Validation
2.3.1. Sensitivity, Linearity, LOD and LOQ
2.3.2. Precision and Recovery
2.3.3. Matrix Effects
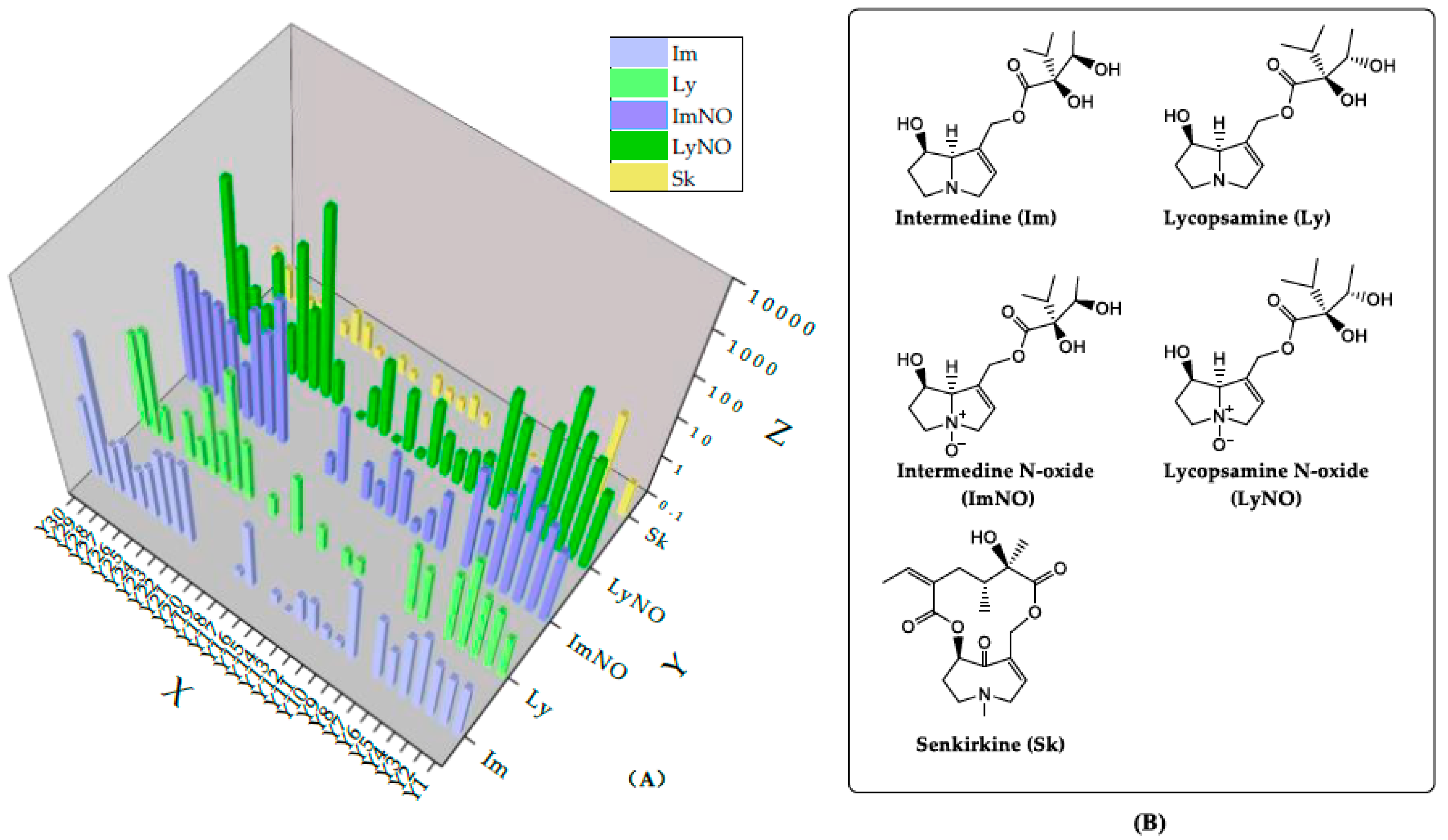
2.4. The Occurrence of PAs in A. capillaris Samples
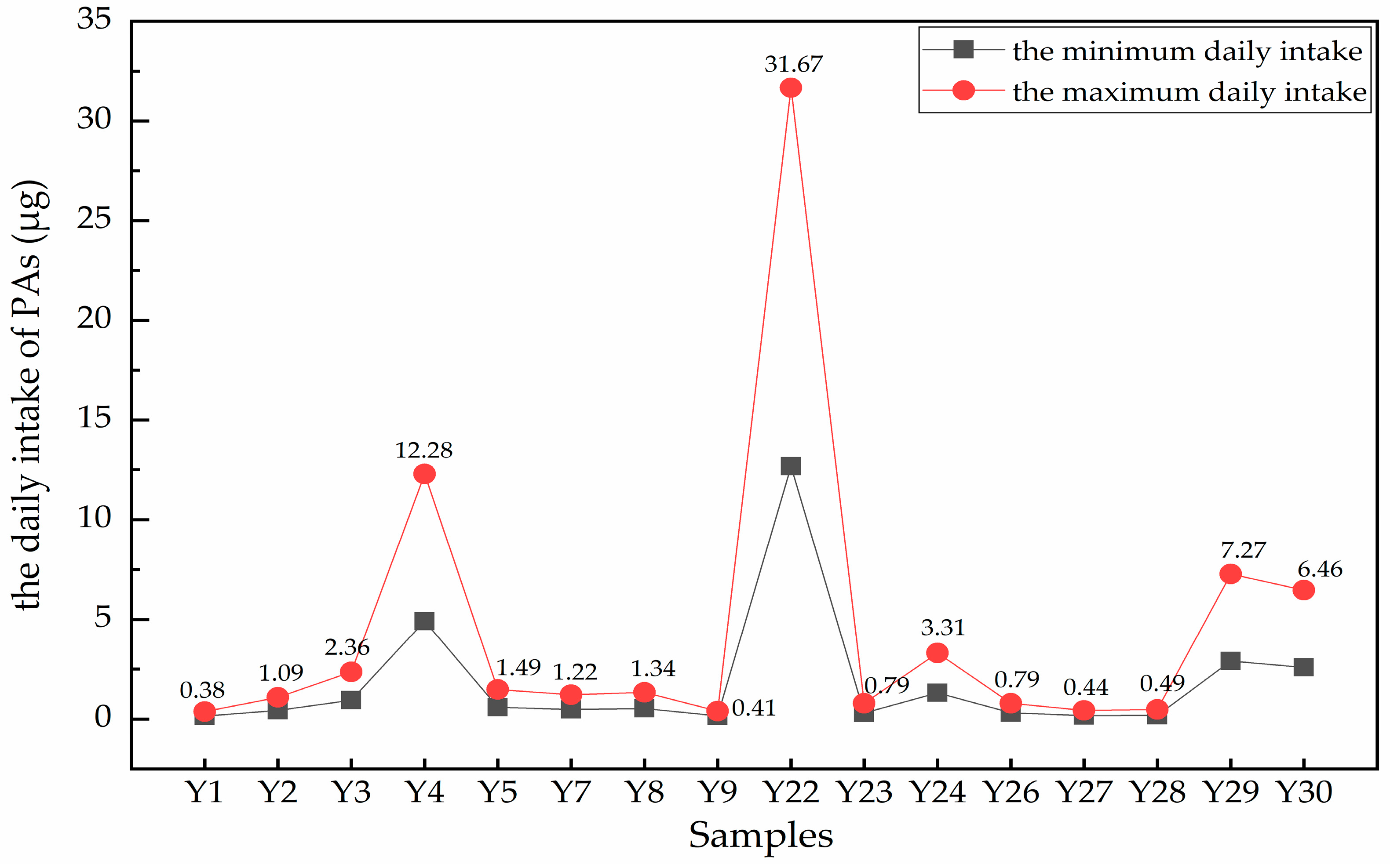
2.5. Risk Assessment for the intakes of PAs in A. capillaris Samples
2.6. Chemometrics Analysis of PAs in A. capillaris Samples
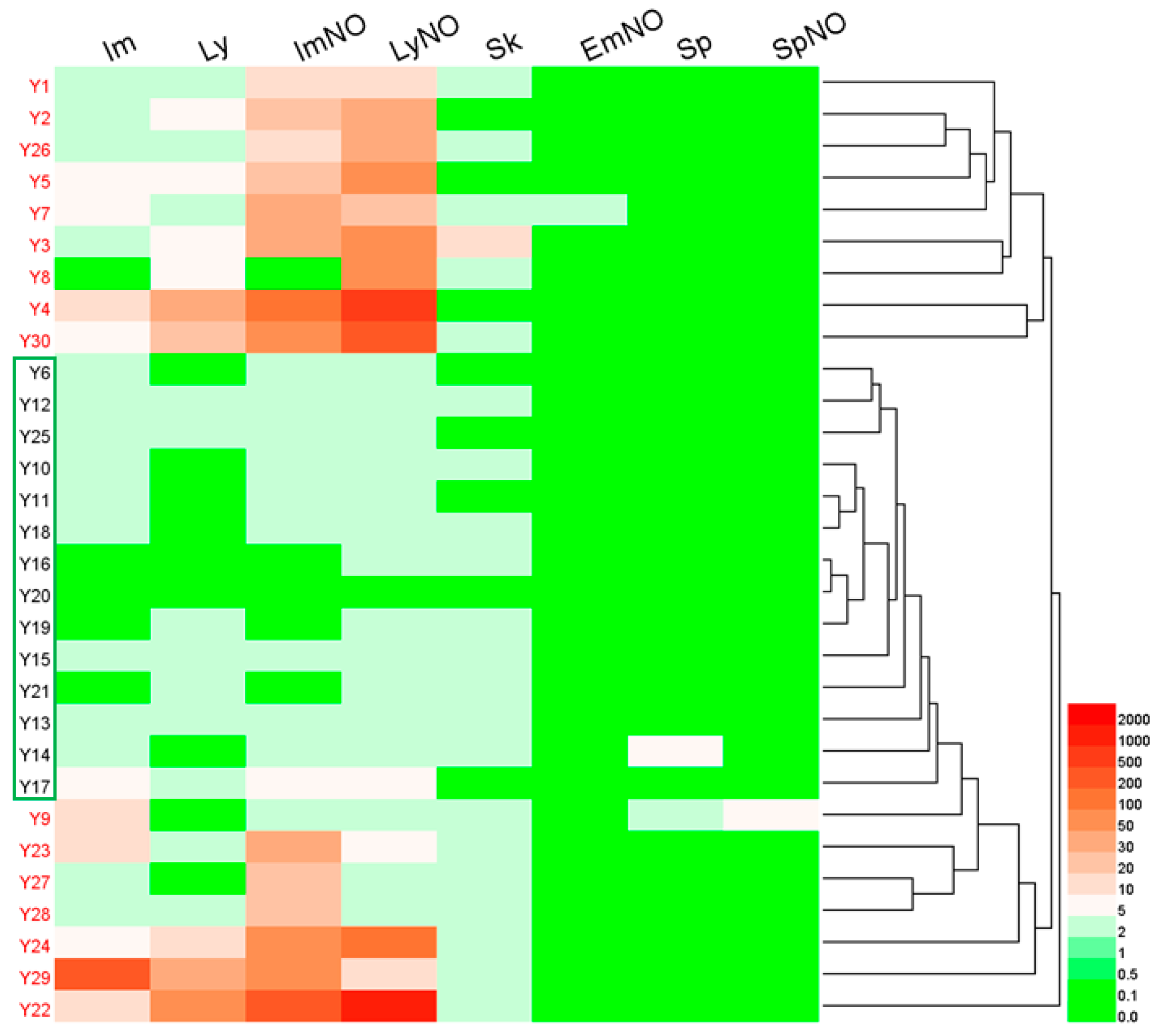
2.6.1. Hierarchical Clustering Analysis
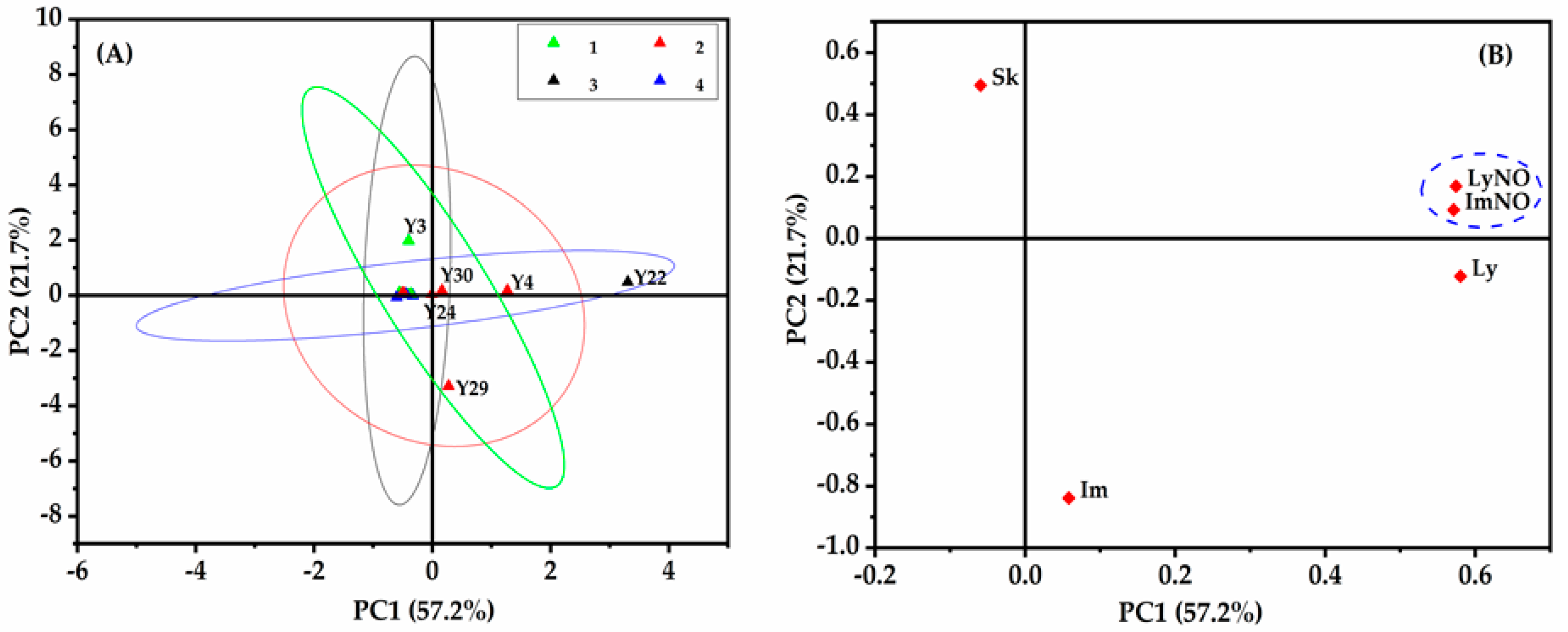
2.6.2. Principal Component Analysis
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Standard Solutions and Sample Preparation
3.2.1. Standard Solutions
3.2.2. Sample Preparation
3.2.3. PCX-SPE Procedure
3.3. Ultra-High Performance Liquid Chromatography-Mass Spectrometry (UPLC-MS/MS) Analysis
3.4. Method Validation
3.5. Method Acquisition and Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jank, B.; Rath, J. The Risk of Pyrrolizidine Alkaloids in Human Food and Animal Feed. Trends Plant Sci. 2017, 22, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Preliasco, M.; Gardner, D.; Moraes, J.; González, A.C.; Uriarte, G.; Rivero, R. Senecio grisebachii Baker: Pyrrolizidine alkaloids and experimental poisoning in calves. Toxicon 2017, 133, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Glück, J.; Ebmeyer, J.; Waizenegger, J.; Luckert, C.; Braeuning, A.; Lampen, A.; Hessel-Pras, S. Hepatotoxicity of pyrrolizidine alkaloids in human hepatocytes and endothelial cells. Toxicol. Lett. 2018, 295, S142. [Google Scholar] [CrossRef]
- Zhu, L.; Xue, J.; Xia, Q.; Fu, P.P.; Lin, G. The long persistence of pyrrolizidine alkaloid-derived DNA adducts in vivo: Kinetic study following single and multiple exposures in male ICR mice. Arch. Toxicol. 2017, 91, 1–17. [Google Scholar] [CrossRef]
