3. Materials and Methods
3.1. General Information
All commercial reagents and solvents were used as received without further purification unless otherwise indicated. Synthetic compounds 3a–d and 4a–l were directly used for reaction without further purification and characterization. Melting points were recorded on an SGW X-4 microscope melting point apparatus (Shanghai Tech Instrument Co., Ltd., Shanghai, China). Infrared spectra (IR) were performed on NICOLET iS10 sepectrometer (Shimazu Co., Ltd., Kyoto, Japan). NMR spectra were recorded on a Bruker Avance 500MHz spectrometer (Bruker Co., Ltd., Zurich, Switzerland) at room temperature with tetramethylsilane (TMS) as an internal standard and CDCl3 or DMSO-d6 as solvents. Mass spectra (MS) were obtained by LCMS-IT-TOF spectrometer (Shimadzu Co., Ltd., Kyoto, Japan) or TSQ Quantum Ultra (Thermo Scientific Co., Ltd., Madison, WI, USA). Elemental analysis for C, H, O, and N were carried out with Elementar VarioMICRO Cube analyzer (Elementar, Frankfurt, Germany).
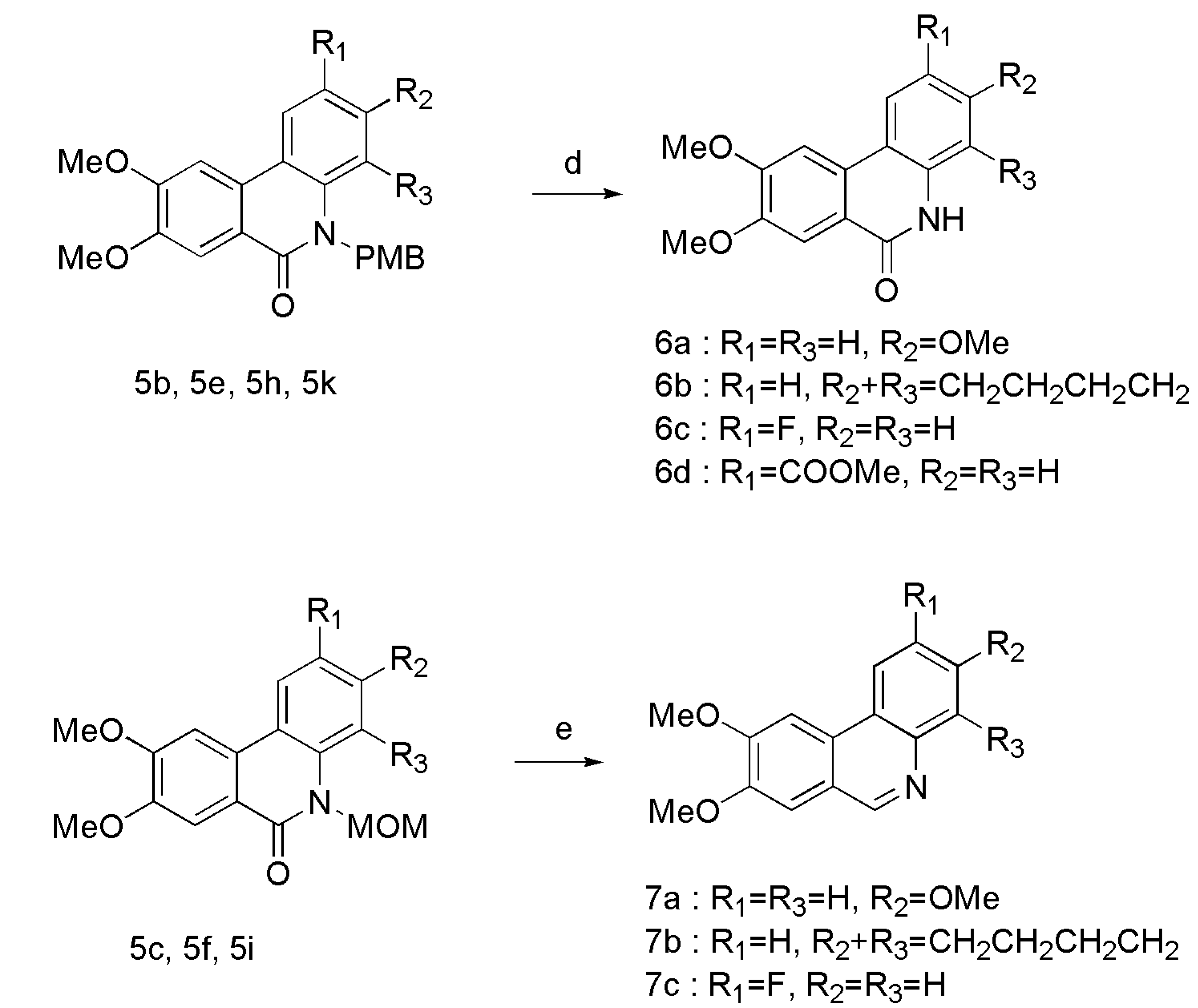
3.2. General Procedure for the Synthesis of Compounds 5a–l
2-bromo-4,5-dimethoxy-N-(3-methoxyphenyl)-N-methylbenzamide (4a) (2.00 g, 5.26 mmol), Pd(oAc)2 (0.118 g, 0.526 mmol), P(o-tol)3 (0.32 g, 1.052 mmol), K2CO3 (2.90 g, 21.0 mmol), and dry DMF (20 mL) were mixed under nitrogen atmosphere, and the mixture was stirred at 100 °C overnight. After cooled to room temperature, the reaction mixture was extracted with CH2Cl2. The organic phase was washed with H2O and brine, and then dried over anhydrous Na2SO4. After being concentrated under reduced pressure, the residue was purified on silica gel column chromatography (PE/EA, v/v = 1:1) to obtain compound 5a. Same method was used to provide compounds 5b–l from 4b–l, respectively. Derivatives 5b, 5c, 5e, 5f, 5h, 5i, 5k, and 5l were used without purification and further characterization.
3,8,9-trimethoxy-5-methyl-5H-phenanthridin-6-one (
5a) White solid, yield 87%, m.p. 191.0–193.0 °C. FTIR (KBr, cm
−1) 3441, 3132, 1643, 1608, 1510, 1463, 1402, 1313, 1262, 1230, 1146, 1032, 872, 821, 775, 726.
1H-NMR (CDCl
3) δ 8.06 (d,
J = 8.6 Hz, 1H), 7.89 (s, 1H), 7.48 (s, 1H), 6.91–6.87 (m, 2H), 4.08 (s, 3H), 4.02 (s, 3H), 3.94 (s, 3H), 3.78 (s, 3H).
13C-NMR (CDCl
3) δ 160.3, 153.5, 149.2, 139.2, 128.8, 128.4, 124.1, 113.0, 109.3, 109.0, 105.5, 102.2, 100.4, 56.3, 56.2, 55.7, 30.1. ESI-MS
m/
z: 338.34 ([M + K]
+). Anal. Calcd for C
17H
17NO
4: C 68.21; H 5.72; N 4.68; O 21.68. Found: C 67.92; H 5.58; N 4.55; O 21.40 (See
Supplementary Materials).
