
Pressure-Induced Polymorphism of Caprolactam: A Neutron Diffraction Study

 ,
, 

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
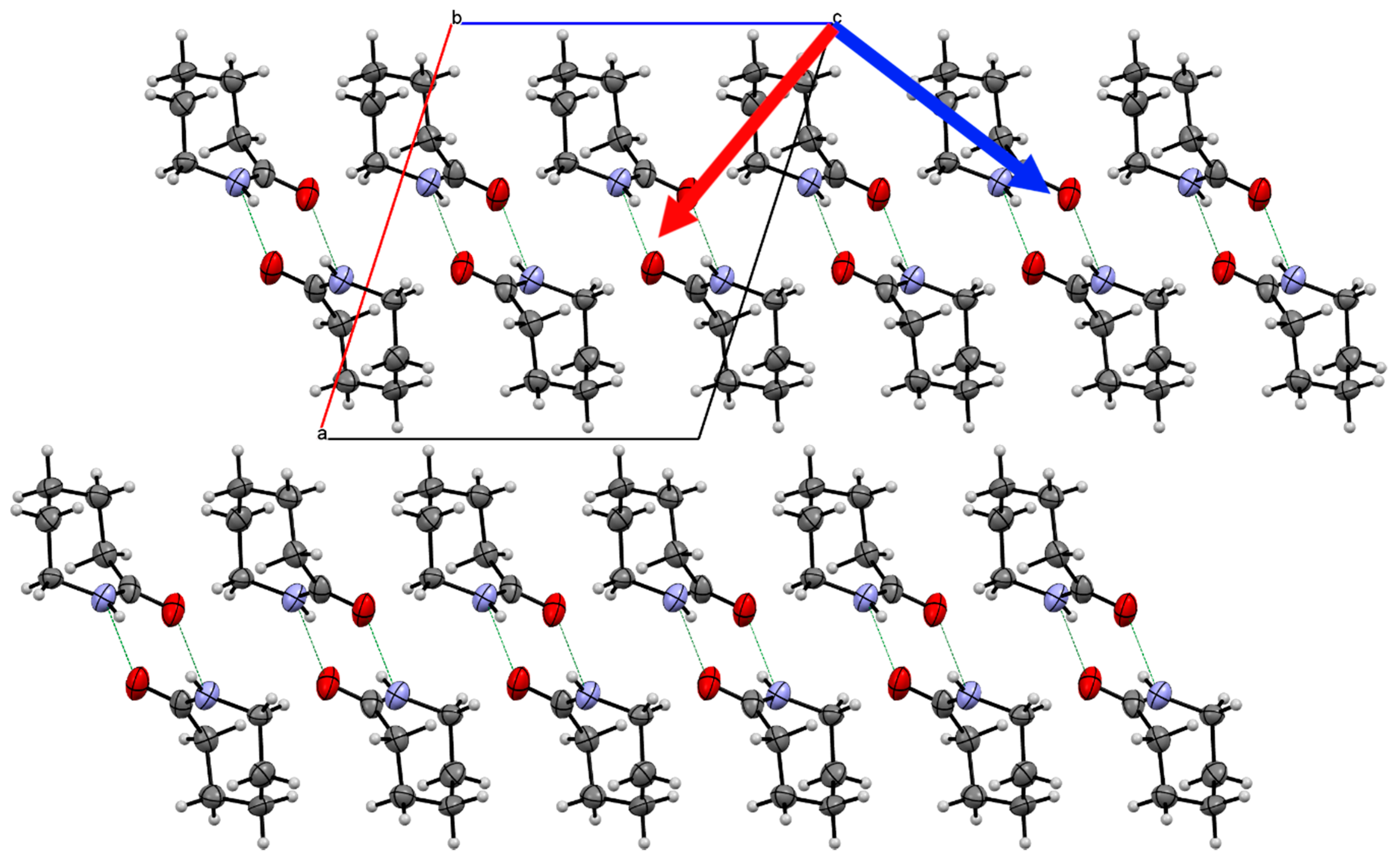
2.1. Caprolactam Form I Crystal Structure
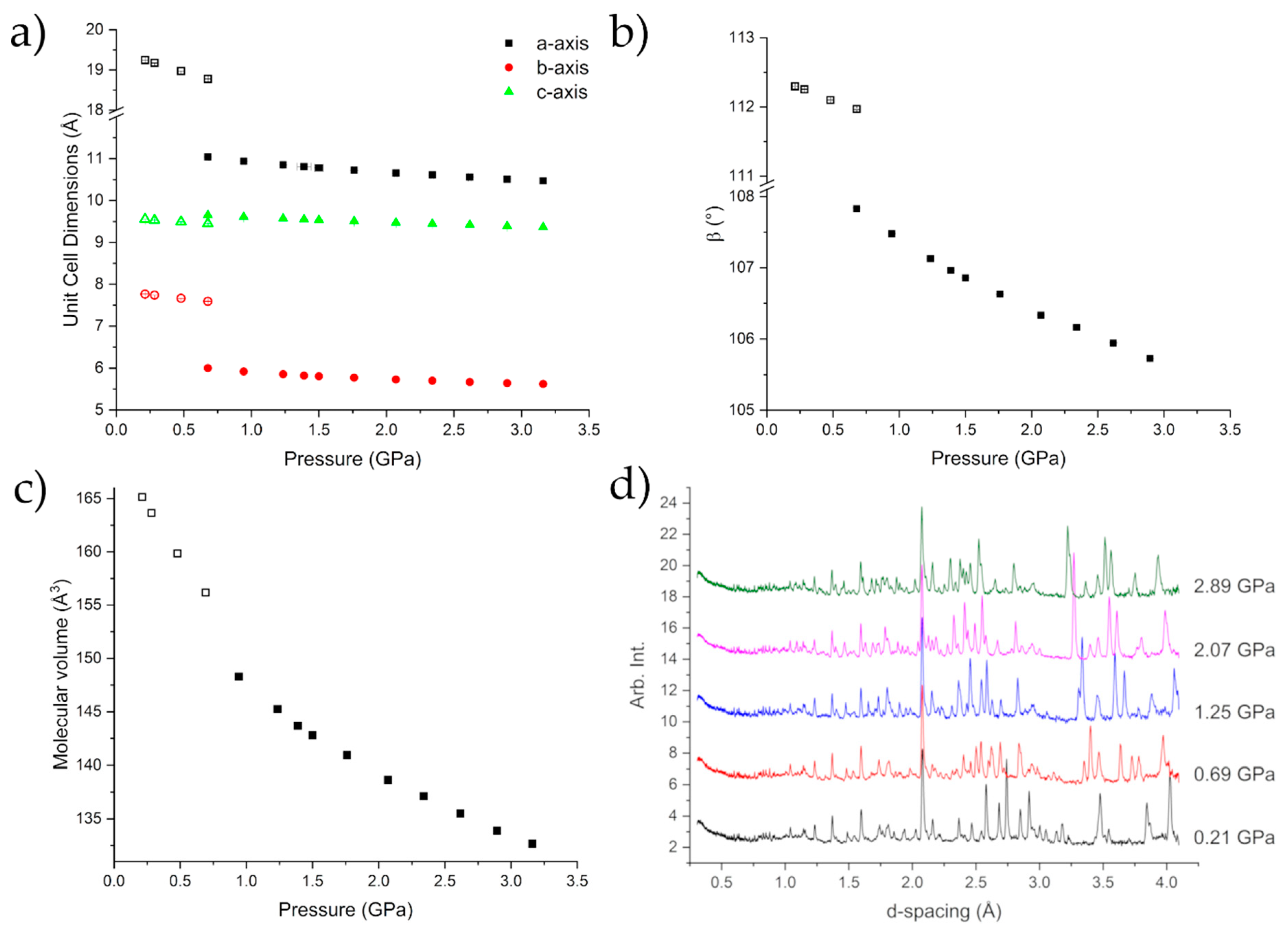
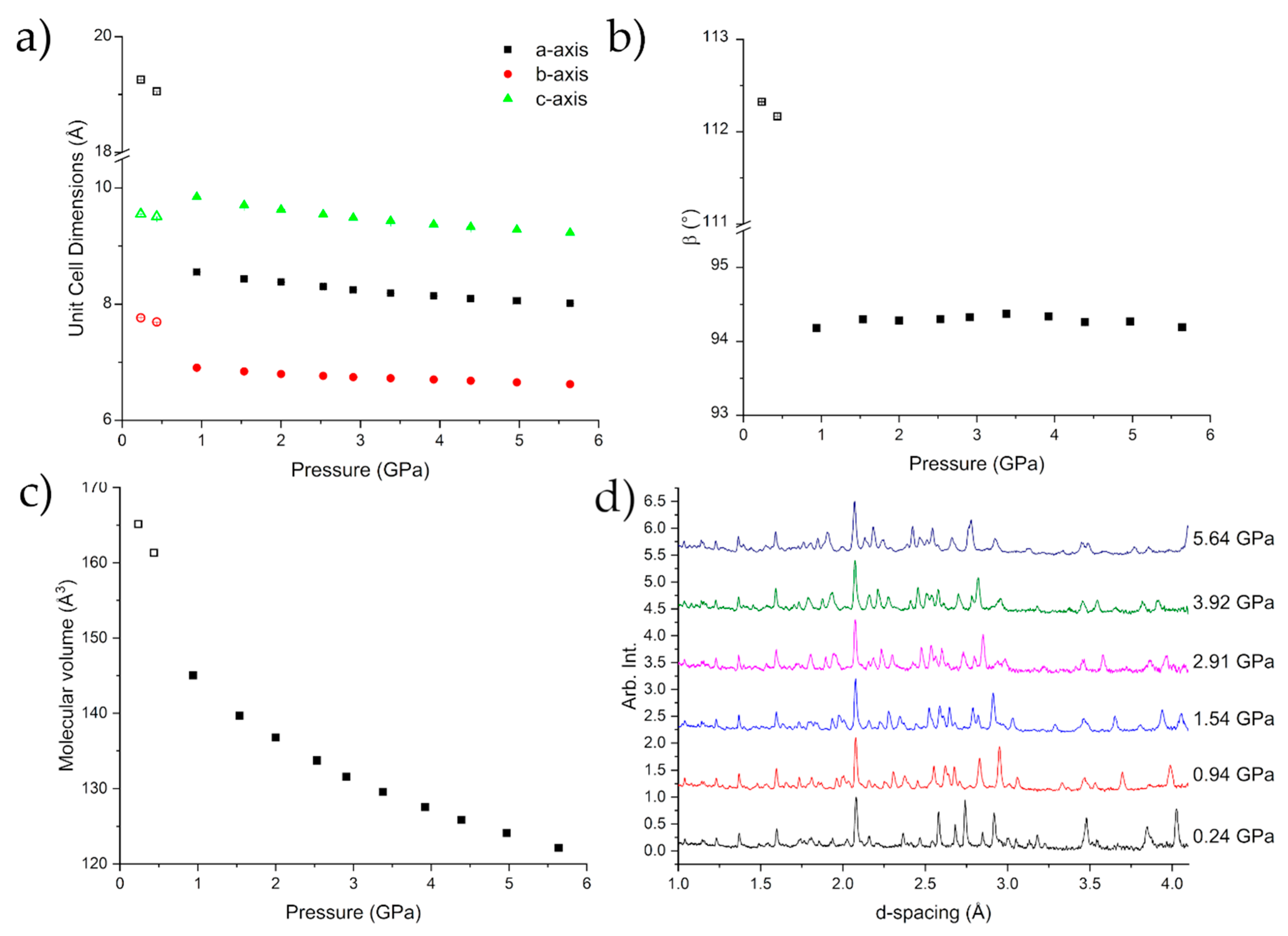
2.2. Diamond Anvil Cell Experiments
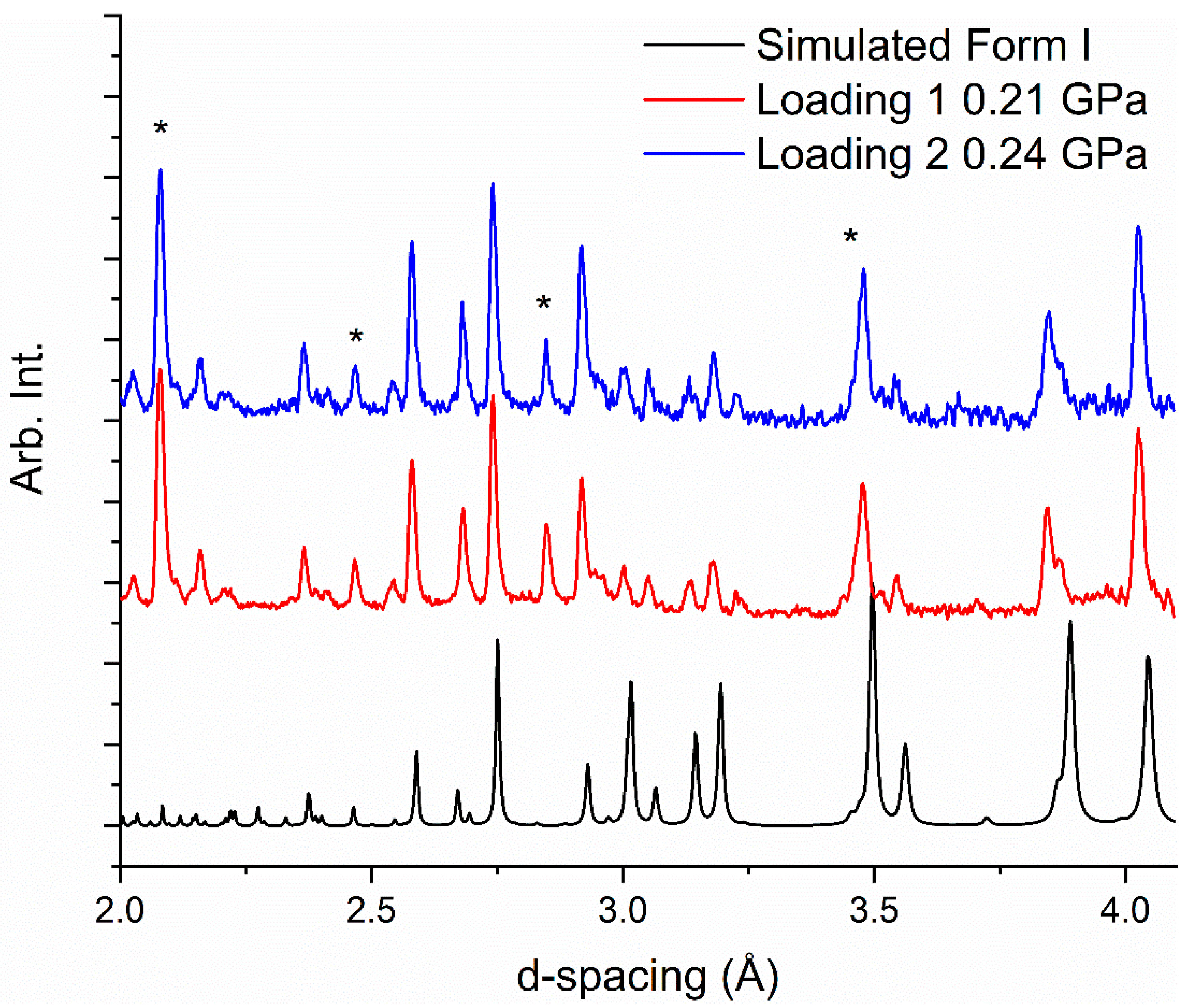
2.3. Neutron Powder Diffraction Studies
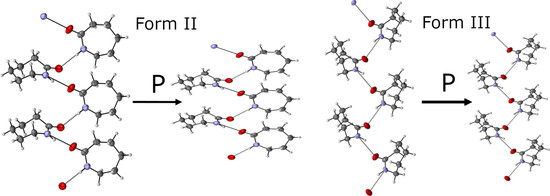
2.4. Loading 1—Neutron Diffraction Study of Caprolactam-d10 Recrystallized from Ethyl Acetate
2.5. Loading 2—Neutron Diffraction Study of Caprolactam Recrystallized from Ethanol
3. Discussion
Crystal Structure Comparison
4. Materials and Methods
4.1. Lab-Based Experiments
4.2. X-ray Diffraction Experiments
4.3. Neutron Diffraction Experiments
4.4. Geometry Optimisation
4.5. Crystal Explorer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murthy, N.S. Hydrogen bonding, mobility, and structural transitions in aliphatic polyamides. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 1763–1782. [Google Scholar] [CrossRef]
- Rahman, M.A.; Renna, L.A.; Venkataraman, D.; Desbois, P.; Lesser, A.J. High crystalline, porous polyamide 6 by anionic polymerization. Polymer 2018, 138, 8–16. [Google Scholar] [CrossRef]
- Fernandes, M.A.; Levendis, D.C. Photodimerisation of the α′-polymorph of ortho-ethoxy-trans-cinnamic acid occurs via a two-stage mechanism at 343 K yielding 100% α-truxillic acid. CrystEngComm 2016, 18, 7363–7376. [Google Scholar] [CrossRef]