- Stegelmeier, B.L.; Colegate, S.M.; Brown, A.W. Dehydropyrrolizidine Alkaloid Toxicity, Cytotoxicity, and Carcinogenicity. Toxins 2016, 8, 356. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.; Ruan, J.Q.; Jie, C.; Na, L.; Ke, C.Q.; Yang, Y.; Ge, L.; Wang, J.Y. Blood pyrrole-protein adducts as a diagnostic and prognostic index in pyrrolizidine alkaloid-hepatic sinusoidal obstruction syndrome. Drug Des. Dev. Ther. 2015, 9, 4861–4868. [Google Scholar] [CrossRef]
- Gao, L.; Rutz, L.; Merz, K.H.; Schrenk, D. Investigation of pyrrolizidine alkaloids—Cytotoxicity tests using primary rat hepatocytes and HepG2 cells: Role of cytochrome P450 3A. Toxicol. Lett. 2018, 295, S139–S140. [Google Scholar] [CrossRef]
- Chen, T.; Mei, N.; Fu, P.P. Genotoxicity of pyrrolizidine alkaloids. J. Appl. Toxicol. 2010, 30, 183–196. [Google Scholar] [CrossRef]
- Segali, H.J.; Wilson, D.W.; Griffin, D.S.; Winter, C.K.; Winter, C.K.; Morin, D.M.; Lame, M.W. The identification, metabolism and toxicity associated with the macrocyclic pyrrolizidine alkaloids. Nat. Toxins 1989, 30–40. [Google Scholar] [CrossRef]
- Moreira, R.; Pereira, D.M.; Valentão, P.; Andrade, P.B. Pyrrolizidine Alkaloids: Chemistry, Pharmacology, Toxicology and Food Safety. Int. J. Mol. Sci. 2018, 19, 1668. [Google Scholar] [CrossRef]
- These, A.; Bodi, D.; Ronczka, S.; Lahrssen-Wiederholt, M.; Preiss-Weigert, A. Structural screening by multiple reaction monitoring as a new approach for tandem mass spectrometry: Presented for the determination of pyrrolizidine alkaloids in plants. Anal. Bioanal. Chem. 2013, 405, 9375–9383. [Google Scholar] [CrossRef] [PubMed]