8,9-dimethoxy-5-methyl-2,3,4,5-tetrahydro-1H-benzo[c]phenanthridin-6-one (5d) White solid, yield 90%, m.p. 167.0–169.0 °C. FTIR (KBr, cm−1) 3441, 3132, 2931, 1640, 1604, 1516, 1493, 1456, 1419, 1374, 1312, 1271, 1234, 1212, 1153, 1028, 907, 768. 1H-NMR (CDCl3) δ 7.85–7.87 (m, 2H), 7.51 (s, 1H), 7.06 (d, J = 8.2 Hz, 1H), 4.06 (s, 3H), 4.02 (s, 3H), 3.74 (s, 3H), 3.60 (s, 1H), 3.33 (s, 1H), 2.95 (t, J = 6.3 Hz, 4H), 1.86 (m, 2H), 1.68 (m, 2H). 13C-NMR (CDCl3) δ 153.4, 149.5, 139.7, 139.2, 129.1, 127.7, 124.7, 119.6, 118.6, 108.8, 102.7, 56.3, 56.2, 39.7, 30.3, 29.6, 23.4, 22.3. ESI-MS: m/z: 324.16 ([M + H]+). Anal. Calcd for C20H21NO3: C 74.28; H 6.55; N 4.33; O 14.84. Found: C 74.03; H 6.68; N 4.24; O 14.92.
2-fluoro-8,9-dimethoxy-5-methyl-5H-phenanthridin-6-one (5g) White solid, yield 87%, m.p. 215.0–216.0 °C. FTIR (KBr, cm−1) 3433, 3134, 1640, 1594, 1515, 1465, 1403, 1320, 1277, 1187, 1027, 856, 808, 780, 616. 1H-NMR (CDCl3) δ 7.94 (s, 1H), 7.82–7.79 (dd, J = 2.7, 6.9 Hz, 1H), 7.47 (s, 1H), 7.38–7.35 (dd, J = 4.4, 4.7 Hz, 1H), 7.25-7.21 (m, 1H), 4.09 (s, 3H), 4.04 (s, 3H), 3.81 (s, 3H). 13C-NMR (CDCl3) δ 160.8, 158.5 (d, J = 241.1 Hz), 153.3, 150.3, 134.0, 127.3, 120.5 (d, J = 7.7 Hz), 120.0, 116.5 (d, J = 9.0 Hz), 115.8 (d, J = 23.0 Hz), 109.2, 108.5 (d, J = 23.5 Hz), 102.7, 56.3, 56.2, 30.2. ESI-MS m/z: 288.12 ([M + H]+). Anal. Calcd for C16H14FNO3: C 66.89; H 4.91; N 4.88; O 16.71. Found: C 66.61; H 4.73; N 4.55; O 16.45.
8,9-dimethoxy-5-methyl-6-oxo-5,6-dihydro-phenanthridine-2-carboxylic acid methyl ester (5j) White solid, yield 84%, m.p. 217.6–218.6 °C. FTIR (KBr, cm−1) 3435, 3134, 1711, 1648, 1613, 1516, 1402, 1314, 1267, 1112, 1032. 1H-NMR (CDCl3) δ 8.84 (d, J = 1.9 Hz, 1H), 8.16-8.14 (dd, J = 1.9, 1.9 Hz, 1H), 7.93 (s, 1H), 7.67 (s, 1H), 7.44 (d, J = 8.9 Hz, 1H), 4.14 (s, 3H), 4.05 (s, 3H), 4.00 (s, 3H), 3.84 (s, 3H). 13C-NMR (CDCl3) δ 166.9, 161.4, 153.8, 150.5, 140.9, 129.6, 128.0, 124.8, 124.0, 119.8, 119.1, 115.1, 109.3, 102.9, 56.5, 56.4, 52.4, 30.4. ESI-MS m/z: 318.12 ([M + H]+). Anal. Calcd for C18H17NO5: C 66.05; H 5.23; N 4.28; O 24.44. Found: C 66.21; H 5.14; N 3.96; O 24.16.
3.3. General Procedure for the Synthesis of Compounds 6a–d
TFA (6 mL) was slowly added to the flask with 5b (1.70 g, 4.193 mmol) under nitrogen atmosphere at 75 °C. The mixture was stirred overnight. After cooling, the reaction mixture was quenched with ethyl acetate and water, and then extracted with ethyl acetate. The organic phase was washed with H2O, aqueous NaHCO3 and brine, and dried over anhydrous Na2SO4. The crude product was purified on silica gel column chromatography (DCM/EA, v/v = 1:1) to obtain compound 6a. The same method was used for 6b–d from reactants 5e, 5h, and 5k.
3,8,9-trimethoxy-5H-phenanthridin-6-one (6a) White solid, yield 69%, m.p. 267.4–269.1 °C. FTIR (KBr, cm−1) 3440, 3160, 1662, 1612, 1503, 1404, 1330, 1258, 1210, 1176, 1098, 1044, 877, 840, 802. 1H-NMR (CDCl3) δ 9.80 (br, 1H), 7.99 (d, J = 8.9 Hz, 1H), 7.51 (s, 1H), 6.89 (d, J = 8.9 Hz, 1H), 6.74 (s, 1H), 4.09 (s, 3H), 4.04 (s, 3H), 3.91 (s, 3H). 13C-NMR (DMSO-d6) δ 160.8, 159.7, 153.4, 148.7, 137.5, 129.5, 124.7, 117.9, 111.4, 109.9, 107.9, 103.6, 99.3, 56.1, 55.6, 55.3. ESI-MS m/z: 286.12 ([M + H]+). Anal. Calcd for C16H15NO4: C 67.36; H 5.30; N 4.91; O 22.43. Found: C 67.07; H 5.38; N 4.76; O 22.24.
8,9-dimethoxy-2,3,4,5-tetrahydro-1H-benzo[c]phenanthridin-6-one (6b) White solid, yield 76%, m.p. 253.0–255.0 °C. FTIR (KBr, cm−1) 3439, 3132, 1651, 1610, 1496, 1398, 1234, 1129, 1079, 836. 1H-NMR (CDCl3) δ 8.56 (br, 1H), 7.87-7.86 (m, 2H), 7.59 (s, 1H), 7.03 (d, J = 8.0 Hz, 1H), 4.09 (s, 3H), 4.04 (s, 3H), 2.88 (t, J = 6.1 Hz, 2H), 2.77 (t, J = 6.4 Hz, 2H), 2.01-1.96 (m, 1H), 1.87-1.83 (m, 2H). 13C-NMR (CDCl3) δ 166.7, 153.9, 149.7, 138.4, 1335, 132.1, 124.0, 121.9, 119.6, 116.0, 108.6, 103.0, 56.4, 56.3, 30.1, 23.6, 22.8, 22.5. ESI-MS m/z: 310.15 ([M + H]+). Anal. Calcd for C19H19NO3: C 73.77; H 6.19; N 4.53; O 15.52. Found: C 73.43; H 6.02; N 4.36; O 15.38.
2-fluoro-8,9-dimethoxy-5H-phenanthridin-6-one (6c) White solid; yield 54%, m.p. 192.1–193.0 °C. FTIR (KBr, cm−1) 3438, 3142, 1643, 1600, 1535, 1510, 1402, 1262, 1213, 1029, 824. 1H-NMR (CDCl3) δ 7.94 (br, 1H), 7.63–7.61 (m, 2H), 7.33 (s, 1H), 7.10–7.05 (m, 2H), 3.93 (s, 3H), 3.92 (s, 1H). 13C-NMR (CDCl3) δ 164.9, 160.0 (d, J = 243.7), 151.4, 148.8, 133.8, 129.0, 122.0, 122.0, 116.0, 116.0, 115.9, 113.4, 110.0, 56.5, 56.4. ESI-MS m/z: 274.35 ([M + H]+). Anal. Calcd for C15H12FNO3: C 65.93; H 4.43; N 5.13; O 17.57. Found: C 65.27; H 4.30; N 5.06; O 17.42.