- Sinnwell, M.A.; Blad, J.N.; Thomas, L.R.; MacGillivray, L.R. Structural flexibility of halogen bonds showed in a single-crystal-to-single-crystal [2 + 2] photodimerization. IUCrJ 2018, 5, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Murli, C.; Song, Y. Pressure-Induced Polymerization of Acrylic Acid: A Raman Spectroscopic Study. J. Phys. Chem. B 2010, 114, 9744–9750. [Google Scholar] [CrossRef] [PubMed]
- Murli, C.; Mishra, A.K.; Thomas, S.; Sharma, S.M. Ring-Opening Polymerization in Carnosine under Pressure. J. Phys. Chem. B 2012, 116, 4671–4676. [Google Scholar] [CrossRef] [PubMed]
- Chelazzi, D.; Ceppatelli, M.; Santoro, M.; Bini, R.; Schettino, V. Pressure-Induced Polymerization in Solid Ethylene. J. Phys. Chem. B 2005, 109, 21658–21663. [Google Scholar] [CrossRef] [PubMed]
- Ciabini, L.; Gorelli, F.A.; Santoro, M.; Bini, R.; Schettino, V.; Mezouar, M. High-pressure and high-temperature equation of state and phase diagram of solid benzene. Phys. Rev. B 2005, 72, 094108. [Google Scholar] [CrossRef]
- Santoro, M.; Ciabini, L.; Bini, R.; Schettino, V. High-Pressure Polymerization of Phenylacetylene and of the Benzene and Acetylene Moieties. J. Raman Spectrosc. 2003, 34, 557–566. [Google Scholar] [CrossRef]
- Ciabini, L.; Santoro, M.; Bini, R.; Schettino, V. High Pressure Reactivity of Solid Benzene Probed by Infrared Spectroscopy. J. Chem. Phys. 2002, 116, 2928–2935. [Google Scholar] [CrossRef]