- Letsyo, E.; Jerz, G.; Winterhalter, P.; Beuerle, T. Toxic pyrrolizidine alkaloids in herbal medicines commonly used in Ghana. J. Ethnopharmacol. 2017, 202, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Roeder, E. Medicinal plants in China containing pyrrolizidine alkaloids. Pharmazie 2000, 55, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Xia, Q.; Fu, P.P.; Lin, G. Pyrrole-protein adducts—A biomarker of pyrrolizidine alkaloid-induced hepatotoxicity. J. Food Drug Anal. 2018, 26, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Schramm, S.; Köhler, N.; Rozhon, W. Pyrrolizidine Alkaloids: Biosynthesis, Biological Activities and Occurrence in Crop Plants. Molecules 2019, 24, 498. [Google Scholar] [CrossRef] [PubMed]
- Diaz, G.J. Toxicosis by Plant Alkaloids in Humans and Animals in Colombia. Toxins 2015, 7, 5408–5416. [Google Scholar] [CrossRef] [PubMed]
- Tamariz, J.; Burgueño-Tapia, E.; Vázquez, M.A.; Delgado, F. Chapter One-Pyrrolizidine Alkaloids. In The Alkaloids: Chemistry and Biology; Hans-Joachim, K., Ed.; Academic Press: Pittsburgh, PA, USA, 2018; Volume 80, pp. 1–314. [Google Scholar]
- Bodi, D.; Ronczka, S.; Gottschalk, C.; Behr, N.; Skibba, A.; Wagner, M.; Lahrssenwiederholt, M.; Preissweigert, A.; These, A. Determination of pyrrolizidine alkaloids in tea, herbal drugs and honey. Food Addit. Contam. 2014, 31, 1886–1895. [Google Scholar] [CrossRef]
- Avula, B.; Sagi, S.; Wang, Y.-H.; Zweigenbaum, J.; Wang, M.; Khan, I.A. Characterization and screening of pyrrolizidine alkaloids and N-oxides from botanicals and dietary supplements using UHPLC-high resolution mass spectrometry. Food Chem. 2015, 178, 136–148. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Chen, H.-W.; Lee, C.-C.; He, B.-J.; Yang, Y.-C. Hepatotoxic pyrrolizidine alkaloids in Emilia sonchifolia from Taiwan. J. Food Compos. Anal. 2015, 42, 1–7. [Google Scholar] [CrossRef]
- Hung, H.Y.; Kuo, S.C. Recent Studies and Progression of Yin Chen Hao, a Long-term Used Traditional Chinese Medicine. J. Tradit. Complem. Med. 2013. [Google Scholar] [CrossRef]