8,9-dimethoxy-6-oxo-5,6-dihydro-phenanthridine-2-carboxylic acid methyl ester (6d) White solid, yield 78%, m.p. 296.5–299.0 °C. FTIR (KBr, cm−1) 3438, 3156, 1709, 1663, 1617, 1514, 1402, 1267, 1092, 1035. 1H-NMR (DMSO-d6) δ 11.93 (s, 1H), 8.84–8.82 (m, 1H), 8.00–7.97 (m, 1H), 7.84 (d, J = 3.0 Hz, 1H), 7.70 (s, 1H), 7.39 (d, J = 9.0 Hz, 1H), 4.06 (s, 3H), 3.91 (s, 3H), 3.90 (s, 3H). 13C-NMR (DMSO-d6) δ 166.1, 161.0, 153.9, 150.3, 140.0, 129.6, 128.8, 125.1, 124.0, 119.8, 117.8, 116.7, 108.3, 104.6, 56.7, 56.1, 52.6. ESI-MS m/z: 314.11 ([M + H]+). Anal. Calcd for C17H15NO5: C 65.17; H 4.83; N 4.47; O 25.53. Found: C 65.62; H 4.91; N 4.19; O 25.17.
3.4. General Procedure for the Synthesis of Compounds 7a–c
LiAlH4 (325 mg, 8.56 mmol) was added to the solution of 5c (940 mg, 2.85 mmol) in dry THF (30 mL) under nitrogen atmosphere at 0 °C, and then the mixture was stirred for 4 h at room temperature. The progress of the reaction was monitored by TLC. The reaction mixture was cooled to room temperature, followed by extraction with DCM. The organic phase was washed with H2O and brine, dried over anhydrous Na2SO4, and then concentrated under reduced pressure. The crude product was purified on silica gel column chromatography (PE/EA, v/v = 1:2) to obtain compound 7a. The same method was used for 7b and 7c from compounds 5f and 5i.
3,8,9-trimethoxyphenanthridine (7a) White solid, yield 55%, m.p. 163.0–165.0 °C. FTIR (KBr, cm−1) 3435, 3141, 1619, 1504, 1398, 1274, 1213, 1162, 1034, 840, 808. 1H-NMR (CDCl3) δ 9.13 (s, 1H), 8.36 (d, J = 8.0 Hz, 1H), 7.81 (s, 1H), 7.63 (s, 1H), 7.35 (s, 1H), 7.33–7.30 (m, 1H), 4.15 (s, 3H), 4.07 (s, 3H), 4.00 (s, 3H). 13C-NMR (CDCl3) δ 159.6, 153.3, 152.1, 149.5, 145.6, 128.8, 123.0, 121.0, 118.2, 118.1, 109.7, 107.9, 101.5, 56.3, 56.2, 55.7. ESI-MS m/z: 270.12 ([M + H]+). Anal. Calcd for C16H15NO3: C 71.36; H 5.61; N 5.20; O 17.82. Found: C 70.90; H 5.43; N 5.15; O 17.55.
8,9-dimethoxy-1,2,3,4-tetrahydrobenzo[c]phenanthridine (7b) White solid, yield 45%, m.p. 178.6–179.8 °C. FTIR (KBr, cm−1) 3437, 3140, 2930, 1612, 1502, 1405, 1269, 1201, 1157, 1027, 846. 1H-NMR (CDCl3) δ 9.14 (s, 1H), 8.18 (d, J = 8.7 Hz, 1H), 7.82 (s, 1H), 7.35 (d, J = 8.7 Hz, 1H), 7.31 (s, 1H), 4.12 (s, 3H), 4.06 (s, 3H), 3.40 (t, J = 5.3 Hz, 2H), 2.97 (t, J = 6.0 Hz, 2H), 1.99–1.96 (m, 2H), 1.93–1.90 (m, 2H). 13C-NMR (CDCl3) δ 152.9, 150.2, 149.7, 142.5, 136.9, 135.6, 128.8, 128.5, 121.5, 121.4, 118.8, 107.7, 101.8, 56.2, 56.2, 30.3, 25.8, 23.3, 23.1. ESI-MS m/z: 294.15 ([M + H]+). Anal. Calcd for C19H19NO2: C 77.79; H 6.53; N 4.77; O 10.91. Found: C 77.34; H 6.38; N 4.52; O 10.94.
2-fluoro-8,9-dimethoxyphenanthridine (7c) White solid, yield 47%, m.p. 178.0–179.0 °C. FTIR (KBr, cm−1) 3436, 3134, 1617, 1511, 1400, 1270, 1195, 1151, 1098, 1028, 848. 1H-NMR (CDCl3) δ 9.11 (s, 1H), 8.16–8.05 (m, 1H), 8.04–8.02 (m, 1H), 7.74 (s, 1H), 7.45–7.41 (m, 1H), 7.37 (s, 1H), 4.15 (s, 3H), 4.08 (s, 3H). 13C-NMR (CDCl3) δ 161.3 (d, J = 247.4), 153.3, 150.9 (d, J = 2.0), 150.7, 140.6, 132.2 (d, J = 9.0), 127.9 (d, J = 4.4), 125.3, 121.9, 117.0 (d, J = 24.4), 108.0, 106.6 (d, J = 23.2), 102.1, 56.4, 56.3, ESI-MS m/z: 258.11 ([M + H]+). Anal. Calcd for C15H12FNO2: C 70.03; H 4.70; N 5.44; O 12.44. Found: C 69.61; H 5.01; N 5.22; O 12.77.
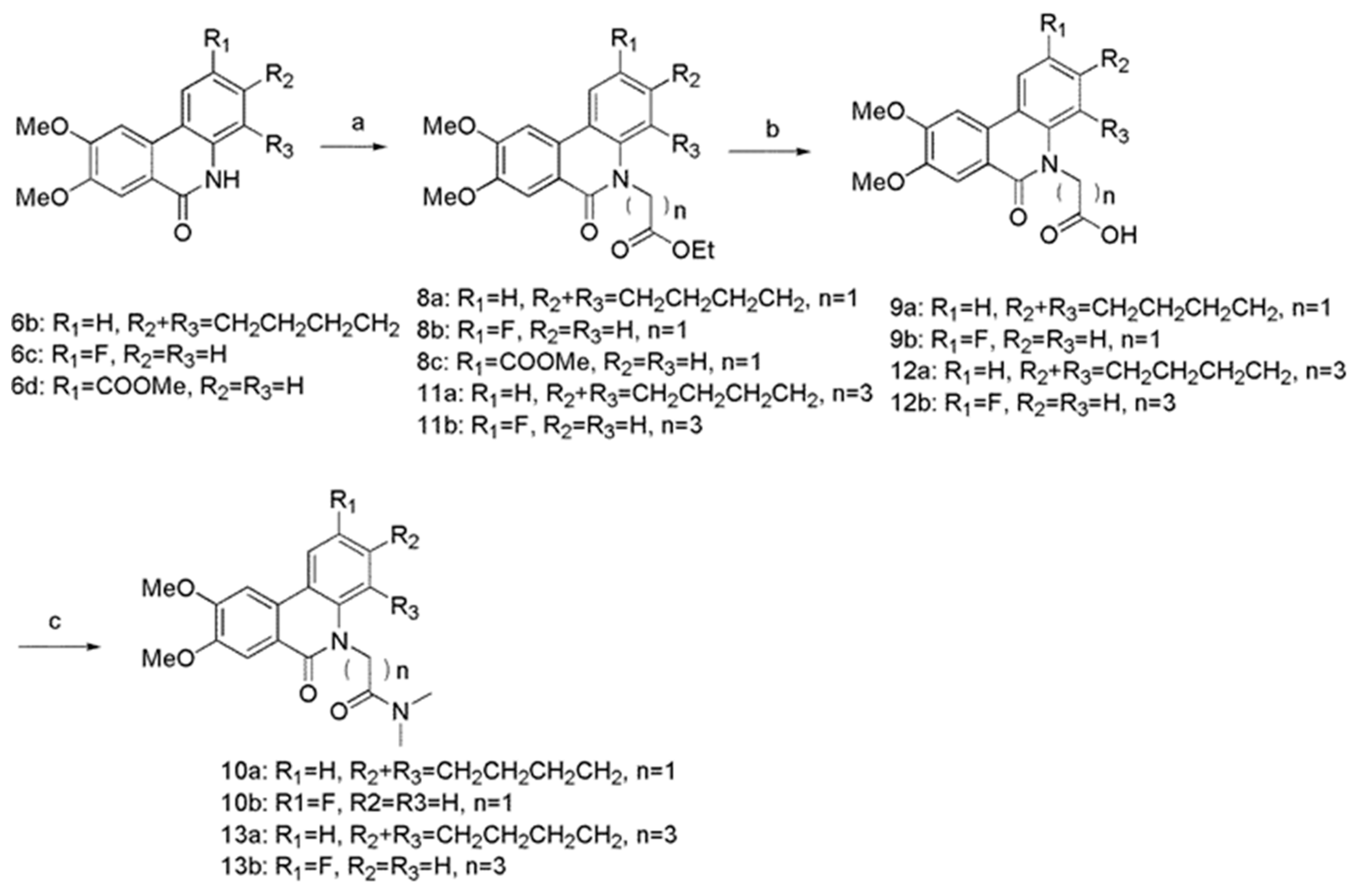
3.5. General Procedure for the Synthesis of Compounds 8a–c and 11a–b
To the solution of 6b (216 mg, 0.70 mmol) in DMF (5 mL), Cs2CO3 (456.83 mg, 1.40 mmol), TBAI (38.84 mg, 0.105 mmol), and ethyl bromoacetate (526.83 mg, 3.15 mmol) were added, and the mixture was stirred for 1 h at 90 °C. After cooled to room temperature, the mixture was extracted with ethyl acetate. The organic phase was washed with H2O and brine, dried over anhydrous Na2SO4, and then concentrated under reduced pressure. The residue was purified on silica gel column chromatography (PE/EA, v/v = 4:1) to obtain compound 8a. Same method was used for compounds 8b, 8c from 6c and 6d. Ethyl 4-bromobutyrate was used instead of ethyl bromoacetate to 11a, 11b from 6b and 6c.