- Guan, J.; Daljeet, R.; Kieran, A.; Song, Y. Pressure-induced amorphization and reactivity of solid dimethyl acetylene probed by in situ FTIR and Raman spectroscopy. J. Phys. Condens. Matter 2018, 30, 224004. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Dong, X.; Wang, Y.; Li, K.; Zheng, H.; Wang, L.; Cody, G.D.; Tulk, C.A.; Molaison, J.J.; Lin, X.; et al. Pressure-Induced Polymerization of Acetylene: Structure-Directed Stereoselectivity and a Possible Route to Graphane. Angew. Chem. Int. Ed. 2017, 56, 6553–6557. [Google Scholar] [CrossRef] [PubMed]
- Delori, A.; Hutchison, I.B.; Bull, C.L.; Funnell, N.P.; Urquhart, A.J.; Oswald, I.D.H. Reaction of Acetylenedicarboxylic Acid Made Easy: High-Pressure Route for Polymerization. Cryst. Growth Des. 2018, 18, 1425–1431. [Google Scholar] [CrossRef]
- Bini, R.; Ceppatelli, M.; Citroni, M.; Schettino, V. From Simple to Complex and Backwards. Chemical Reactions under Very High Pressure. Chem. Phys. 2012, 398, 262–268. [Google Scholar] [CrossRef]
- Dziubek, K.F.; Ende, M.; Scelta, D.; Bini, R.; Mezouar, M.; Garbarino, G.; Miletich, R. Crystalline polymeric carbon dioxide stable at megabar pressures. Nat. Commun. 2018, 9, 3148. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, C.; Boyd, S.A.; Chawda, S.; Fowler, F.W.; Goroff, N.S.; Halada, G.P.; Grey, C.P.; Lauher, J.W.; Luo, L.; Martin, C.D.; et al. Pressure-Induced Polymerization of Diiodobutadiyne in Assembled Cocrystals. J. Am. Chem. Soc. 2008, 130, 4415–4420. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Plonka, A.M.; Parise, J.B.; Goroff, N.S. Pressure Induced Topochemical Polymerization of Diiodobutadiyne: A Single-Crystal-to-Single-Crystal Transformation. CrystEngComm 2013, 15, 3106–3110. [Google Scholar] [CrossRef]
