12N-Substituted Matrinol Derivatives Inhibited the Expression of Fibrogenic Genes via Repressing Integrin/FAK/PI3K/Akt Pathway in Hepatic Stellate Cells

Abstract
1. Introduction
2. Results and Discussion
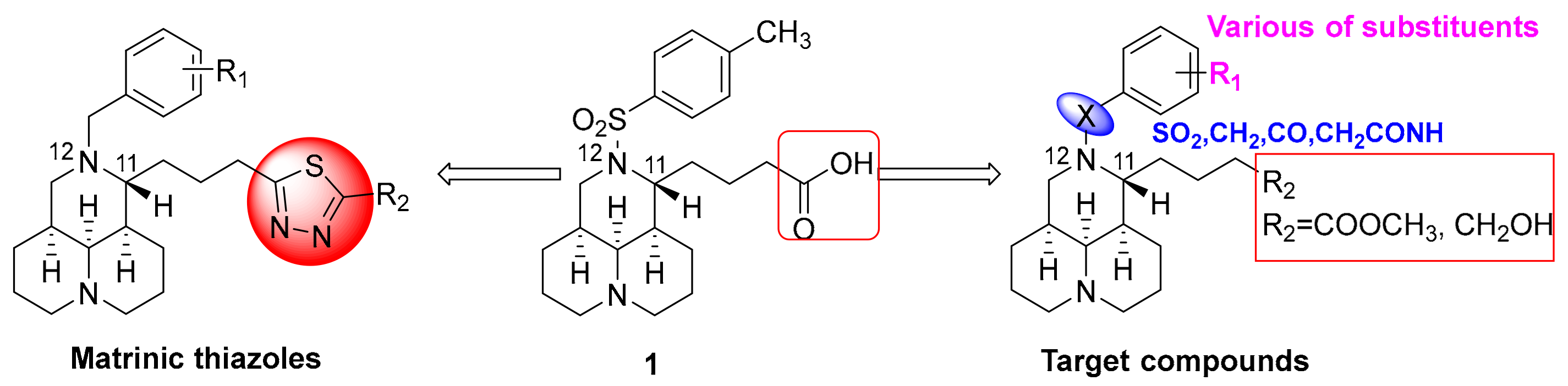
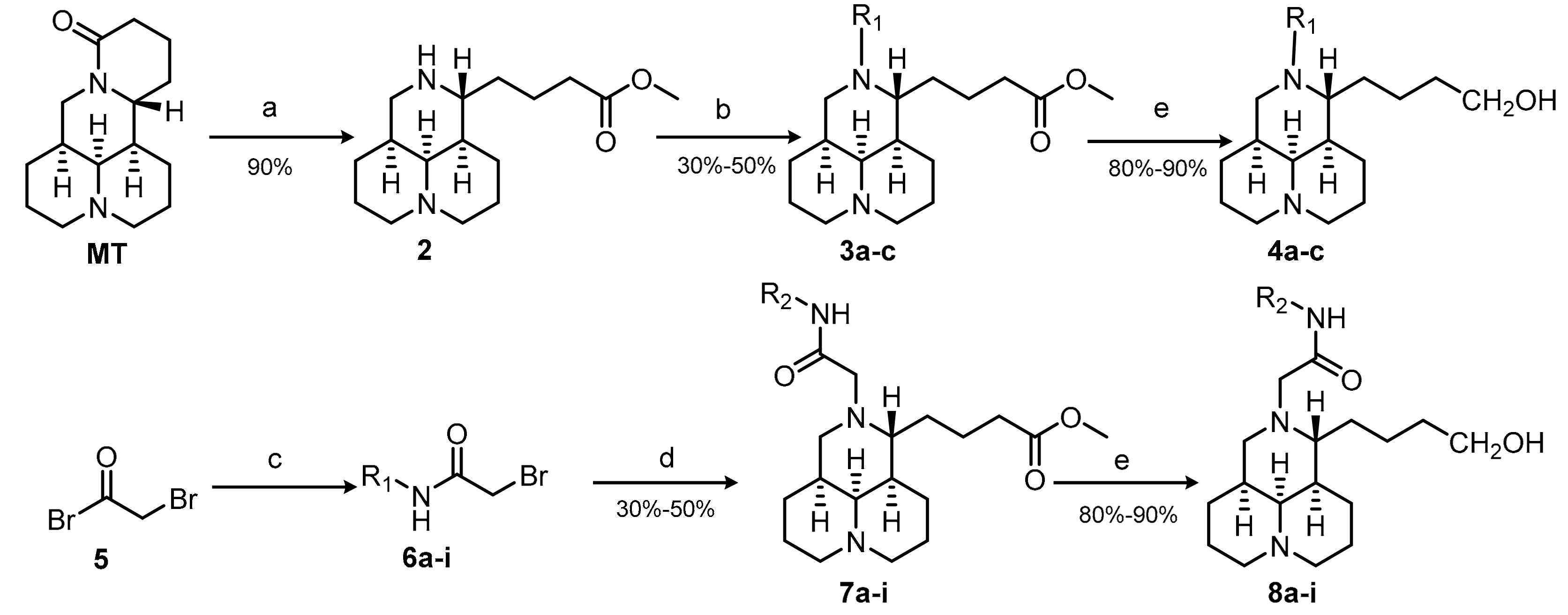
2.1. Chemistry
2.2. Target Compounds Inhibited the Expresion of COL1A1 Promotor in Human Hepatic Stellate LX-2 Cells
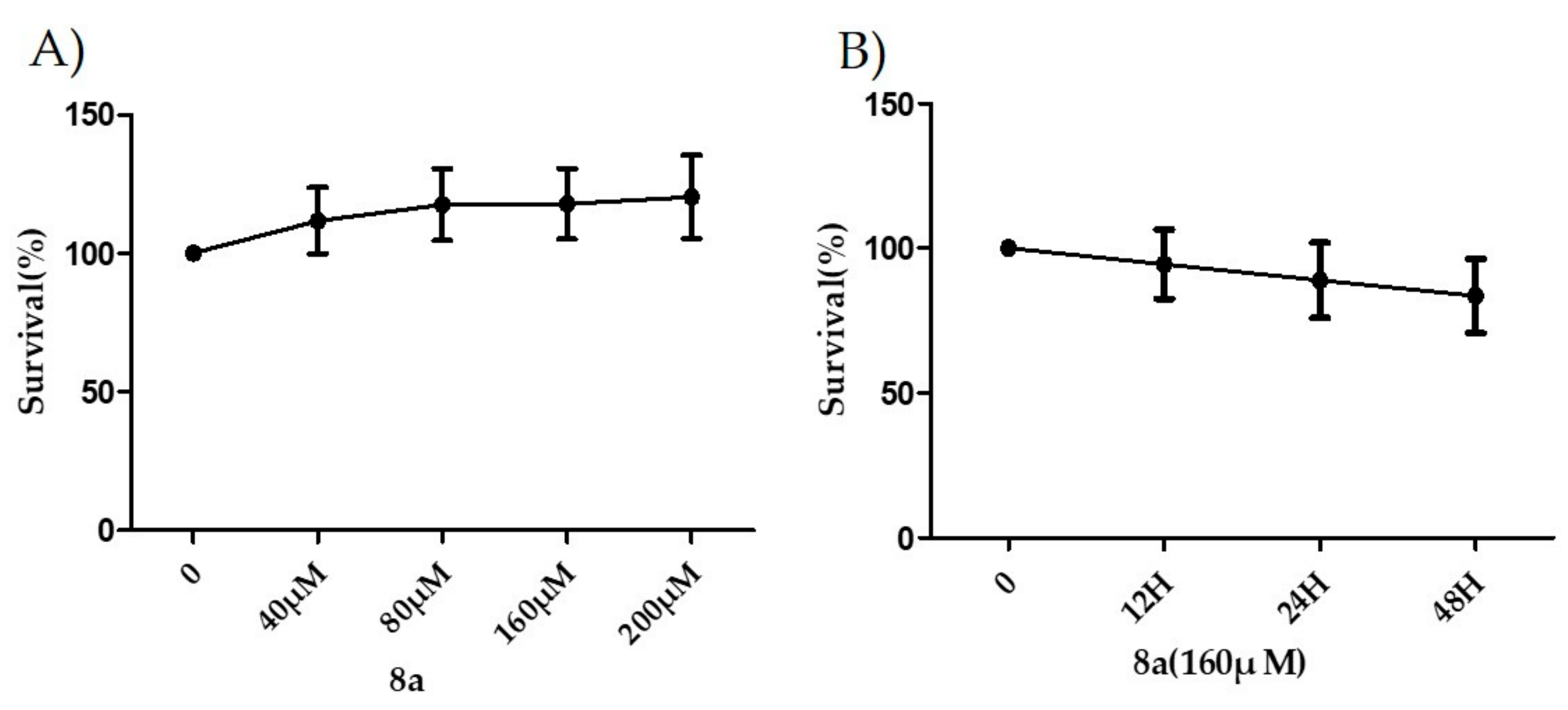
2.3. 8a Held a Good Safe Profile in LX-2 Cells
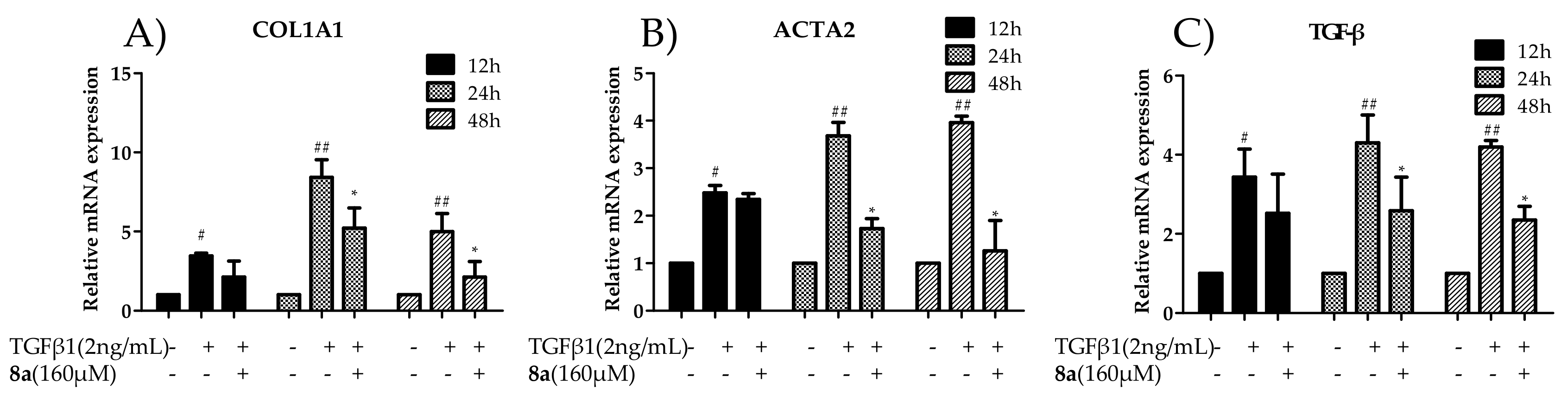
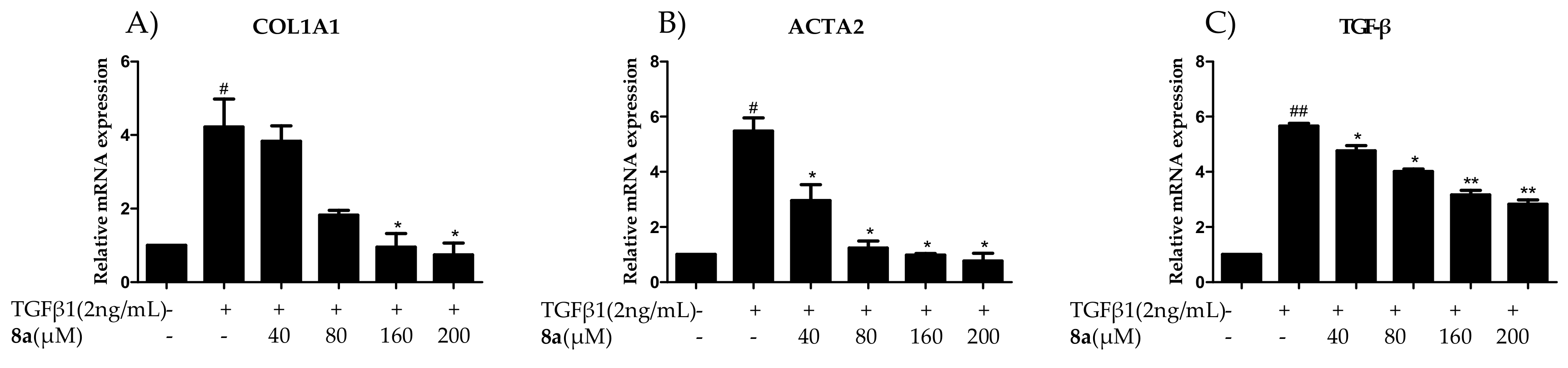
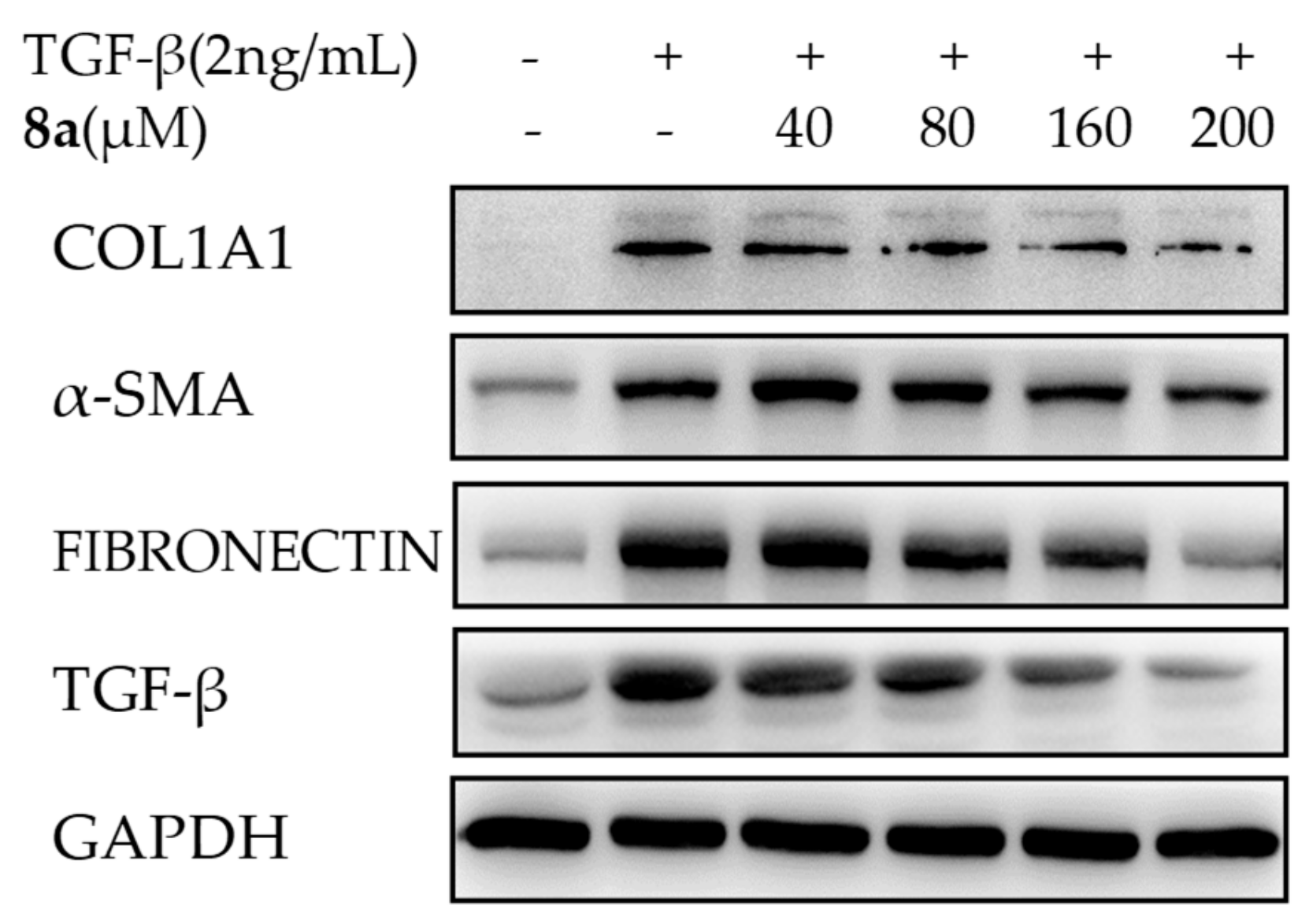
2.4. 8a Inhibited the Expressions of COL1A1 on mRNA and Protein Levels in Dose- and Time-dependent Manners
2.5. 8a Inhibited the Expressions of α-SMA on mRNA and Protein Levels in Time- and Dose-dependent Manner
2.6. 8a Inhibited the Expressions of Fibronectin on Protein Level in a Dose-Dependent Manner
2.7. 8a Inhibited the Expressions of TGFβ on mRNA and Protein Levels in Time and Dose-Dependent Manner
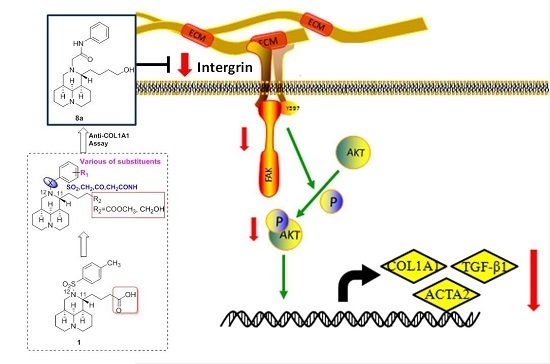
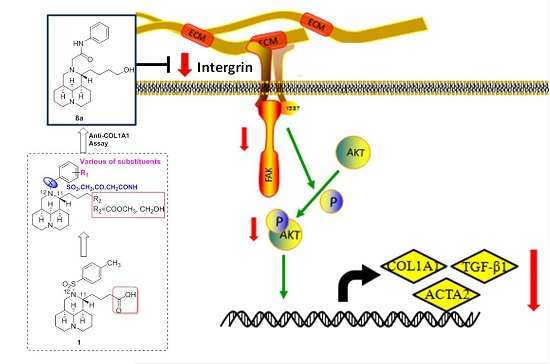
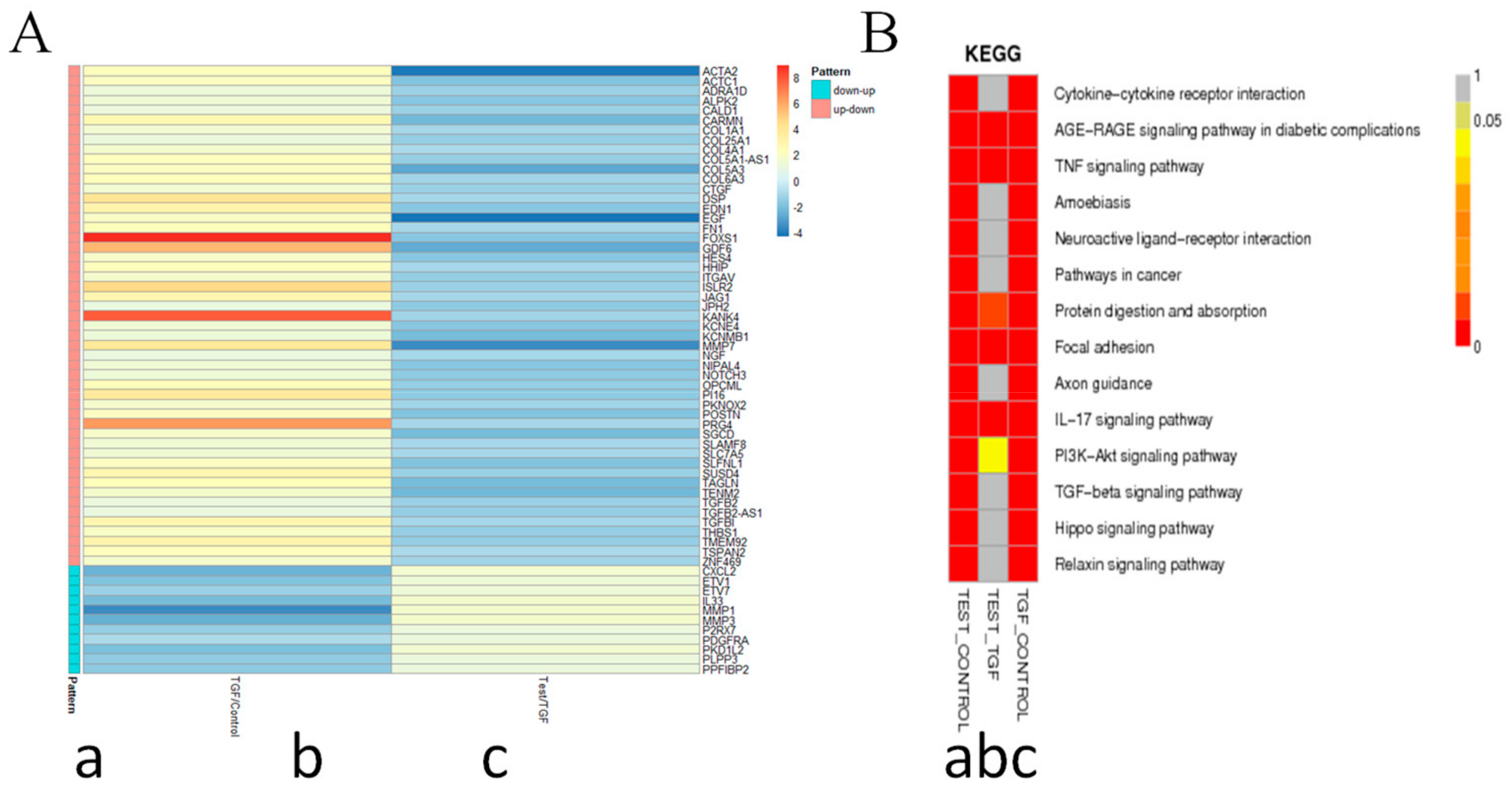
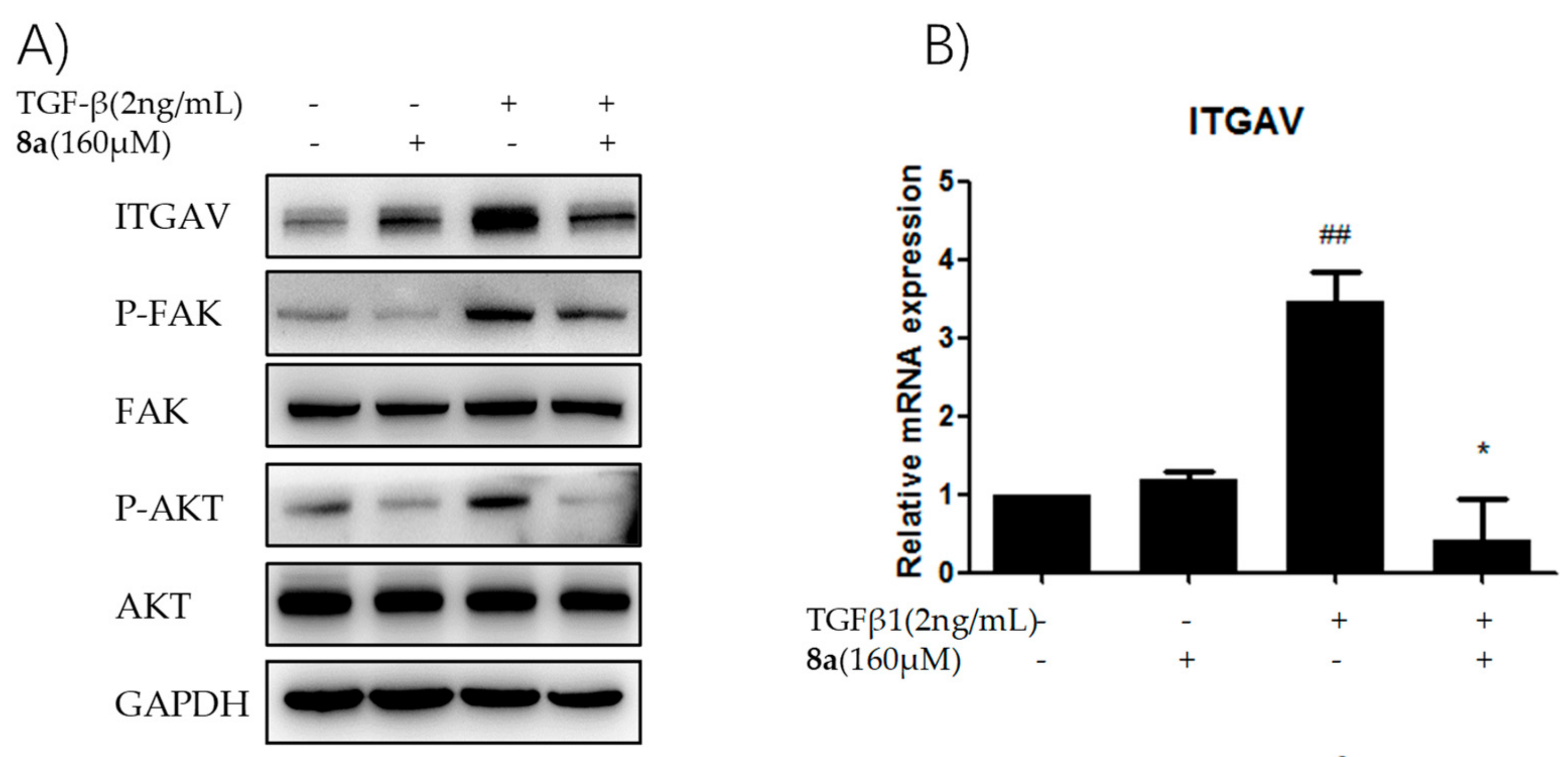
