
Synthesis and Cytostatic Effect of 3’-deoxy-3’-C-Sulfanylmethyl Nucleoside Derivatives with d-xylo Configuration


Abstract
1. Introduction
2. Results and Discussion
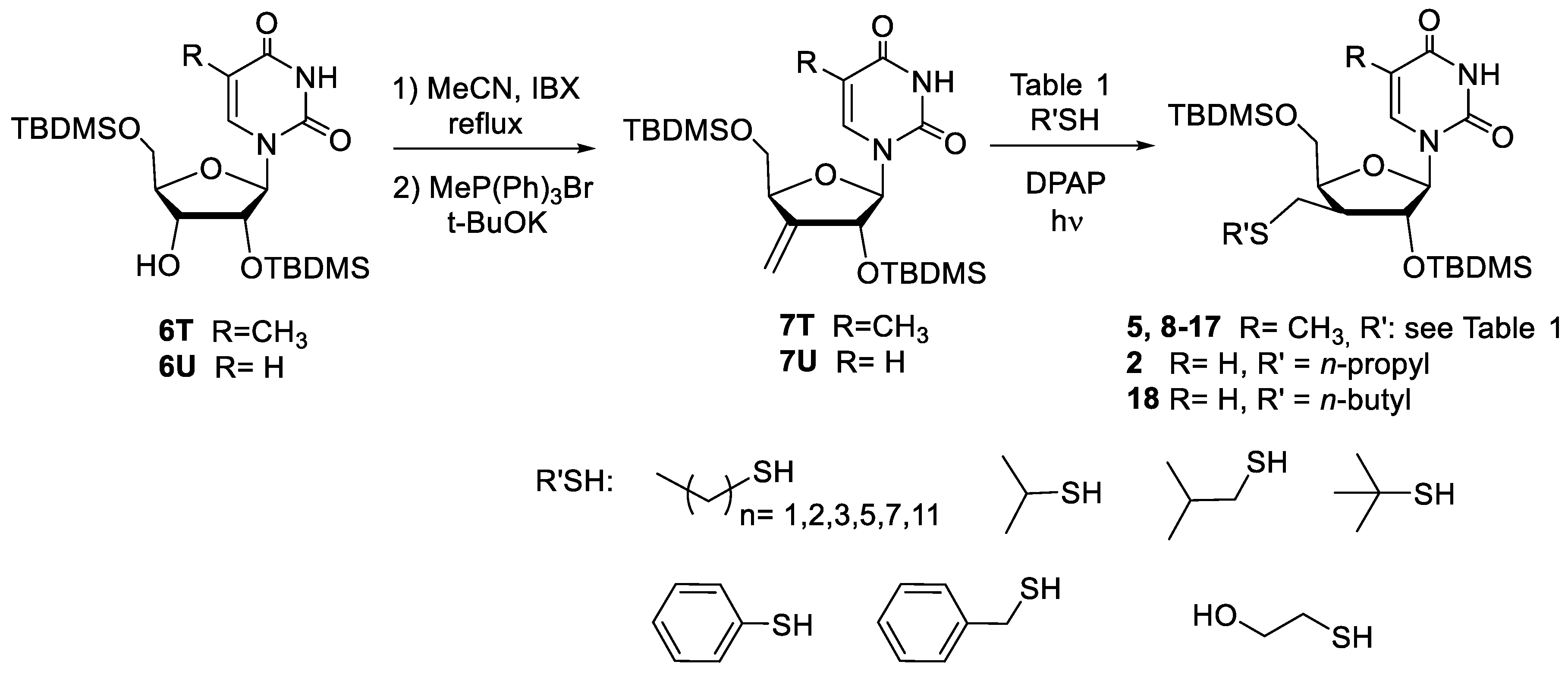
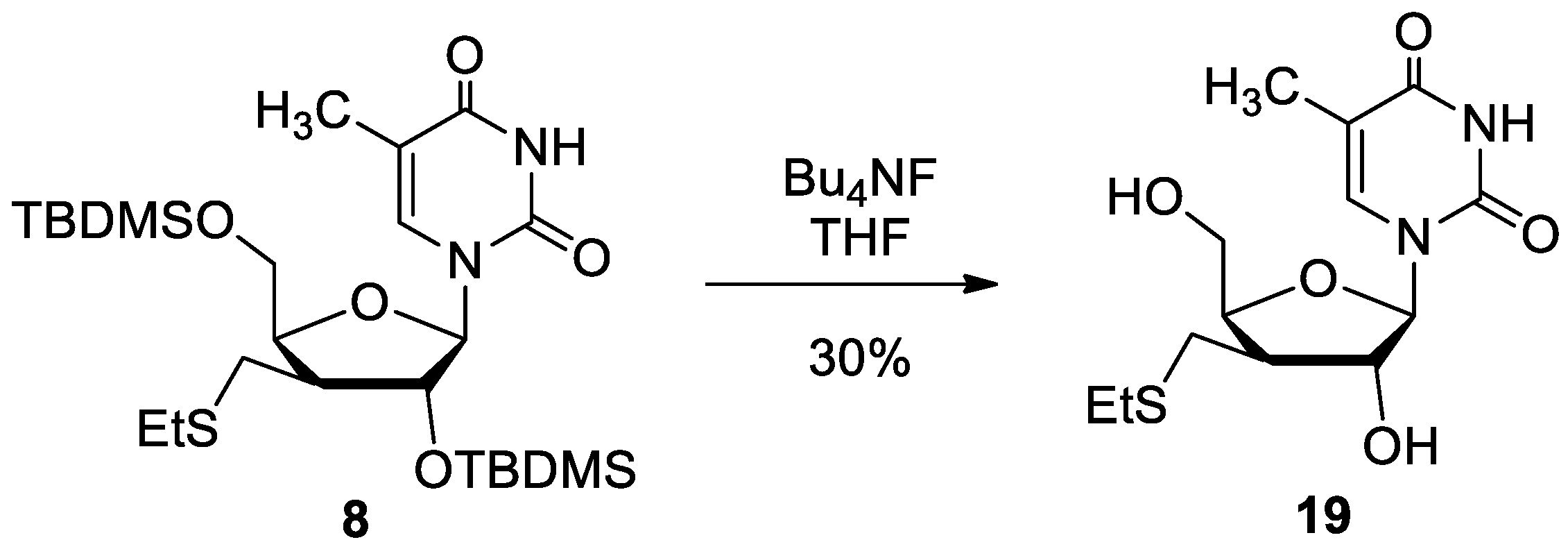
2.1. Chemistry
2.2. Biological Evaluation
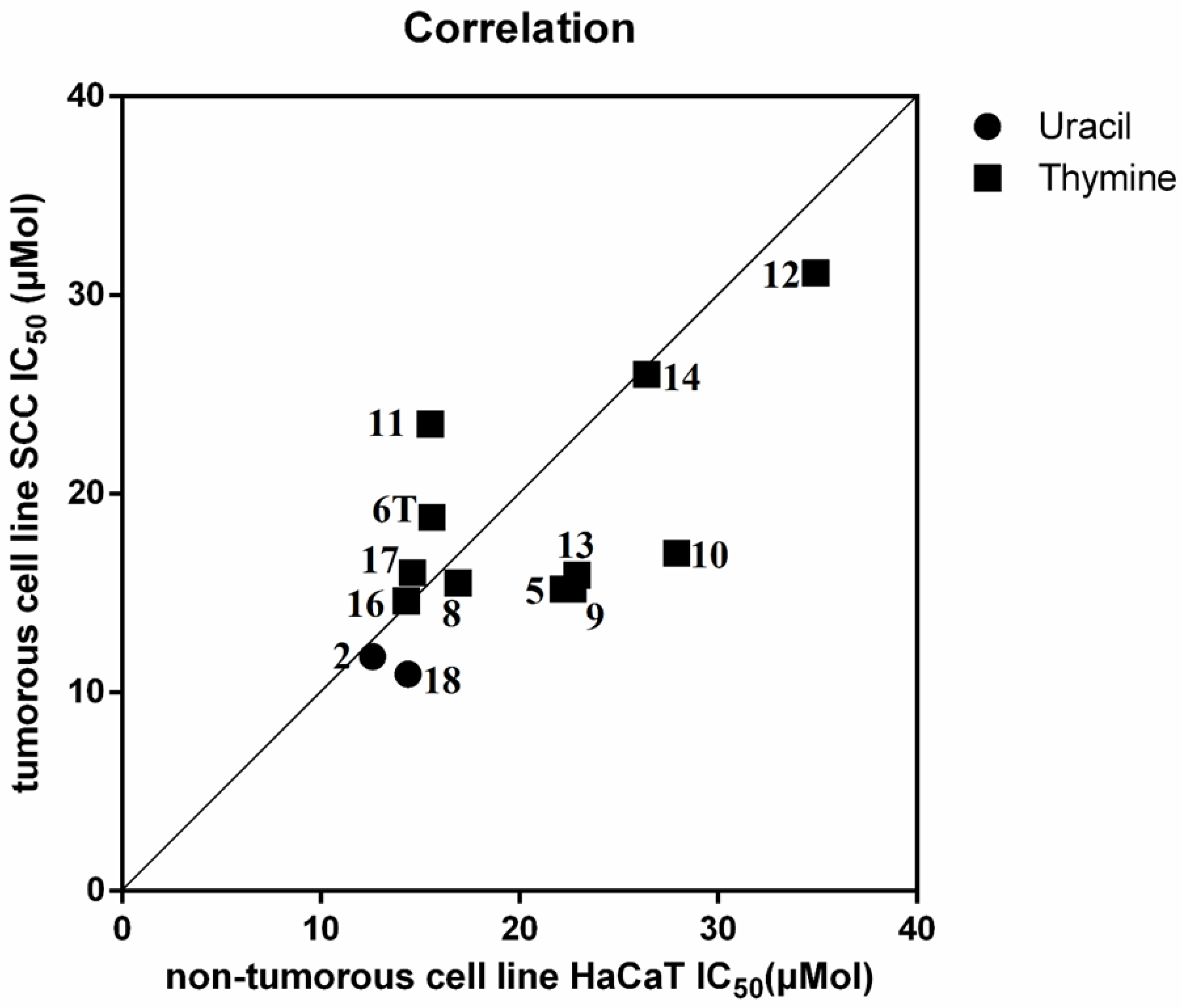