- Tan, X.J.; Li, Q.; Chen, X.H.; Wang, Z.W.; Shi, Z.Y.; Bi, K.S.; Ying, J. Simultaneous determination of 13 bioactive compounds in Herba Artemisiae Scopariae (Yin Chen) from different harvest seasons by HPLC-DAD. J. Pharm. Biomed. Anal. 2008, 47, 847–853. [Google Scholar] [CrossRef]
- Verma, R.S.; Rahman, L.; Chanotiya, C.S.; Verma, R.K.; Chauhan, A.; Yadav, A.; Yadav, A.K.; Singh, A. Chemical composition of volatile fraction of fresh and dry Artemisia capillaris Thunb. from Kumaon Himalaya. J. Essent. Oil Bear. Plants 2010, 13, 118–122. [Google Scholar] [CrossRef]
- Tian, M.; Row, K.H. Separation of four bioactive compounds from Herba artemisiae scopariae by HPLC with ionic liquid-based silica column. J. Anal. Chem. 2011, 66, 580–585. [Google Scholar] [CrossRef]
- Kundan Singh, B.; Anupam, S. The genus Artemisia: A comprehensive review. Pharm. Biol. 2011, 49, 101–109. [Google Scholar] [CrossRef]
- Crews, C.; Berthiller, F.; Krska, R. Update on analytical methods for toxic pyrrolizidine alkaloids. Anal. Bioanal. Chem. 2010, 396, 327–338. [Google Scholar] [CrossRef]
- Griffin, C.T.; Danaher, M.; Elliott, C.T.; Glenn Kennedy, D.; Furey, A. Detection of pyrrolizidine alkaloids in commercial honey using liquid chromatography-ion trap mass spectrometry. Food Chem. 2013, 136, 1577–1583. [Google Scholar] [CrossRef]
- Ma, C.; Liu, Y.; Zhu, L.; Ji, H.; Song, X.; Guo, H.; Yi, T. Determination and regulation of hepatotoxic pyrrolizidine alkaloids in food: A critical review of recent research. Food Chem. Toxicol. 2018, 119, 50–60. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on Pyrrolizidine alkaloids in food and feed. EFSA J. 2011, 9, 134. [Google Scholar] [CrossRef]
- Mulder, P.P.J.; Sánchez, P.L.; Anja, T. Occurrence of Pyrrolizidine Alkaloids in Food; Efsa Supporting Publications: Parma, Italy, 2015. [Google Scholar] [CrossRef]