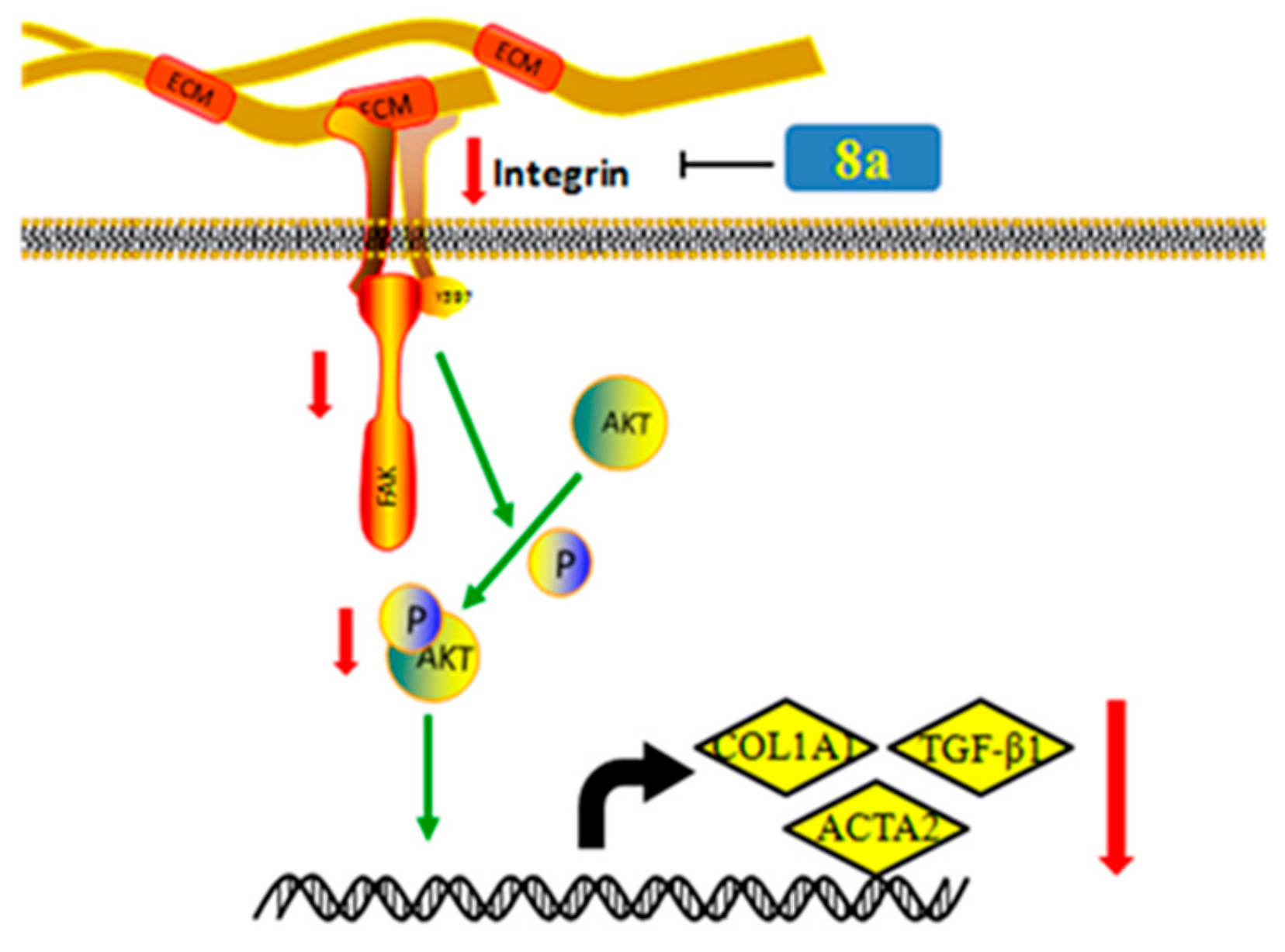
2.8. 8a Supressed the Intergrin-Focal Adhesion Pathway
3. Experimental Section
3.1. Apparatus, Materials, and Analysis Reagents
3.2. Chemistry
3.2.1. Synthesis of 12N-Benzoyl Matrinic Methyl Butyrate Hydrochloride (3b)
3.2.2. Synthesis of 12N-Benzenesulfonyl Matrinol Hydrochloride (4c)
3.2.3. General Procedure for the Synthesis of Compounds 7a–i
3.2.4. General Procedure for the Synthesis of Compounds 8a–i
3.3. Biology Assay
3.3.1. Cell Culture and Screening of Compounds
3.3.2. Sulforhodamine B (SRB) Assay
3.3.3. RT-qPCR Assay
3.3.4. Western Blot
3.3.5. Total mRNA Extraction
3.3.6. Genome Sequencing and Annotation
3.3.7. Gene Network/Pathway Analysis
3.3.8. Statistics
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campana, L.; Iredale, J.P. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar] [PubMed]
- Elpek, G.O. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef] [PubMed]
- Zoubek, M.E.; Trautwein, C.; Strnad, P. Reversal of liver fibrosis: From fiction to reality. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.; Lawlor, D.A.; Fraser, A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef]