(8,9-dimethoxy-6-oxo-1,3,4,6-tetrahydro-2H-benzo[c]phenanthridin-5-yl)-acetic acid ethyl ester (8a) White solid, yield 87.%, m.p. 152.3–153.7 °C. FTIR (KBr, cm−1) 3440, 3132, 2927, 1753, 1599, 1525, 1401, 1333, 1266, 1203, 1175, 1125, 1087, 1042, 860, 767. 1H-NMR (CDCl3) δ 8.05 (d, J = 8.2 Hz, 1H), 7.77 (s, 1H), 7.73 (s, 1H), 7.19 (d, J = 8.2 Hz, 1H), 5.10 (s, 2H), 4.25 (q, J = 7.1 Hz, 2H), 4.11 (s, 3H), 4.06 (s, 3H), 3.16 (t, J = 6.1 Hz, 2H), 2.92 (t, J = 5.9 Hz, 2H), 1.93–1.86 (m, 4H), 1.28 (t, J = 6.3 Hz, 3H). 13C-NMR (CDCl3) δ 169.7, 155.8, 152.8, 149.4, 140.5, 137.1, 134.1, 131.3, 126.0, 120.1, 118.6, 113.2, 104.8, 102.4, 63.1, 61.0, 56.2, 56.2, 30.2, 25.2, 23.4, 23.3, 14.4. ESI-MS m/z: 396.17 ([M + H]+). Anal. Calcd for C23H25NO5: C 69.86; H 6.37; N 3.54; O 20.23. Found: C 69.71; H 6.20; N 3.42; O 20.89.
(2-fluoro-8,9-dimethoxy-6-oxo-6H-phenanthridin-5-yl)-acetic acid ethyl ester (8b) White solid, yield 95%, m.p. 152.3–153.7 °C. FTIR (KBr, cm−1) 3439, 3128, 1744, 1659, 1600, 1509, 1399, 1258, 1208, 1024, 845. 1H-NMR (CDCl3) δ 7.28–7.25 (m, 1H), 6.90–6.87 (m, 2H), 6.84 (s, 1H), 6.65 (s, 1H), 4.55 (s, 2H), 4.25 (q, J = 7.2 Hz, 2H), 3.80 (s, 3H), 3.71 (s, 3H), 1.31 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3) δ 169.1, 169.0, 161.6 (d, J = 240 Hz), 150.0, 148.0, 138.7, 138.6, 129.4, 129.4, 116.1, 116.0, 115.4, 112.0, 110.6, 61.7, 56.2, 56.2, 51.5, 29.8, 14.3. ESI-MS m/z: 360.09 ([M + H]+). Anal. Calcd for C19H18FNO5: C 63.50; H 5.05; N 3.90; O 22.26. Found: C 63.04; H 4.85; N 3.62; O 21.10.
5-ethoxycarbonylmethyl-8,9-dimethoxy-6-oxo-5,6-dihydro-phenanthridine-2-carboxylic acid methyl ester (8c) White solid, yield 87%, m.p. 211.9–214.0 °C. FTIR (KBr, cm−1) 3432, 3134, 1740, 1711, 1648, 1614, 1517, 1400, 1322, 1270, 1215, 1118, 1046, 1017, 835, 769. 1H-NMR (CDCl3) δ 8.85 (s, 1H), 8.10 (d, J = 8.9 Hz, 1H), 7.91 (s, 1H), 7.67 (s, 1H), 7.17 (d, J = 8.9 Hz, 1H), 5.21 (s, 2H), 4.25 (q, J = 7.2 Hz, 2H), 4.14 (s, 3H), 4.04 (s, 3H), 3.99 (s, 3H), 1.27 (t, J = 7.2 Hz, 4H). 13C-NMR (CDCl3) δ 168.3, 166.7, 154.2, 150.5, 140.1, 129.7, 128.4, 125.2, 124.4, 119.3, 119.2, 114.6, 109.4, 103.1, 62.0, 56.6, 56.4, 52.4, 44.7, 14.3. ESI-MS m/z: 400.13 ([M + H]+). Anal. Calcd for C21H21NO7: C 63.15; H 5.30; N 3.51; O 28.04. Found: C 63.27; H 5.41; N 3.49; O 27.86.
4-(8,9-dimethoxy-6-oxo-1,3,4,6-tetrahydro-2H-benzo[c]phenanthridin-5-yl)-butyric acid ethyl ester (11a) White solid, yield 76%, m.p. 104.8–106.8 °C. FTIR (KBr, cm−1) 3434, 3137, 2921, 1728, 1595, 1528, 1403, 1322, 1267, 1228, 1176, 1035, 859, 766. 1H-NMR (CDCl3) δ 8.05 (d, J = 8.4 Hz, 1H), 7.78 (s, 1H), 7.64 (s, 1H), 7.18 (d, J = 8.4 Hz, 1H), 4.70 (t, J = 6.2 Hz, 2H), 4.14–4.10 (m, 5H), 4.06 (s, 3H), 3.25 (t, J = 6.1 Hz, 2H), 2.93 (t, J = 6.1 Hz, 2H), 2.58 (t, J = 7.4 Hz, 2H), 2.33–2.27 (m, 2H), 1.95–1.87 (m, 4H), 1.22 (t, J = 7.6 Hz, 3H). 13C-NMR (CDCl3) δ 173.6, 156.9, 152.6, 149.3, 141.1, 137.0, 134.1, 131.0, 125.6, 119.7, 118.5, 113.8, 104.7, 102.4, 65.0, 60.5, 56.2, 56.2, 31.7, 30.3, 25.4, 24.8, 23.5, 23.3, 14.3. ESI-MS m/z: 424.20 ([M + H]+). Anal. Calcd for C25H29NO5: C 70.90; H 6.90; N 3.31; O 18.89. Found: C 70.82; H 6.95; N 3.38; O 18.77.