- Bolechová, M.; Čáslavský, J.; Pospíchalová, M.; Kosubová, P. UPLC–MS/MS method for determination of selected pyrrolizidine alkaloids in feed. Food Chem. 2015, 170, 265–270. [Google Scholar] [CrossRef]
- Martinello, M.; Borin, A.; Stella, R.; Bovo, D.; Biancotto, G.; Gallina, A.; Mutinelli, F. Development and validation of a QuEChERS method coupled to liquid chromatography and high resolution mass spectrometry to determine pyrrolizidine and tropane alkaloids in honey. Food Chem. 2017, 234, 295–302. [Google Scholar] [CrossRef]
- Kaltner, F.; Rychlik, M.; Gareis, M.; Gottschalk, C. Influence of Storage on the Stability of Toxic Pyrrolizidine Alkaloids and Their N-Oxides in Peppermint Tea, Hay, and Honey. J. Agric. Food. Chem. 2018, 66, 5221–5228. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Kim, M.-S.; Kim, S.H.; Park, H.M.; Pyo, H.; Lee, Y.M.; Lee, K.-T.; Hong, J. Effective application of freezing lipid precipitation and SCX-SPE for determination of pyrrolizidine alkaloids in high lipid foodstuffs by LC-ESI-MS/MS. J. Chromatogr. B 2015, 992, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Mroczek, T.; Glowniak, K.; Wlaszczyk, A. Simultaneous determination of N-oxides and free bases of pyrrolizidine alkaloids by cation-exchange solid-phase extraction and ion-pair high-performance liquid chromatography. J. Chromatogr. A 2002, 949, 249–262. [Google Scholar] [CrossRef]
- Swc, C.; Lam, C.H. Development of an analytical method for analyzing pyrrolizidine alkaloids in different groups of foods by UPLC-MS/MS. J. Agric. Food. Chem. 2018, 66, 3009–3018. [Google Scholar] [CrossRef]
- World Health Organization. Pyrrolizidine Alkaloids Health and Safety Guide; World Health Organization: Geneva, Switzerland, 1989; Available online: http://www.inchem.org/documents/hsg/hsg/hsg026.htm (accessed on 5 February 2019).