2.2.1. Cell Viability Study
2.2.2. Live Cell Imaging via Time-Lapse Microscopy
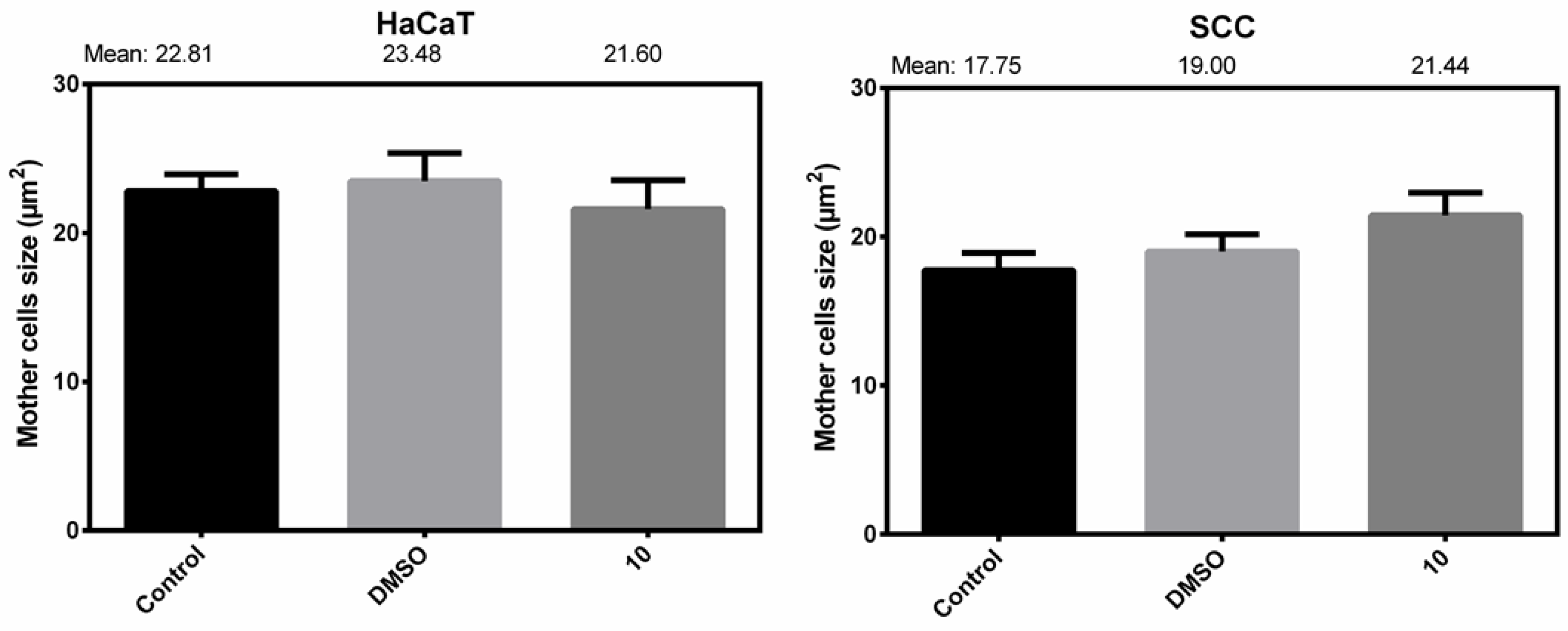
Mother Cell Size Changes
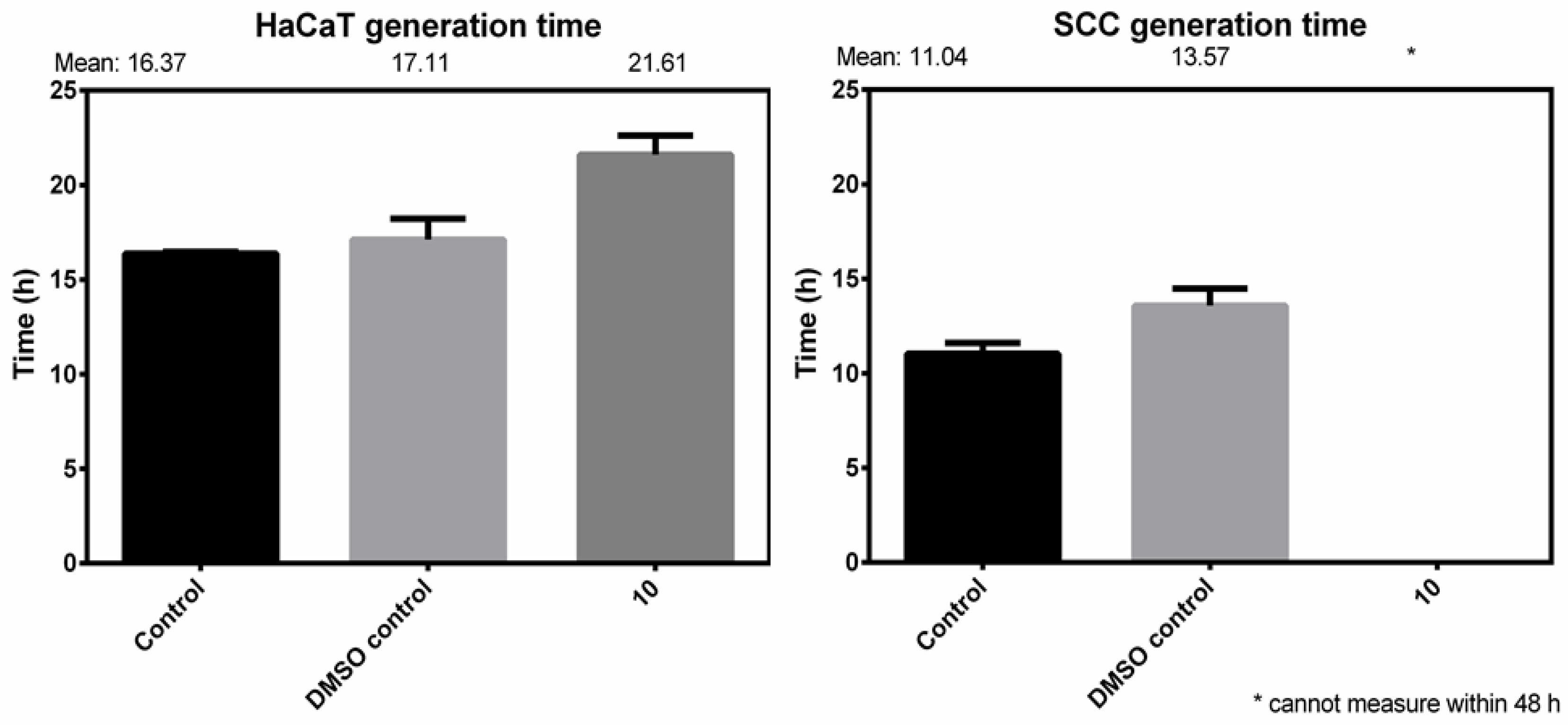
Generation Time
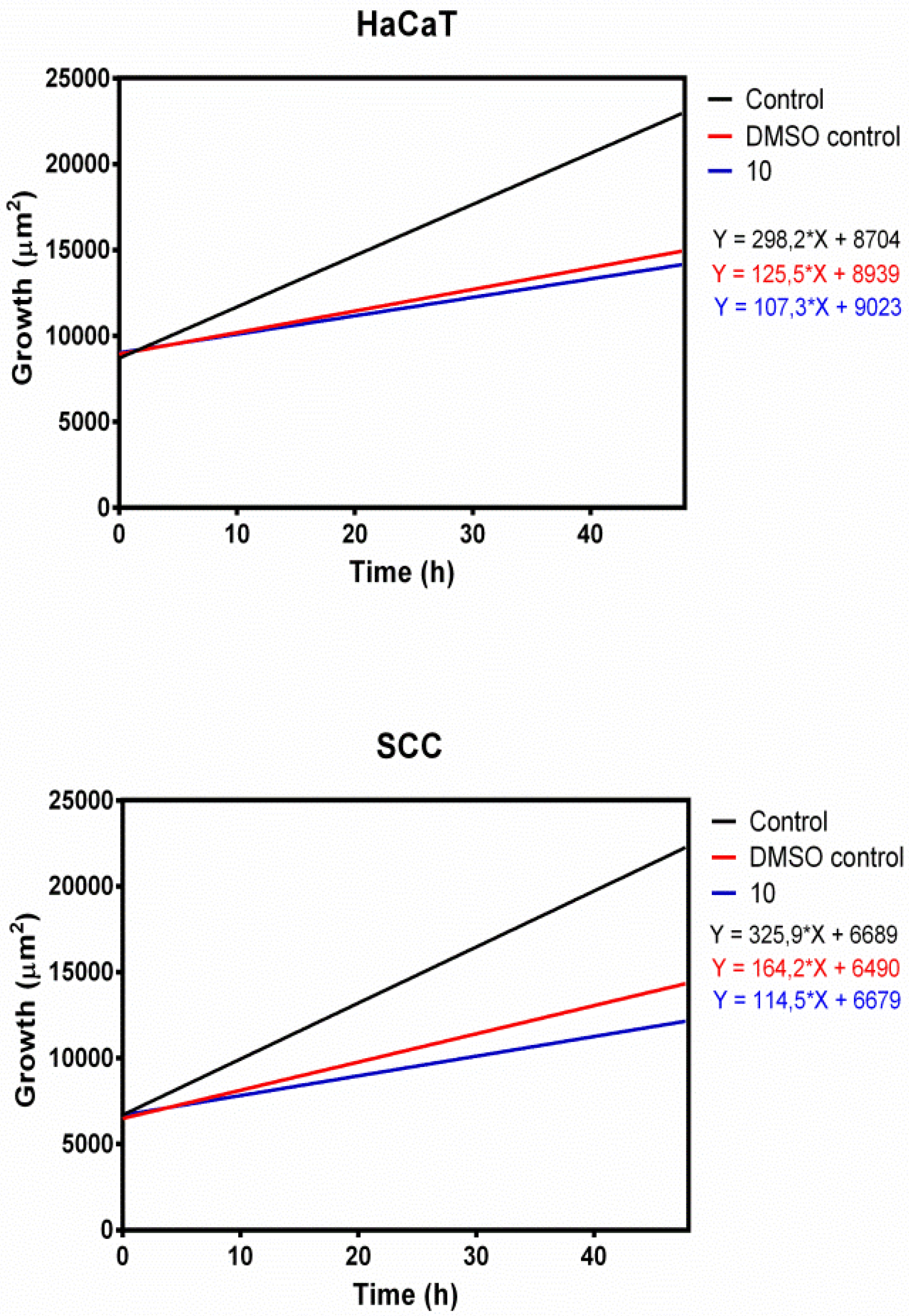
Growth Inhibition
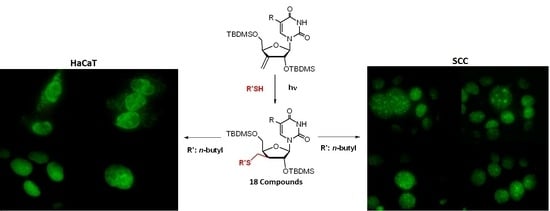
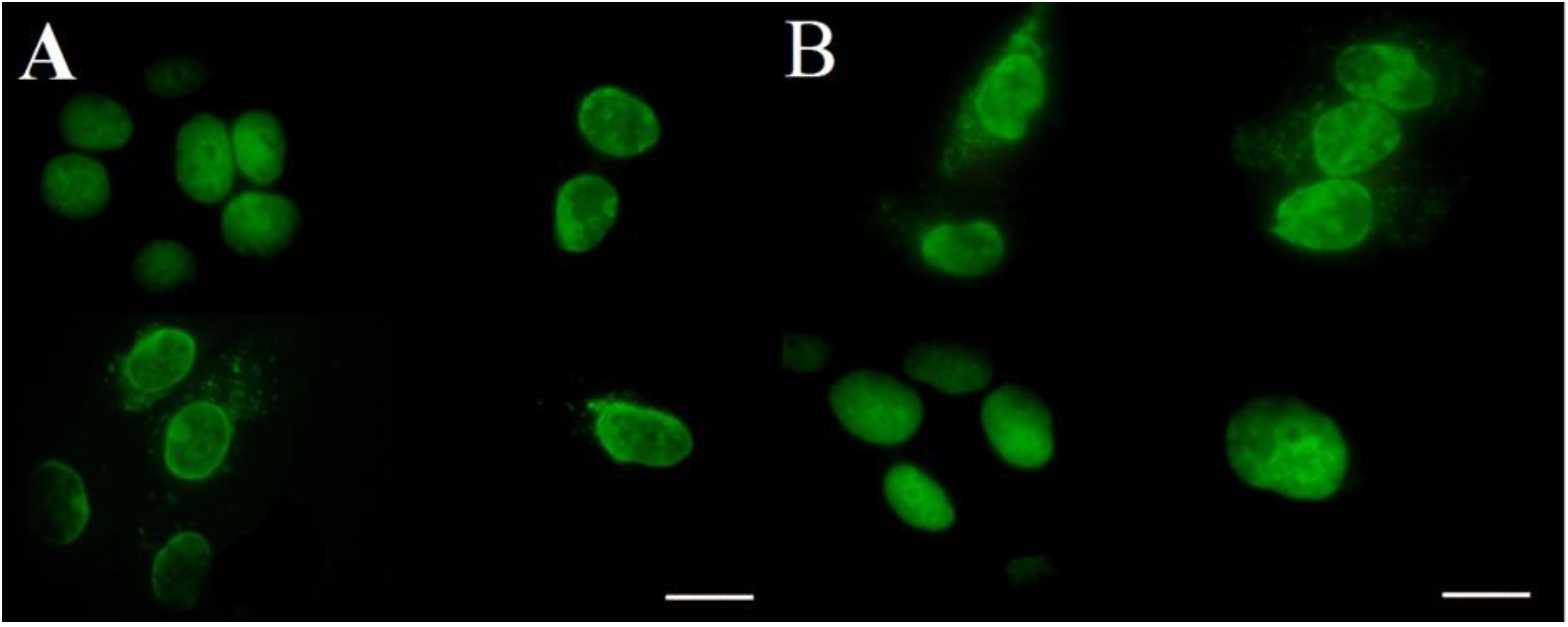