- Harris, R.; Harman, D.J.; Card, T.R.; Aithal, G.P.; Guha, I.N. Prevalence of clinically significant liver disease within the general population, as defined by non-invasive markers of liver fibrosis: A systematic review. Lancet Gastroenterol. Hepatol. 2017, 2, 288–297. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Sutariya, B.; Jhonsa, D.; Saraf, M.N. TGF-β: The connecting link between nephropathy and fibrosis. Immunopharmacol. Immunotoxicol. 2016, 38, 39–49. [Google Scholar] [CrossRef]
- Altrock, E.; Sens, C.; Wuerfel, C.; Vasel, M.; Kawelke, N.; Dooley, S.; Sottile, J.; Nakchbandi, I.A. Inhibition of fibronectin deposition improves experimental liver fibrosis. J. Hepatol. 2015, 62, 625–633. [Google Scholar] [CrossRef]
- Abd-Elhakim, Y.M.; Moustafa, G.G.; Hashem, M.M.; Ali, H.A.; Abo-El-Sooud, K.; El-Metwally, A.E. Influence of the long-term exposure to tartrazine and chlorophyll on the fibrogenic signalling pathway in liver and kidney of rats: The expression patterns of collagen 1-α, TGFβ-1, fibronectin, and caspase-3 genes. Environ. Sci. Pollut. Res. Int. 2019, 26, 12368–12378. [Google Scholar] [CrossRef]