- Langer, R.; Vacanti, J.P. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, I.B.; Delori, A.; Wang, X.; Kamenev, K.V.; Urquhart, A.J.; Oswald, I.D.H. Polymorphism of a polymer precursor: Metastable glycolide polymorph recovered via large scale high-pressure experiments. CrystEngComm 2015, 17, 1778–1782. [Google Scholar] [CrossRef]
- Hutchison, I.B.; Bull, C.L.; Marshall, W.G.; Parsons, S.; Urquhart, A.J.; Oswald, I.D.H. Compression of glycolide-h4to 6GPa. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 1151–1157. [Google Scholar] [CrossRef]
- Oya, K.P.; Myasnikova, R.M. Crystals of binary molecular compounds formed by hydrogen bonds—III. Crystal structure of e{open}-caprolactam. J. Struct. Chem. 1974, 15, 578–583. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Assarsson, P. Space group of ɛ-caprolactam (2-oxohexamethylenimine). J. Polym. Sci. 1962, 56, S30–S31. [Google Scholar] [CrossRef]
- Winkler, F.K.; Dunitz, J.D. Medium-Ring Compounds. XIX. Caprolactam: Structure Refinement. Acta Crystallogr. 1975, 31, 268–269. [Google Scholar] [CrossRef]
- Thomas, S.P.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Accurate Lattice Energies for Molecular Crystals from Experimental Crystal Structures. J. Chem. Theory Comput. 2018, 14, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova, I.A.; Lokshin, B.V. Hydrogen bonding in ε-caprolactam dimer: A quantum-chemical study. Russ. Chem. Bull. 2004, 53, 1894–1902. [Google Scholar] [CrossRef]