4-(2-fluoro-8,9-dimethoxy-6-oxo-6H-phenanthridin-5-yl)-butyric acid ethyl ester (11b) Yellow oil, yield 79.%. FTIR (KBr, cm−1) 3437, 3120, 2934, 1729, 1643, 1598, 1508, 1444, 1405, 1323, 1259, 1210, 1184, 1092, 1023, 848, 787. 1H-NMR (CDCl3) δ 7.15–7.12 (m, 2H), 6.91–6.87 (m, 1H), 6.81 (s, 1H), 6.54 (s, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.94 (t, J = 6.7 Hz, 1H), 3.78 (s, 3H), 3.71 (s, 3H), 2.45 (t, J = 6.8 Hz, 1H), 2.00–1.94 (m, 2H), 1.24 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3) δ 173.1, 168.7, 161.4 (d, J = 248.0), 149.7, 148.0, 138.0, 138.0, 129.5, 129.4, 116.2, 116.0, 115.3, 111.7, 110.5, 60.6, 56.2, 56.2, 489, 31.7, 23.1, 14.3. ESI-MS m/z: 388.12 ([M + H]+). Anal. Calcd for C21H22FNO5: C 65.11; H 5.72; N 3.62; O 20.65. Found: C 64.94; H 5.63; N 3.65; O 20.32.
3.6. General Procedure for the Synthesis of Compounds 9a–b and 12a–b
To the solution of 8a (107 mg, 0.27 mmol) in MeOH (27 mL) was added the solution of 10N NaOH (18 mL). The reaction mixture was refluxed for 16 h at room temperature. And then 2 M HCl was added to adjust pH~2. The aqueous layer was extracted with ethyl acetate. The combined organic phase was washed with H2O and brine, dried over anhydrous Na2SO4, and then concentrated under reduced pressure. The residue was purified on silica gel column chromatography (DCM/MeOH, v/v = 15:1) to obtain compound 9a. The same method was used for 9b, 12a, and 12b from reactants 8b, 11a, and 11b.
(8,9-dimethoxy-6-oxo-1,3,4,6-tetrahydro-2H-benzo[c]phenanthridin-5-yl)-acetic acid (9a) White solid, yield 93%, m.p. 181.3–182.3 °C. FTIR (KBr, cm−1) 3130, 2932, 1724, 1599, 1402, 1330, 1261, 1211, 1173, 1127, 1036. 1H-NMR (CDCl3) δ 8.09 (d, J = 8.4 Hz, 1H), 7.80 (s, 1H), 7.68 (s, 1H), 7.27 (d, J = 8.4, 1H), 5.18 (s, 2H), 4.13 (s, 3H), 4.06 (s, 3H), 3.17 (t, J = 6.0 Hz, 2H), 2.94 (t, J = 6.0 Hz, 2H), 1.95–1.86 (m, 4H). 13C-NMR (CDCl3) δ 157.1, 153.4, 149.7, 137.8, 133.6, 131.7, 126.7, 120.3, 118.7, 112.9, 104.5, 102.4, 65.4, 56.3, 56.3, 30.3, 29.9, 25.2, 23.2, 23.1. ESI-MS m/z: 368.14 ([M + H]+). Anal. Calcd for C21H21NO5: C 68.65; H 5.76; N 3.81; O 21.77. Found: C 68.50; H 5.34; N 3.73; O 21.52.
(2-fluoro-8,9-dimethoxy-6-oxo-6H-phenanthridin-5-yl)-acetic acid (9b) White solid, yield 96%, m.p. 107.0–108.8 °C. FTIR (KBr, cm−1) 3439, 3132, 1638, 1509, 1400, 1258, 1212, 1014, 844. 1H-NMR (CDCl3) δ 7.25 (m, 1H), 6.90–6.88 (m, 2H), 6.84 (s, 1H), 6.65 (s, 1H), 4.61 (s, 2H), 3.80 (s, 3H), 3.72 (s, 3H). 13C-NMR (CDCl3) δ 169.7, 162.5, 160.5, 150.0, 148.0, 138.6, 138.6, 129.3, 129.2, 116.1, 115.9, 115.2, 112.1, 110.4, 56.3, 56.1, 29.8. ESI-MS m/z: 332.12 ([M + H]+). Anal. Calcd for C17H14FNO5: C 61.63; H 4.26; N 4.23; O 24.15. Found: C 61.42; H 4.55; N 4.05; O 23.80.
4-(8,9-dimethoxy-6-oxo-1,3,4,6-tetrahydro-2H-benzo[c]phenanthridin-5-yl)-butyric acid (12a) White solid, yield 95%, m.p. 209.0–210.0 °C. FTIR (KBr, cm−1) 3443, 3130, 2931, 1703, 1594, 1503, 1401, 1325, 1270, 1208, 1040, 1001, 864, 774. 1H-NMR (DMSO-d6) δ 8.33 (d, J = 8.4 Hz, 1H), 8.01 (s, 1H), 7.58 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 4.59 (t, J = 7.1 Hz, 2H), 4.03 (s, 3H), 3.93 (s, 3H), 3.16 (t, J = 7.7 Hz, 2H), 2.88 (t, J = 4.8 Hz, 2H), 2.48 (t, J = 6.7 Hz, 2H), 2.16–2.11 (m, 2H), 1.87-1.81 (m, 4H). 13C-NMR (DMSO-d6) δ 174.7, 156.8, 153.2, 149.6, 140.7, 136.8, 133.2, 130.8, 125.8, 119.9, 119.7, 113.1, 104.5, 103.5, 65.3, 56.4, 56.1, 31.3, 29.9, 25.2, 24.6, 23.3, 23.2. ESI-MS m/z: 396.18 ([M + H]+). Anal. Calcd for C23H25NO5: C 69.86; H 6.37; N 3.54; O 20.23. Found: C 69.88; H 6.46; N 3.29; O 20.51.
4-(2-fluoro-8,9-dimethoxy-6-oxo-6H-phenanthridin-5-yl)-butyric acid (12b) White solid, yield 98. %, m.p. 47.5–49.6 °C. FTIR (KBr, cm−1) 3440, 3132, 1644, 1509, 1400, 1257, 1213, 1024, 841. 1H-NMR (CDCl3) δ 7.14 (dd, J = 8.7, 4.8 Hz, 2H), 6.91 (t, J = 8.4 Hz, 1H), 6.82 (s, 1H), 6.53 (s, 1H), 3.97 (d, J = 6.3 Hz, 2H), 3.79 (s, 3H), 3.70 (s, 3H), 2.54 (t, J = 7.3 Hz, 2H), 2.00–1.95 (m, 2H). 13C-NMR (CDCl3) δ 176.7, 169.2, 161.5 (d, J = 251.0 Hz), 149.9, 148.1, 137.8, 130.2, 129.4 (d, J = 8.7 Hz), 116.2 (d, J = 22 Hz), 115.3, 111.6, 110.6, 100.1, 56.3, 56.2, 48.8, 31.3, 23.0. ESI-MS m/z: 360.13 ([M + H]+). Anal. Calcd for C19H18FNO5: C 63.50; H 5.05; N 3.90; O 22.26. Found: C 63.24; H 4.92; N 3.68; O 22.01.
3.7. General Procedure for the Synthesis of Compounds 10a–b and 13a–b
Compound 9a (36.8 mg, 0.1 mmol) was dissolved in THF (10 mL) and CH2Cl2 (4 mL) at 0 °C. Triethylamine (20 μL) and isobutyl chloroformate (18 μL) were added (18 μL), and the mixture was stirred for 20 min. Then dimethylamine (0.5 mL) was added at 0 °C and the mixture was stirred for 30 min at room temperature. After evaporation of solvent under reduced pressure, the mixture was extracted with ethyl acetate. The combined organic phase was washed with H2O and brine, dried over anhydrous Na2SO4, and then concentrated under reduced pressure. The crude product was purified on silica gel column chromatography (DCM/MeOH, v/v = 30:1) to obtain compound 10a. The same method was used for 10b, 13a, and 13b from 9b, 12a, and 12b.