- ANZFA Australia. Pyrrolizidine Alkaloids in Food: A Toxicological Review and Risk Assessment; Technical Report Series No. 2; Australia New Zealand Food Authority: Canberra, Australia, November 2001. Available online: http://www.foodstandards.gov.au/publications/Documents/TR2.pdf (accessed on 5 February 2019).
- National Toxicology Program (NTP). Final Report on Carcinogens Background Document for Riddelliine; U.S. Department of Health and Human Services: Washington, DC, USA, 11 August 2008. Available online: https://ntp.niehs.nih.gov/ntp/roc/twelfth/2010/finalbds/riddelliine_final_508.pdf (accessed on 5 February 2019).
- Committee on Toxicity (COT). Statement on Pyrrolizidine Alkaloids in Food; Committee on Toxicity of Chemicals in Food, Consumer, Products and the Environment: 2008. Available online: https://cot.food.gov.uk/sites/default/files/cot/cotstatementpa200806.pdf (accessed on 5 February 2019).
- Das Bundesinstitut für Risikobewertung (BfR). Analytik und Toxizität von Pyrrolizidinalkaloiden sowie eine Einschätzung des Gesundheitlichen Risikos durch deren Vorkommen in Honig; Stellungnahme Nr. 038/2011; Das Bundesinstitut für Risikobewertung (BfR): Berlin, Germany, 2011; Available online: https://www.bfr.bund.de/cm/343/analytik-und-toxizitaet-von-pyrrolizidinalkaloiden.pdf (accessed on 5 February 2019).
- Committee on Herbal Medicinal Products (HMPC). Public Statement on the Use of Herbal Medicinal Products Containing Toxic, Unsaturated Pyrrolizidine Alkaloids (PAs) EMA/HMPC/893108/2011; European Medicines Agency: London, UK, 24 November 2014. Available online: http://www.ehtpa.eu/pdf/EMA%20statement%202014.pdf (accessed on 5 February 2019).
- Zhao, J.C.; Jianjun, Y.; Christine, R.; Longtao, W.; Ming, H.; Dayong, W.; Liu, J.S.; Qianben, W.; Qin, Z.S.; Jindan, Y. Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012, 22, 322–331. [Google Scholar] [CrossRef]
- Kempf, M.; Beuerle, T.; Bühringer, M.; Denner, M.; Trost, D.; von der Ohe, K.; Bhavanam, V.B.R.; Schreier, P. Pyrrolizidine alkaloids in honey: Risk analysis by gas chromatography-mass spectrometry. Mol. Nutr. Food Res. 2008, 52, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Fan, Y.; He, W.; Hu, D.; Wu, A.; Wu, W. Development and Application of a QuEChERS-Based Liquid Chromatography Tandem Mass Spectrometry Method to Quantitate Multi-Component Alternaria Toxins in Jujube. Toxins 2018, 10, 382. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Statement on the risks for human health related to the presence of pyrrolizidine alkaloids in honey, tea, herbal infusions and food supplements. EFSA J. 2017, 15, 34. [Google Scholar] [CrossRef]
- Merz, K.-H.; Schrenk, D. Interim relative potency factors for the toxicological risk assessment of pyrrolizidine alkaloids in food and herbal medicines. Toxicol. Lett. 2016, 263, 44–57. [Google Scholar] [CrossRef]
- Wu, X.W.; Zhang, Y.B.; Zhang, L.; Yang, X.W. Simultaneous quantification of 33 active components in Notopterygii Rhizoma et Radix using ultra high performance liquid chromatography with tandem mass spectrometry. J. Chromatogr. B 2018, 1092, 244–251. [Google Scholar] [CrossRef]
- Mordehai, A.; Fjeldsted, J. Agilent Jet Stream Thermal Gradient Focusing Technology 2009. Available online: https://www.agilent.com/cs/library/technicaloverviews/public/5990-3494en_lo%20CMS.pdf (accessed on 7 March 2019).