- Bull, C.L.; Funnell, N.P.; Tucker, M.G.; Hull, S.; Francis, D.J.; Marshall, W.G. PEARL: The high pressure neutron powder diffractometer at ISIS. High Press. Res. 2016, 36, 493–511. [Google Scholar] [CrossRef]
- Fabbiani, F.P.A.; Allan, D.R.; Dawson, A.; David, W.I.F.; McGregor, P.A.; Oswald, I.D.H.; Parsons, S.; Pulham, C.R. Pressure-induced formation of a solvate of paracetamol. Chem. Commun. 2003, 3, 3004–3005. [Google Scholar] [CrossRef]
- Oswald, I.D.H.; Pulham, C.R. Co-crystallisation at high pressure—An additional tool for the preparation and study of co-crystals. CrystEngComm 2008, 10, 1114–1116. [Google Scholar] [CrossRef]
- Fabbiani, F.P.A.; Buth, G.; Levendis, D.C.; Cruz-Cabeza, A.J. Pharmaceutical hydrates under ambient conditions from high-pressure seeds: A case study of GABA monohydrate. Chem. Commun. 2014, 50, 1817–1819. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Platas, J.; Alvaro, M.; Nestola, F.; Angel, R. EosFit7-GUI: A new graphical user interface for equation of state calculations, analyses and teaching. J. Appl. Crystallogr. 2016, 49, 1377–1382. [Google Scholar] [CrossRef]
- Funnell, N.P.; Dawson, A.; Marshall, W.G.; Parsons, S. Destabilisation of hydrogen bonding and the phase stability of aniline at high pressure. CrystEngComm 2013, 15, 1047–1060. [Google Scholar] [CrossRef]
- Cliffe, M.J.; Goodwin, A.L. PASCal: A principal axis strain calculator for thermal expansion and compressibility determination. J. Appl. Crystallogr. 2012, 45, 1321–1329. [Google Scholar] [CrossRef]