2-(8,9-dmethoxy-6-oxo-1,3,4,6-tetrahydro-2H-benzo[c]phenanthridin-5-yl)-N,N-dimethyl-acetamide (10a) White solid, yield 85%, m.p. 196.0–197.8 °C. FTIR (KBr, cm−1) 3435, 3134, 2931, 1667, 1601, 1502, 1400, 1324, 1269, 1215, 1175, 1122, 1035, 769. 1H-NMR (CDCl3) δ 8.05 (d, J = 8.4 Hz, 1H), 7.78 (s,1H), 7.77 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 5.27 (s, 2H), 4.11 (s, 3H), 4.06 (s, 3H), 3.19 (s, 3H), 3.18 (t, J = 6.5 Hz, 2H), 3.04 (s, 3H), 2.92 (t, J = 6.0 Hz, 2H), 1.94–1.85 (m, 4H). 13C-NMR (CDCl3) δ 168.6, 156.0, 152.8, 149.4, 140.7, 137.0, 133.9, 131.3, 125.9, 120.1, 118.6, 113.4, 104.8, 102.4, 63.4, 56.3, 56.2, 36.6, 35.8, 30.2, 25.2, 23.5, 23.2. ESI-MS m/z: 395.19 ([M + H]+). Anal. Calcd for C23H26N2O5: C 70.03; H 6.64; N 7.10; O 16.22. Found: C 69.71; H 6.55; N 6.96; O 16.19.
2-(2-fluoro-8,9-dimethoxy-6-oxo-6H-phenanthridin-5-yl)-N,N-dimethyl-acetamide (10b) White solid; yield 83%, m.p. 57.0–59.0 °C. FTIR (KBr, cm−1) 3442, 3123, 2934, 1652, 1600, 1508, 1399, 1325, 1257, 1212, 1155, 1032, 927, 847, 790. 1H-NMR (CDCl3) δ 7.37–7.34 (m, 2H), 6.87–6.82 (m, 2H), 6.75 (s, 1H), 4.63 (s, 2H), 3.79 (s, 3H), 3.71 (s, 3H), 3.07 (s, 3H), 3.02 (s, 3H). 13C-NMR (CDCl3) δ 169.1, 167.4, 149.9, 148.1, 130.0, 129.6, 129.6, 115.9, 115.7, 115.2, 114.4, 112.3, 110.4, 108.4, 56.3, 56.2, 51.5, 36.6, 36.1. ESI-MS m/z: 361.17 ([M + H]+). Anal. Calcd for C19H19FN2O4: C 63.68; H 5.34; N 7.82; O 17.86. Found: C 63.27; H 5.09; N 7.72; O 17.48.
4-(8,9-dimethoxy-6-oxo-1,3,4,6-tetrahydro-2H-benzo[c]phenanthridin-5-yl)-N,N-dimethyl-butyramide (13a) White solid, yield 84.%, m.p. 249.2–251.5 °C. FTIR (KBr, cm−1) 3436, 3135, 2924, 1643, 1597, 1501, 1403, 1320, 1264, 1229, 1205, 1174, 1034, 996, 868, 776. 1H-NMR (CDCl3) δ 8.05 (d, J = 8.3 Hz, 1H), 7.78 (s, 1H), 7.65 (s, 1H), 7.18 (d, J = 8.3 Hz, 1H), 4.70 (t, J = 6.2 Hz, 2H), 4.11 (s, 3H), 4.05 (s, 3H), 3.25 (t, J = 5.9 Hz, 2H), 2.99 (s, 3H), 2.96 (s, 3H), 2.58 (t, J = 7.6 Hz, 2H), 2.58 (t, J = 7.5 Hz, 2H), 2.35-2.29 (m, 2H), 1.94-1.88 (m, 4H). 13C-NMR (CDCl3) δ 172.7, 156.9, 152.5, 149.2, 141.2, 136.9, 134.1, 131.0, 125.5, 119.6, 118.5, 113.8, 104.6, 102.4, 66.3, 56.2, 56.1, 37.3, 35.6, 30.3, 30.2, 25.4, 25.0, 23.4, 23.3. ESI-MS m/z: 423.22 ([M + H]+). Anal. Calcd for C25H30N2O4: C 71.07; H 7.16; N 6.63; O 15.15. Found: C 70.82; H 7.22; N 6.37; O 15.31.
4-(2-fluoro-8,9-dimethoxy-6-oxo-6H-phenanthridin-5-yl)-N,N-dimethyl-butyramide (13b) White solid, yield 82.%, m.p. 48.9–50.5 °C. FTIR (KBr, cm−1) 3437, 3132, 1646, 1508, 1400, 1258, 1211, 1024, 846. 1H-NMR (CDCl3) δ 7.18–7.05 (m, 1H), 6.88 (t, J = 8.5 Hz, 1H), 6.81 (s, 1H), 6.54 (s, 1H), 3.95 (t, J = 6.9 Hz, 2H), 3.78 (s, 3H), 3.69 (s, 3H), 3.02(s, 3H), 2.94 (s, 3H), 2.49 (t, J = 7.3 Hz, 2H), 2.02-1.97 (m, 2H). 13C-NMR (CDCl3) δ 172.3, 168.8, 149.7, 148.0, 138.2, 130.6, 129.6, 129.5, 116.2, 116.0, 115.3, 111.8, 110.5, 100.1, 56.2, 49.3, 37.4, 35.6, 30.8, 23.4. ESI-MS m/z: 387.16 ([M + H]+). Anal. Calcd for C21H23FN2O4: C 65.27; H 6.00; N 7.25; O 16.56. Found: C 64.92; H 5.97; N 6.97; O 16.37.