- Wankun, D.; Yongbo, W.; Zexian, L.; Han, C.; Yu, X. HemI: A toolkit for illustrating heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef]
- Vercruysse, M.; Fauvart, M.; Jans, A.; Beullens, S.; Braeken, K.; Cloots, L.; Engelen, K.; Marchal, K.; Michiels, J. Stress response regulators identified through genome-wide transcriptome analysis of the (p)ppGpp-dependent response in Rhizobium etli. Genome Biol. 2011, 12, R17. [Google Scholar] [CrossRef]
- Farag, M.A.; Ali, S.E.; Hodaya, R.H.; El-Seedi, H.R.; Sultani, H.N.; Laub, A.; Eissa, T.F.; Abou-Zaid, F.O.F.; Wessjohann, L.A. Phytochemical Profiles and Antimicrobial Activities of Allium cepa Red cv. and A. sativum Subjected to Different Drying Methods: A Comparative MS-Based Metabolomics. Molecules 2017, 22, 761. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | PAs Abbr. | LOD μg/kg | LOQ μg/kg | Recoveries (%) Mean ± SD, n = 3 | Intra-Day RSD%, n = 6 | Inter-Day RSD%, n = 6 | Matrix Effects (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 μg/kg | 10 μg/kg | 100 μg/kg | |||||||
| 1 | Ret | 0.10 | 0.50 | 68.10 ± 0.35 | 75.38 ± 1.16 | 74.78 ± 2.12 | 4.54 | 3.32 | 47.22 |
| 2 | Em | 0.05 | 0.20 | 74.78 ± 0.18 | 79.41 ± 0.54 | 85.55 ± 1.16 | 0.94 | 5.04 | 96.91 |
| 3 | EmNO | 0.10 | 0.20 | 74.96 ± 0.21 | 91.38 ± 0.41 | 89.86 ± 0.78 | 1.51 | 7.64 | 96.48 |
| 4 | Er | 0.02 | 0.20 | 67.84 ± 1.55 | 71.82 ± 2.31 | 74.57 ± 1.02 | 0.59 | 4.37 | 90.16 |
| 5 | ErNO | 0.10 | 0.50 | 71.41 ± 1.24 | 87.15 ± 2.03 | 86.34 ± 1.43 | 1.78 | 3.27 | 102.36 |
| 6 | Eu | 0.01 | 0.10 | 73.33 ± 1.57 | 97.25 ± 1.14 | 91.47 ± 2.18 | 1.04 | 2.55 | 98.82 |
| 7 | EuNO | 0.02 | 0.10 | 82.25 ± 0.78 | 95.74 ± 2.24 | 97.40 ± 1.52 | 2.32 | 2.35 | 107.90 |
| 8 | He | 0.01 | 0.10 | 73.34 ± 0.35 | 92.67 ± 0.23 | 90.08 ± 0.80 | 0.65 | 2.81 | 103.98 |
| 9 | HeNO | 0.10 | 0.20 | 79.93 ± 0.46 | 93.88 ± 0.33 | 93.00 ± 2.25 | 1.86 | 3.76 | 95.06 |
| 10 | Im | 0.01 | 0.10 | 81.97 ± 1.34 | 91.45 ± 1.07 | 92.80 ± 1.11 | 0.79 | 4.72 | 87.77 |
| 11 | ImNO | 0.05 | 0.20 | 96.15 ± 1.47 | 96.50 ± 3.01 | 91.15 ± 2.38 | 6.88 | 4.04 | 114.20 |
| 12 | Jb | 0.10 | 0.50 | 77.63 ± 0.42 | 80.18 ± 1.12 | 78.82 ± 3.46 | 1.73 | 4.72 | 96.82 |
| 13 | JbNO | 0.10 | 0.50 | 71.64 ± 1.53 | 80.67 ± 2.45 | 81.99 ± 3.12 | 3.99 | 3.71 | 88.31 |
| 14 | Lc | 0.01 | 0.05 | 71.75 ± 0.57 | 89.46 ± 0.53 | 84.90 ± 5.12 | 1.89 | 4.48 | 100.80 |
| 15 | LcNO | 0.05 | 0.20 | 73.70 ± 0.95 | 97.82 ± 2.58 | 91.51 ± 0.31 | 2.43 | 5.05 | 97.42 |
| 16 | Ly | 0.01 | 0.10 | 76.47 ± 0.07 | 85.91 ± 1.65 | 89.52 ± 1.26 | 3.32 | 4.45 | 94.85 |
| 17 | LyNO | 0.05 | 0.2 | 101.58 ± 1.43 | 98.71 ± 1.53 | 93.08 ± 2.03 | 2.81 | 3.23 | 93.07 |
| 18 | Mc | 0.02 | 0.10 | 74.01 ± 1.14 | 88.38 ± 2.14 | 89.62 ± 1.68 | 0.38 | 4.35 | 91.92 |
| 19 | McNO | 0.20 | 0.50 | 84.62 ± 0.50 | 101.59 ± 1.09 | 92.46 ± 1.34 | 2.30 | 2.86 | 100.10 |
| 20 | Re | 0.05 | 0.50 | 78.97 ± 0.78 | 87.07 ± 0.55 | 88.80 ± 4.61 | 2.11 | 5.09 | 92.57 |