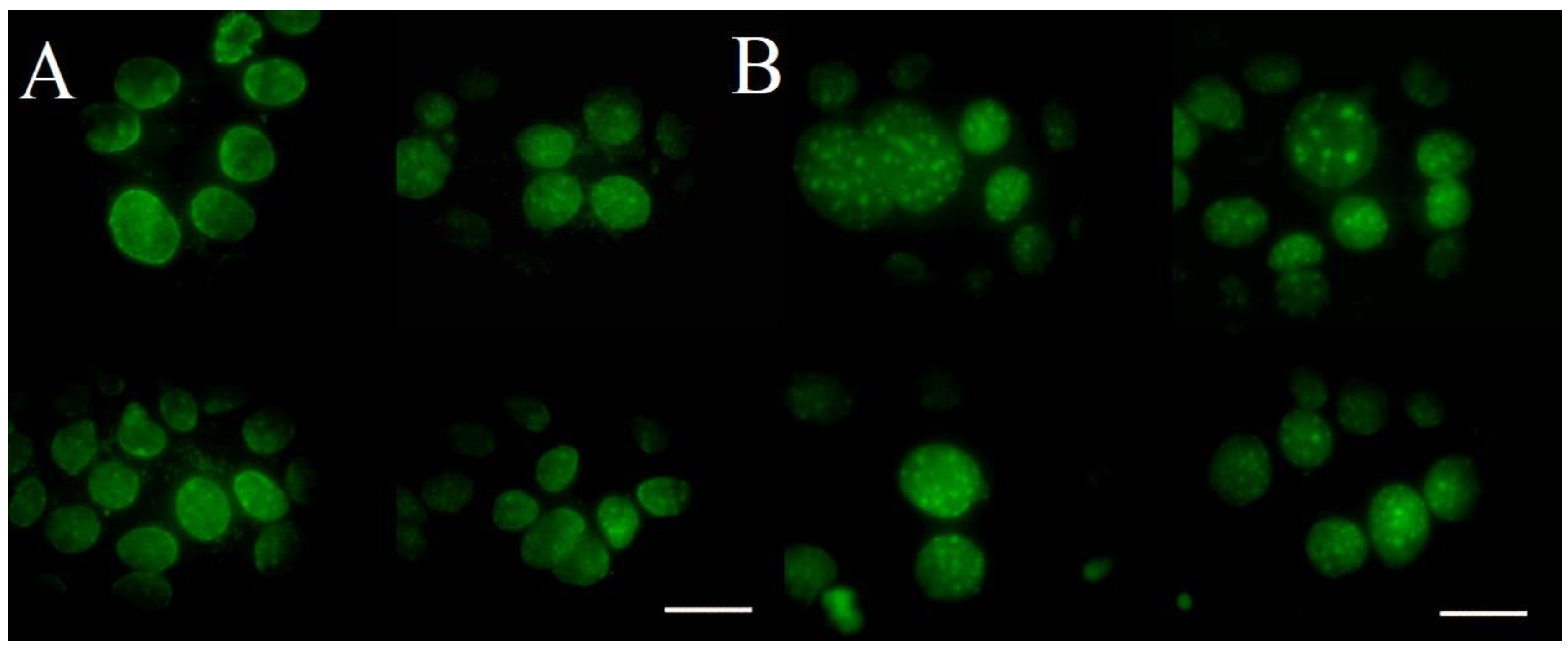
2.2.3. Fluorescent Microscopy
3. Conclusions
4. Materials and Methods
4.1. General Informations
4.2. Synthesis of Nucleoside Derivatives
4.3. Cell Lines and Cell Culture Conditions
4.4. MTT Assay
4.5. Time-Lapse Image Video-Microscopy and Image Analysis
- Inverse microscopes sitting in the incubator CO2 (SANYOMCO18-AC, Wood Dale, USA).
- Illumination under minimized heat- and phototoxicity, operated in the near-infrared range (940 nM), with light emitting diodes synchronized by short (1s) image-acquisition periods. Light intensity/energy was limited to the lowest possible level for image acquisition. Cells were only illuminated during image acquisition periods.
- Opening the image sequence in 8-bit format.
- Stack Deflicker: The Stack Deflicker calculates the average grey value for each frame and normalizes all frames so that they have the same average grey level as a specified frame of the stack. This plugin is very useful to remove flickering in movies caused by frame rates different from the frequency of AC used for the light-source that illuminate the scene. An input value of −1 corresponds to the brightest frame while an input value of zero corresponds to the faintest frame. If a region of the stack is selected the average frame intensity will be calculated from this region [56].