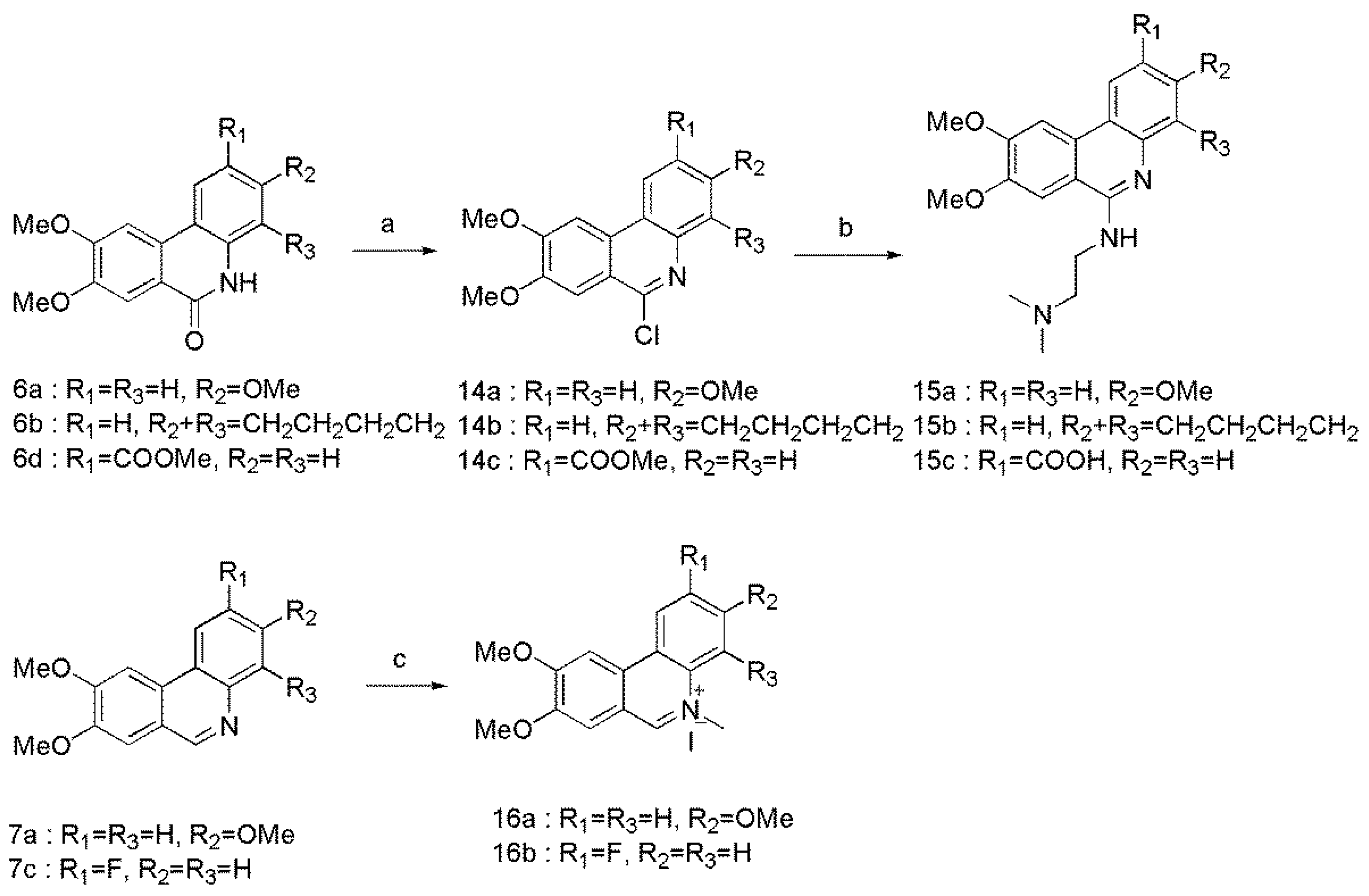
3.8. General Procedure for the Synthesis of Compounds 14a–c
POCl3 (0.64 mL) was added to compound 6a (100 mg, 0.35 mmol) in flask. Then the reaction mixture was stirred for 2 h at 105 °C. After cooled to room temperature, the mixture was poured carefully into a beaker filled with ice water. Concentrated ammonia water was added untill pH > 7. The precipitation was washed with water and purified on silica gel column chromatography (PE/DCM, v/v = 1:1) to obtain compound 14a. The same method was used for 14b and 14c from compounds 6b and 6d.
6-chloro-3,8,9-trimethoxyphenanthridine (14a) White solid, yield 83%, m.p. 174.1–175.6 °C. FTIR (KBr, cm−1) 3443, 3133, 1617, 1579, 1503, 1400, 1313, 1234, 1213, 1155, 1113, 1038, 912, 845. 1H-NMR (CDCl3) δ 8.29 (d, J = 9.1 Hz, 1H), 7.77 (s, 1H), 7.72 (s, 1H), 7.47 (d, J = 2.6 Hz, 1H), 7.29 (d, J = 2.6 Hz), 4.14 (s, 3H), 4.09 (s, 3H), 3.96 (s, 3H). 13C-NMR (CDCl3) δ 160.1, 153.8, 150.4, 149.8, 144.5, 130.9, 123.0, 119.0, 118.5, 118.0, 109.0, 107.1, 101.8, 56.4, 56.3, 55.8. ESI-MS m/z: 304.08 ([M + H]+). Anal. Calcd for C16H14ClNO3: C 63.27; H 4.65; N 4.61; O 15.80. Found: C 63.41; H 4.80; N 4.39; O 16.26.
6-chloro-8,9-dimethoxy-1,2,3,4-tetrahydrobenzo[c]phenanthridine (14b) White solid, yield 85%, m.p. 201.3–202.3 °C. FTIR (KBr, cm−1) 3434, 3133, 2928, 1613, 1578, 1523, 1501, 1465, 1402, 1299, 1249, 1206, 1160, 1081, 1043, 953, 840. 1H-NMR (CDCl3) δ 8.13 (d, J = 8.4 Hz, 1H), 7.83 (s, 1H), 7.72 (s, 1H), 7.34 (d, J = 8.4 Hz, 1H), 4.14 (s, 3H), 4.09 (s, 3H), 3.35 (t, J = 6.1 Hz, 2H), 2.96 (t, J = 6.0 Hz, 2H), 1.96-1.89 (m, 4H)). 13C-NMR (CDCl3) δ 153.2, 149.0, 148.5, 141.9, 137.6, 135.3, 130.9, 128.7, 121.5, 119.6, 118.6, 106.9, 102.2, 56.3, 56.3, 30.2, 25.3, 23.1, 23.1. ESI-MS m/z: 328.11 ([M + H]+). Anal. Calcd for C19H18ClNO2: C 69.62; H 5.53; N 4.27; O 9.76. Found: C 69.62; H 5.48; N 4.17; O 9.45.
9-chloro-6,7-dimethoxy-phenanthrene-3-carboxylic acid methyl ester (14c) White solid, yield 83.%, m.p. 194.9–196.6 °C. FTIR (KBr, cm−1) 3444, 3135, 1716, 1615, 1514, 1400, 1254, 1163, 1101, 1037, 849. 1H-NMR (CDCl3) δ 9.08 (d, J = 1.4 Hz, 1H), 8.26 (dd, J = 8.6, 1.7 Hz, 1H), 8.06 (d, J = 8.6 Hz, 1H), 7.91 (s, 1H), 7.74 (s, 1H), 4.19 (s, 3H), 4.10 (s, 3H), 4.03 (s, 3H). 13C-NMR (CDCl3) δ 167.0, 154.1, 152.4, 150.9, 145.5, 130.6, 129.6, 128.5, 128.3, 124.6, 123.4, 120.4, 107.3, 102.5, 56.7, 56.4, 52.7. ESI-MS m/z: 332.07 ([M + H]+). Anal. Calcd for C17H14ClNO4: C 61.55; H 4.25; N 4.22; O 19.29. Found: C 61.76; H 4.72; N 4.02; O 19.77.
3.9. General Procedure for the Synthesis of Compounds 15a–c
To compound 14a (60 mg, 0.20 mmol) was added N,N-dimethylaminoethylamine (696.5 mg, 7.90 mmol) under nitrogen atmosphere, and the reaction mixture was stirred at 105 °C for 6 h. After cooled to room temperature, the solvent was removed under reduced pressure and extracted with CH2Cl2. The organic phase was washed with 5% NaOH and H2O, dried over anhydrous Na2SO4, and then concentrated. The crude product was purified on silica gel column chromatography (DCM/MeOH, v/v = 5:1) to obtain compound 15a. The same method was use for 15b and 15c from 14b and 14c.
N,N-dimethyl-N’-(3,8,9-trimethoxy-phenanthridin-6-yl)-ethane-1,2-diamine (15a) Yellow solid, yield 59.%, m.p. 126.0–128.0 °C. FTIR (KBr, cm−1) 3414, 3138, 1617, 1592, 1400, 1306, 1209, 1171, 1039, 841, 801. 1H-NMR (CDCl3) δ 8.08 (d, J = 8.8 Hz, 1H), 7.67 (d, J = 2.4 Hz, 1H), 7.34 (s, 1H), 7.16 (s, 1H), 6.94 (dd, J = 8.8, 2.3 Hz, 1H), 6.41 (br, 1H), 4.07 (s, 3H), 4.05 (s, 3H), 3.93 (s, 3H), 3.81 (t, J = 6.2 Hz, 2H), 2.74 (t, J = 6.2 Hz, 2H), 2.37 (s, 6H). 13C-NMR (CDCl3) δ 160.2, 153.7, 153.6, 149.8, 129.4, 122.3, 114.4, 112.8, 112.1, 106.5, 104.8, 102.3, 59.4, 57.7, 56.2, 55.6, 45.9, 44.6, 39.8. ESI-MS m/z: 356.19 ([M + H]+). Anal. Calcd for C20H25N3O3: C 67.58; H 7.09; N 11.82; O 13.50. Found: C 67.92; H 7.15; N 11.70; O 13.26.