| 21 | ReNO | 0.10 | 0.50 | 74.40 ± 0.62 | 95.93 ± 2.32 | 86.89 ± 6.07 | 2.42 | 6.87 | 91.56 |
| 22 | Sn | 0.10 | 0.50 | 71.52 ± 0.85 | 87.74 ± 0.76 | 84.53 ± 7.43 | 1.24 | 1.10 | 106.26 |
| 23 | SnNO | 0.10 | 0.50 | 73.22 ± 0.98 | 94.08 ± 1.04 | 89.74 ± 0.87 | 0.47 | 3.58 | 108.66 |
| 24 | Sp | 0.10 | 0.50 | 69.65 ± 1.34 | 81.92 ± 4.34 | 80.32 ± 2.24 | 1.36 | 3.51 | 105.05 |
| 25 | SpNO | 0.20 | 0.50 | 73.51 ± 1.42 | 79.47 ± 1.21 | 80.19 ± 1.05 | 2.44 | 4.20 | 106.86 |
| 26 | Sv | 0.05 | 0.20 | 83.60 ± 0.25 | 82.63 ± 1.11 | 82.90 ± 1.14 | 0.75 | 2.52 | 103.74 |
| 27 | SvNO | 0.20 | 0.50 | 78.02 ± 0.34 | 89.94 ± 2.53 | 90.67 ± 5.21 | 4.29 | 3.27 | 112.64 |
| 28 | Ic | 0.01 | 0.10 | 81.97 ± 1.34 | 91.45 ± 1.07 | 92.80 ± 1.11 | 0.79 | 4.72 | 87.77 |
| 29 | IcNO | 0.05 | 0.20 | 96.15 ± 1.47 | 96.50 ± 3.01 | 91.15 ± 2.38 | 6.88 | 4.04 | 114.20 |
| 30 | 7-Im | 0.01 | 0.10 | 70.00 ± 1.52 | 81.60 ± 1.26 | 82.67 ± 1.26 | 1.17 | 0.14 | 110.29 |
| 31 | 7-ImNO | 0.05 | 0.20 | 70.45 ± 1.29 | 71.42 ± 0.94 | 71.52 ± 0.75 | 4.42 | 4.50 | 93.34 |
| 32 | Sk | 0.02 | 0.10 | 102.96 ± 3.07 | 100.08 ± 2.35 | 90.05 ± 1.56 | 1.19 | 1.43 | 97.50 |
| 33 | Td | 0.05 | 0.20 | 70.62 ± 0.24 | 90.04 ± 0.04 | 84.26 ± 0.17 | 3.33 | 3.95 | 101.22 |
| 34 | 7-Ly | 0.01 | 0.10 | 70.65 ± 1.53 | 83.40 ± 1.48 | 80.49 ± 0.04 | 0.45 | 1.04 | 95.62 |
| PAs | Im | Ly | ImNO | LyNO | Sk | EmNO | Sp | SpNO |
|---|---|---|---|---|---|---|---|---|
| Positive Samples | 25 | 22 | 25 | 29 | 22 | 1 | 2 | 1 |
| Range(μg/kg) | 0.11-383.28 | 0.29-92.05 | 0.20-255.46 | 0.21-1750.99 | 0.10-15.81 | 3.25 | 1.35-5.14 | 5.57 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.-H.; Wang, J.-C.; Guo, Q.-L.; Qiao, Y.; Wang, H.-J.; Liao, Y.-H.; Sun, D.-A.; Si, J.-Y. Simultaneous Determination and Risk Assessment of Pyrrolizidine Alkaloids in Artemisia capillaris Thunb. by UPLC-MS/MS Together with Chemometrics. Molecules 2019, 24, 1077. https://doi.org/10.3390/molecules24061077
Chen L-H, Wang J-C, Guo Q-L, Qiao Y, Wang H-J, Liao Y-H, Sun D-A, Si J-Y. Simultaneous Determination and Risk Assessment of Pyrrolizidine Alkaloids in Artemisia capillaris Thunb. by UPLC-MS/MS Together with Chemometrics. Molecules. 2019; 24(6):1077. https://doi.org/10.3390/molecules24061077
Chicago/Turabian StyleChen, Li-Hua, Jun-Chi Wang, Qi-Lei Guo, Yue Qiao, Hui-Juan Wang, Yong-Hong Liao, Di-An Sun, and Jian-Yong Si. 2019. "Simultaneous Determination and Risk Assessment of Pyrrolizidine Alkaloids in Artemisia capillaris Thunb. by UPLC-MS/MS Together with Chemometrics" Molecules 24, no. 6: 1077. https://doi.org/10.3390/molecules24061077
APA StyleChen, L.-H., Wang, J.-C., Guo, Q.-L., Qiao, Y., Wang, H.-J., Liao, Y.-H., Sun, D.-A., & Si, J.-Y. (2019). Simultaneous Determination and Risk Assessment of Pyrrolizidine Alkaloids in Artemisia capillaris Thunb. by UPLC-MS/MS Together with Chemometrics. Molecules, 24(6), 1077. https://doi.org/10.3390/molecules24061077