- Buck, M.; Kim, D.J.; Houglum, K.; Hassanein, T.; Chojkier, M. c-Myb modulates transcription of the α-smooth muscle actin gene in activated hepatic stellate cells. Am. J. Physiol. Gastrointest Liver Physiol. 2000, 278, G321. [Google Scholar] [CrossRef]
- Christner, P.J.; Ayitey, S. Extracellular matrix containing mutated fibrillin-1 (Fbn1) down regulates Col1a1, Col1a2, Col3a1, Col5a1, and Col5a2 mRNA levels in Tsk/+ and Tsk/Tsk embryonic fibroblasts. Amino Acid. 2006, 30, 445. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.S.; Wang, J.X.; Wang, Y.C.; Shao, R.G.; He, H.W. Establishment and application of a high-throughput drug screening model based on COL1A1 promoter for anti-liver fibrosis. Acta Pharm. Sin. 2015, 50, 169–173. [Google Scholar]
- Niu, T.; Niu, W.; Bao, Y.; Liu, T.; Song, D.; Li, Y.; He, H. Discovery of Matrinic Thiadiazole Derivatives as a Novel Family of Anti-Liver Fibrosis Agents via Repression of the TGFβ/Smad Pathway. Molecules 2018, 23, 1644. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Liu, H.; Zhang, Y.; Li, N.; Zhao, S.; Zhao, W.; Zhen, Y.; Yu, J.; He, H.; Shao, R.G. The anti-hepatic fibrosis effects of dihydrotanshinone I are mediated by disrupting the yes-associated protein and transcriptional enhancer factor D2 complex and stimulating autophagy. Br. J. Pharmacol. 