- Arlin, J.-B.; Price, L.S.; Price, S.L.; Florence, A.J.; Price, S.L.; Dunitz, J.D.; Scheraga, H.A.; Byrn, S.R.; Pfeiffer, R.R.; Stephenson, G.; et al. A strategy for producing predicted polymorphs: Catemeric carbamazepine form V. Chem. Commun. 2011, 47, 7074. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Allen, F.H.; Baalham, C.A.; Lommerse, J.P.M.; Raithby, P.R. Carbonyl–Carbonyl Interactions can be Competitive with Hydrogen Bonds. Acta Crystallogr. Sect. B Struct. Sci. 1998, 54, 320–329. [Google Scholar] [CrossRef]
- Wood, P.A.; Haynes, D.A.; Lennie, A.R.; Samuel, W.D.; Parsons, S.; Pidcock, E.; Warren, J.E. The Anisotropic Compression of the Crystal Structure of 3-Aza-bicyclo(3.3.1)nonane-2,4-dione to 7.1 GPa. Cryst. Growth Des. 2008, 8, 549–558. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, Programs for Scaling and Absorption Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 2008. [Google Scholar]
- Sheldrick, G.M. IUCr SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Coelho, A. TOPAS—Academic: General Profile and Structure Analysis Software for Powder Diffraction Data; Bruker AXS: Karlsruhe, Germany, 2012. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Delley, B. An All-Electron Numerical-Method for Solving the Local Density Functional for Polyatomic-Molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- BIOVIA. D.S. Materials Studio 2018; BIOVIA: San Diego, CA, USA, 2018. [Google