- Changing the brightness/contrast, if it is neccesary.
- Subtrack background: Removes smooth continuous backgrounds from gels and other images. Based on the concept of the ‘rolling ball’ algorithm described by Sternberg Stanley. Imagine that the 2D grayscale image has a third dimension (height) by the image value at every point in the image, creating a surface. A ball of given radius is rolled over the bottom side of this surface; the hull of the volume reachable by the ball is the background to be subtracted [57].
- Treshold: Use this tool to automatically or interactively set lower and upper threshold values, segmenting grayscale images into features of interest and background [58].
- Divided cells were selected on the binary image sequence based on their circularity determined by area/perimeter ratio.
- Bigger pre-division mother cells were separated from smaller post-division daughter cells.
- Pixel size was calibrated with Burker chamber.
- Area calculation from pixel2 to µm2.
4.5.1. Determination of generation time
4.5.2. Confluence
4.5.3. Statistical analysis
4.5.4. Fluorescent microscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Clercq, E.; Holy, A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 2: Complex modifications to the nucleoside scaffold. Antivir. Res. 2019, 162, 5–21. [Google Scholar]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogues. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.B.; Secrist, J.A., III; Waud, W.R. Purine Nucleoside Antimetabolites in Development for the Treatment of Cancer. Curr. Opin. Investig. Drugs 2004, 5, 592–596. [Google Scholar]
- Larson, R.A. Three New Drugs for Acute Lymphoblastic Leukemia: Nelarabine, Clofarabine, and Forodesine. Semin. Oncol. 2007, 34, S13–S20. [Google Scholar] [CrossRef]
- Jacobs, A.D. Gemcitabine-Based Therapy in Pancreas Cancer. Gemcitabine-Docetaxel and Other Novel Combinations. Cancer 2002, 95, 923–927. [Google Scholar] [CrossRef]
- Burkes, R.L.; Shepherd, F.A. Gemcitabine in the Treatment of Non-small-cell Lung Cancer. Ann. Oncol. 1995, 6, S57–S60. [Google Scholar] [CrossRef]
- King, R.S. Gemcitabine. New First-line Therapy for Pancreatic Cancer. Cancer Pract. 1996, 4, 353–354. [Google Scholar]
- Shimma, N.; Umeda, I.; Arasaki, M.; Murasaki, C.; Masubuchi, K.; Kohchi, Y.; Miwa, M.; Ura, M.; Sawada, N.; Tahara, H.; et al. The design and synthesis of a new tumor-selective fluoropyrimidine carbamate, capecitabine. Bioorg. Med. Chem. 2000, 8, 1697–1706. [Google Scholar] [CrossRef]
- Bentley, H.R.; Cunningham, K.G.; Spring, F.S. 509. Cordycepin, a metabolic product from cultures of cordyceps militaris(Linn.)Link. Part II. The structure of cordycepin. J. Chem. Soc. 1951, 0, 2301–2305. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Cortes, J. Evaluation of the L-stereoisomeric nucleoside analog troxacitabine for the treatment of acute myeloid leukemia. Expert Opin. Investig. Drugs 2007, 16, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Garcia-Manero, G.; Cortes, J.E.; Baker, S.D.; Miller, C.B.; O’Brien, S.M.; Thomas, D.A.; Andreeff, M.; Bivins, C.; Jolivet, J.; et al. Phase II study of troxacitabine, a novel dioxolane nucleoside analog, in patients with refractory leukemia. J. Clin. Oncol. 2002, 20, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, R.; Letourneau, R.; Steward, W.; Hawkins, R.E.; Batist, G.; Vincent, M.; Whittom, R.; Eatock, M.; Jolivet, J.; Moore, M. Phase II study of troxacitabine in chemotherapy-naive patients with advanced cancer of the pancreas: Gastrointestinal tumors. Ann. Oncol. 2005, 16, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Townsley, C.A.; Chi, K.; Ernst, D.S.; Belanger, K.; Tannock, I.; Bjarnason, G.A.; Stewart, D.; Goel, R.; Ruether, J.D.; Siu, L.L.; et al. Phase II study of troxacitabine (BCH-4556) in patients with advanced and/or metastatic renal cell carcinoma: A trial of the National Cancer Institute of Canada-Clinical Trials Group. J. Clin. Oncol. 2003, 21, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Tuli, H.S.; Sandhu, S.S.; Sharma, A.K. Pharmacological and therapeutical potential of Cordyceps with special reference to Cordycepin. 3 Biotech 2014, 4, 1–12. [Google Scholar] [CrossRef]