2017, 174, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Li, Y.; Bao, Y.; Dai, Z.; Niu, T.; Wang, K.; He, H.; Song, D. Novel cytisine derivatives exert anti-liver fibrosis effect via PI3K/Akt/Smad pathway. Bioorg. Chem. 2019, 90, 103032. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.W.; Sun, G. Advances in research on anti-fibrosis mechanism of matrine components. Food Drug 2006, 8, 14–16. [Google Scholar]
- Huang, G.C. Clinical observation on the effect of Matrine on hepatic fibrosis. Pract. Chin. West. Med. Comb. Clin. 2005, 5, 17. [Google Scholar]
- Yu, D.K.; Zhang, C.X.; Zhao, S.S.; Zhang, S.H.; Zhang, H.; Cai, S.Y.; Shao, R.G.; He, H.W. The anti-fibrotic effects of epigallocatechin-3-gallate in bile duct-ligated cholestatic rats and human hepatic stellate LX-2 cells are mediated by the PI3K/Akt/Smad pathway. Acta Pharmacol. Sin. 2015, 36, 473–482. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Tamaki, Z.; Wang, W.; Hinchcliff, M.; Hoover, P.; Getsios, S.; White, E.S.; Varga, J. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci. Transl. Med. 2014, 6, 232ra50. [Google Scholar] [CrossRef]