Scholar]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, Molecules, Soilds, and Surfaces—Applications of the Generalized Gradient Approximation for Exchange and Correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 73005. [Google Scholar] [CrossRef] [PubMed]
- Oswald, I.D.H.; Connor, L.E.; Marshall, W.G.; Hutchison, I.B.; Bull, C.L.; Urquhart, A.J. Investigation of Caprolactam-D11 at High Pressure. Available online: https://data.isis.stfc.ac.uk/doi/investigation/73945863 (accessed on 3 June 2019).
Sample Availability: Not Available. |









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutchison, I.B.; Bull, C.L.; Marshall, W.G.; Urquhart, A.J.; Oswald, I.D.H. Pressure-Induced Polymorphism of Caprolactam: A Neutron Diffraction Study. Molecules 2019, 24, 2174. https://doi.org/10.3390/molecules24112174
Hutchison IB, Bull CL, Marshall WG, Urquhart AJ, Oswald IDH. Pressure-Induced Polymorphism of Caprolactam: A Neutron Diffraction Study. Molecules. 2019; 24(11):2174. https://doi.org/10.3390/molecules24112174
Chicago/Turabian StyleHutchison, Ian B., Craig L. Bull, William G. Marshall, Andrew J. Urquhart, and Iain D.H. Oswald. 2019. "Pressure-Induced Polymorphism of Caprolactam: A Neutron Diffraction Study" Molecules 24, no. 11: 2174. https://doi.org/10.3390/molecules24112174
APA StyleHutchison, I. B., Bull, C. L., Marshall, W. G., Urquhart, A. J., & Oswald, I. D. H. (2019). Pressure-Induced Polymorphism of Caprolactam: A Neutron Diffraction Study. Molecules, 24(11), 2174. https://doi.org/10.3390/molecules24112174