N’-(8,9-dimethoxy-1,2,3,4-tetrahydro-benzo[c]phenanthridin-6-yl)-N,N-dimethyl-ethane-1,2-diamine (15b) Yellow solid, yield 78.%, m.p. 121.6–123.0 °C. FTIR (KBr, cm−1) 3430, 3134, 2926, 1593, 1530, 1488, 1401, 1262, 1253, 1207, 1037, 837, 784. 1H-NMR (CDCl3) δ 7.98 (d, J = 8.3 Hz, 1H), 7.77 (s, 1H), 7.42 (s, 1H), 7.06 (d, J = 8.3 Hz, 1H), 6.40 (br, 1H), 4.09 (s, 3H), 4.08 (s, 3H), 3.88 (t, J = 5.6 Hz, 2H), 3.21 (t, J = 6.2 Hz, 2H), 2.91 (t, J = 6.2 Hz, 2H), 2.84 (t, J = 6.7 Hz, 2H), 2.42 (s, 6H), 1.96–1.85 (m, 4H). 13C-NMR (CDCl3) δ 152.0, 151.9, 149.1, 141.8, 136.8, 132.5, 129.8, 123.9, 118.5, 117.9, 113.1, 103.8, 103.0, 58.7, 56.5, 56.0, 45.4, 39.2, 30.3, 25.4, 23.6, 23.4. ESI-MS m/z: 380.22 ([M + H]+). Anal. Calcd for C23H29N3O2: C 72.79; H 7.70; N 11.07; O 8.43. Found: C 72.24; H 7.85; N 10.83; O 8.91.
6-(2-dimethylamino-ethylamino)-8,9-dimethoxy-phenanthridine-2-carboxylic acid (15c) Yellow solid, yield 69%, m.p. 192.4–193.5 °C. FTIR (KBr, cm−1) 3451, 3142, 1687, 1619, 1590, 1511, 1398, 1262, 1209, 1115, 1033, 843. 1H-NMR (CDCl3) δ 8.95 (s, 1H), 8.13-8.11 (m, 1H), 7.89 (s, 1H), 7.73 (d, J = 8.3, Hz, 1H), 7.23 (s, 1H), 6.24 (br, 1H), 4.14 (s, 3H), 4.06 (s, 3H), 3.83-3.79 (m, 2H), 2.70 (t, J = 5.3 Hz, 2H), 2.34 (s, 6H). 13C-NMR (CDCl3) δ 167.4, 154.4, 152.4, 149.9, 148.1, 129.4, 128.4, 126.8, 124.4, 123.6, 120.1, 113.8, 103.4, 103.3, 58.2, 56.4, 56.4, 45.4, 38.9, 29.8. ESI-MS m/z: 384.17([M + H]+). Anal. Calcd for C21H25N3O4: C 65.78; H 6.57; N 10.96; O 16.69. Found: C 65.59; H 6.71; N 10.95; O 16.45.
3.10. General Procedure for the Synthesis of Compounds 16a–b
CH3I (158.2 mg, 1.11 mmol) was added to the suspension of compound 7a (100 mg, 0.37 mmol) in dry toluene, and the mixture was stirred for 16 h at 80 °C. After cooled to room temperature, the solution was filtrated and washed with toluene and ether to obtain compound 16a. The same method was used for 16b from compound 7c.
3,8,9-trimethoxy-5-methyl-phenanthridinium iodide (16a) Yellow solid, yield 84%, m.p. 245.2–247.2 °C. FTIR (KBr, cm−1) 3423, 3131, 1624, 1501, 1402, 1294, 1232, 1172, 1033, 836. 1H-NMR (DMSO-d6) δ 9.81 (s, 1H), 9.09 (d, J = 9.2 Hz, 1H), 8.30 (s, 1H), 7.82 (s, 1H), 7.71–7.66 (m, 2H), 4.55 (s, 3H), 4.19 (s, 3H), 4.09 (s, 3H), 4.00 (s, 3H). 13C-NMR (DMSO-d6) δ 161.8, 158.8, 151.5, 151.0, 135.9, 132.9, 127.0, 120.0, 119.3, 118.5, 109.9, 103.2, 101.0, 57.7, 56.9, 56.7, 45.8. ESI-MS m/z: 284.14 ([M + H]+). Anal. Calcd for C17H18NO3: C 49.65; H 4.41; N 3.41; O 11.67. Found: C 49.52; H 4.32; N 3.49; O 12.17.
2-fluoro-8,9-dimethoxy-5-methyl-phenanthridinium iodide (16b) Yellow solid, yield 75%, m.p. 246.7–247.7 °C.FTIR (KBr, cm−1) 3438, 3120, 3016, 1606, 1516, 1474, 1438, 1400, 1279, 1205, 1131, 1036, 988, 876. 1H-NMR (DMSO-d6) δ 9.89 (s, 1H), 9.08 (d, J = 10.3 Hz, 1H), 8.55–8.52 (m, 1H), 8.40 (s, 1H), 8.03–7.99 (m, 1H), 7.92 (s, 1H), 4.59 (s, 3H), 4.20 (s, 3H), 4.03 (s, 3H). 13C-NMR (DMSO-d6) δ 162.1 (d, J = 249.6 Hz), 158.6, 152.0, 151.9, 131.7 (d, J = 3.8 Hz), 130.0, 127.2 (d, J = 9.9 Hz), 123.3 (d, J = 9.6 Hz), 120.3 (d, J = 25.1 Hz), 119.6, 110.7, 110.4 (d, J = 25.0 Hz), 104.7, 58.0, 56.8, 46.1. ESI-MS m/z: 272.11 ([M + H]+). Anal. Calcd for C16H15FINO2: C 48.14; H 3.79; N 3.51; O 8.02. Found: C48.30; H 3.72; N 3.62; O 8.43.
3.11. Cytotoxicity Assay In Vitro
All cell lines used in this study, including HepG2, A549, NCI-H460, and CNE1, were purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China) and cultured at 37 °C in a humidified atmosphere containing 5% CO2. HepG2, A549 and CNE1 cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% (v/v) FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. NCI-H460 cell line was cultured in ATCC Modified 1640 Medium supplemented with 10% (v/v) FBS, 100 U/mL penicillin and 100 μg/mL streptomycin.
The antitumor activity of new compounds was evaluated by MTT assay in vitro. 5 × 10
3 cells in 100 μL medium were plated to each well of a 96-well flat-bottom microtiter plate and cultured overnight. Then the cells were treated with serial diluted candidate compounds. The control was treated with equivalent DMSO. After 48 h, 10 μL MTT stock (5 mg/mL) was added to each well and incubated for 2 h. Then, the supernatants were discarded and 100 μL DMSO was added to each well. After 10 minutes’ shaking, the optical density at the wavelengths of 490 nm (OD
490) was measured on a SPARK microplate reader (Tecan, Männedorf, Switzerland). All samples were repeated 3 times and each time tested in triplicate. The cell inhibition rate of each sample was calculated by the following formula. The IC
50 values of the compounds were calculated using SPSS 17.0 software.







{kind=link}
{kind=link}
{kind=link}
{kind=link}