- Iredale, J.P. Hepatic stellate cell behavior during resolution of liver injury. Semin. Liver Dis. 2001, 21, 427–436. [Google Scholar] [CrossRef]
- Zhao, X.K.; Yu, L.; Cheng, M.L.; Che, P.; Lu, Y.Y.; Zhang, Q.; Mu, M.; Li, H.; Zhu, L.L.; Zhu, J.J.; et al. Focal Adhesion Kinase Regulates Hepatic Stellate Cell Activation and Liver Fibrosis. Sci. Rep. 2017, 7, 4032. [Google Scholar] [CrossRef] [PubMed]
- Vitillo, L.; Kimber, S.J. Integrin and FAK Regulation of Human Pluripotent Stem Cells. Curr. Stem Cell Rep. 2017, 3, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Guan, J.L.; Shalloway, D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature 1992, 358, 690–692. [Google Scholar] [CrossRef]
- Zhang, B.C.; Sun, Z.Q.; Lv, M.; Xu, H. Semi-synthesis of matrinic acid/alcohol/ester derivatives, their pesticidal activities, and investigation of mechanisms of action against tetranychus cinnabarinus. Agric. Food Chem. 2018, 66, 12898–12910. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3a–c, 4a–c, 7a–i and 8a–i are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Code | X | R1 | R2 | Inhibition Rate a |
|---|---|---|---|---|
| 1 | - | - | - | 18.5% ± 1.1% |
| 3a | CH2 | H | COOCH3 | 24.7% ± 10.0% |
| 3b | CO | H | COOCH3 | 13.9% ± 8.3% |
| 3c | SO2 | H | COOCH3 | 30.6% ± 18.1% |
| 4a | CH2 | H | CH2OH | 31.5% ± 7.5% |
| 4b | CO | H | CH2OH | 21.6% ± 5.2% |
| 4c | SO2 | H | CH2OH | 13.9% ± 2.0% |
| 7a | NHCOCH2 | H | COOCH3 | 39.5% ± 7.1% |
| 7b | CH2NHCOCH2 | H | COOCH3 | 22.7% ± 8.5% |
| 7c | NHCOCH2 | p-F | COOCH3 | 45.2% ± 3.6% |
| 7d | NHCOCH2 | p-Cl | COOCH3 | 45.9% ± 2.2% |
| 7e | NHCOCH2 | p-Br | COOCH3 | 51.9% ± 3.1% |
| 7f | NHCOCH2 | p-CH3 | COOCH3 | 47.0% ± 9.8% |
| 7g | NHCOCH2 | p-CF3 | COOCH3 | 43.4% ± 11.2% |
| 7h | NHCOCH2 | p-NO2 | COOCH3 | 29.8% ± 9.2% |
| 7i | NHCOCH2 | 2,6-di-CH3 | COOCH3 | 24.0% ± 6.9% |
| 8a | NHCOCH2 | H | CH2OH | 51.0% ± 2.8% |
| 8b | CH2NHCOCH2 | H | CH2OH | 40.4% ± 5.2% |
| 8c | NHCOCH2 | p-F | CH2OH | 43.6% ± 1.8% |
| 8d | NHCOCH2 | p-Cl | CH2OH | 42.0% ± 19.2% |
| 8e | NHCOCH2 | p-Br | CH2OH | 50.6% ± 17.1% |
| 8f | NHCOCH2 | p-CH3 | CH2OH | 25.6% ± 5.6% |
| 8g | NHCOCH2 | p-CF3 | CH2OH | 32.3% ± 32.3% |
| 8h | NHCOCH2 | p-NO2 | C H2OH | 22.5% ± 13.0% |
| 8i | NHCOCH2 | 2,6-di-CH3 | CH2OH | 6.0% ± 14.5% |
| EGCG b | - | - | - | 27.5% ± 7.9% |
| DMSO | - | - | - | 2.9% ± 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, Y.; Pang, Y.; Tang, S.; Niu, T.; Guo, Z.; He, H.; Li, Y.; Song, D. 12N-Substituted Matrinol Derivatives Inhibited the Expression of Fibrogenic Genes via Repressing Integrin/FAK/PI3K/Akt Pathway in Hepatic Stellate Cells. Molecules 2019, 24, 3748. https://doi.org/10.3390/molecules24203748
Bao Y, Pang Y, Tang S, Niu T, Guo Z, He H, Li Y, Song D. 12N-Substituted Matrinol Derivatives Inhibited the Expression of Fibrogenic Genes via Repressing Integrin/FAK/PI3K/Akt Pathway in Hepatic Stellate Cells. Molecules. 2019; 24(20):3748. https://doi.org/10.3390/molecules24203748
Chicago/Turabian StyleBao, Yunyang, Yudong Pang, Sheng Tang, Tianyun Niu, Zhihao Guo, Hongwei He, Yinghong Li, and Danqing Song. 2019. "12N-Substituted Matrinol Derivatives Inhibited the Expression of Fibrogenic Genes via Repressing Integrin/FAK/PI3K/Akt Pathway in Hepatic Stellate Cells" Molecules 24, no. 20: 3748. https://doi.org/10.3390/molecules24203748
APA StyleBao, Y., Pang, Y., Tang, S., Niu, T., Guo, Z., He, H., Li, Y., & Song, D. (2019). 12N-Substituted Matrinol Derivatives Inhibited the Expression of Fibrogenic Genes via Repressing Integrin/FAK/PI3K/Akt Pathway in Hepatic Stellate Cells. Molecules, 24(20), 3748. https://doi.org/10.3390/molecules24203748