- Franchetti, P.; Cappellacci, L.; Pasqualini, M.; Petrelli, R.; Vita, P.; Jayaram, H.N.; Horvath, Z.; Szekeres, T.; Grifantini, M. Antitumor activity of C-methyl-beta-D-ribofuranosyladenine nucleoside ribonucleotide reductase inhibitors. J. Med. Chem. 2005, 48, 4983–4989. [Google Scholar] [CrossRef]
- Cappellacci, L.; Franchetti, P.; Petrelli, R.; Riccioni, S.; Vita, P.N.; Jayaram, H.; Grifantini, M. Purine and Pyrimidine Nucleoside Analogs of 3′-C-Methyladenosine as Antitumor Agents. Collect. Czechoslov. Chem. Commun. 2006, 71, 1088–1098. [Google Scholar] [CrossRef]
- Cappellacci, L.; Franchetti, P.; Vita, P.; Petrelli, R.; Lavecchia, A.; Jayaram, H.N.; Saiko, P.; Graser, G.; Szekeres, T.; Grifantini, M. Ribose-Modified Purine Nucleosides as Ribonucleotide Reductase Inhibitors. Synthesis, Antitumor Activity, and Molecular Modeling of N6-Substituted 3′-C-Methyladenosine Derivatives. J. Med. Chem. 2008, 51, 4260–4269. [Google Scholar] [CrossRef]
- Hattori, H.; Tanaka, M.; Fukushima, M.; Sasaki, T.; Matsuda, A. Nucleosides and nucleotides. 158. 1-(3-C-Ethynyl-β-D-ribo-pentofuranosyl)-cytosine, 1-(3-C-Ethynyl-β-D-ribo-pentofuranosyl)uracil, and their nucleobase analogues as new potential multifunctional antitumor nucleosides with a broad spectrum of activity. J. Med. Chem. 1996, 39, 5005–5011. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, A.; Fukushima, M.; Wataya, Y.; Sasaki, T. A new antitumor nucleoside, 1-(3-C-ethynyl-β-D-ribo-pentofuranosyl)cytosine (ECyd), is a potent inhibitor of RNA synthesis. Nucleosides Nucleotides 1999, 18, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Hulpia, F.; Noppen, S.; Schols, D.; Andrei, G.; Snoeck, R.; Liekens, S.; Vervaeke, P.; Van Calenbergh, S. Synthesis of a 3’-C-ethynyl-β-D-ribofuranose purine nucleoside library: Discovery of C7-deazapurine analogs as potent antiproliferative nucleosides. Eur. J. Med. Chem. 2018, 157, 248–267. [Google Scholar] [CrossRef] [PubMed]
- Hulpia, F.; Balzarini, J.; Schols, D.; Andrei, G.; Snoeck, R.; Van Calenbergh, S. Exploring the purine core of 3′-C-ethynyladenosine (EAdo) in search of novel nucleoside therapeutics. Bioorg. Med. Chem. Lett. 2016, 26, 1970–1972. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, A.; Sasaki, T. Antitumor activity of sugar-modified cytosine nucleosides. Cancer Sci. 2004, 95, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.E.; Bowman, C.N. Thiol-Ene Click Chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Marra, A. Recent applications of thiol–ene coupling as a click process for glycoconjugation. Chem. Soc. Rev. 2012, 41, 573–586. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, L.; Dénès, F.; Scanlan, E.M. Thiyl-Radical Reactions in Carbohydrate Chemistry: From Thiosugars to Glycoconjugate Synthesis. Eur. J. Org. Chem. 2016, 12, 2080–2095. [Google Scholar] [CrossRef]
- Lázár, L.; Csávás, M.; Herczeg, M.; Herczegh, P.; Borbás, A. Synthesis of S-Linked Glycoconjugates and S-Dissacharides by Thiol-Ene Coupling Reaction of Enoses. Org. Lett. 2012, 14, 4650–4653. [Google Scholar] [CrossRef]
- Lázár, L.; Csávás, M.; Tóth, M.; Somsák, L.; Borbás, A. Thio-click approach to the synthesis of stable glycomymetics. Chem. Pap. 2015, 69, 889–895. [Google Scholar] [CrossRef]
- Bege, M.; Bereczki, I.; Herczeg, M.; Kicsák, M.; Eszenyi, D.; Herczegh, P.; Borbás, A. A low-temperature photoinduced thiol-ene click reaction: A method for the synthesis of sugar modified nucleosides. Org. Biomol. Chem. 2017, 15, 9226–9233. [Google Scholar] [CrossRef] [PubMed]
- Eszenyi, D.; Kelemen, V.; Balogh, F.; Bege, M.; Csávás, M.; Herczegh, P.; Borbás, A. Promotion of a Reaction by Cooling: Stereoselective 1,2-cis-α-Thioglycoconjugation by Thiol-Ene Coupling at −80 °C. Chem. Eur. J. 2018, 24, 4532–4536. [Google Scholar] [CrossRef] [PubMed]
- Lázár, L.; Csávás, M.; Hadházi, Á.; Herczeg, M.; Tóth, M.; Somsák, L.; Barna, T.; Herczegh, P.; Borbás, A. Systematic study on free radical hydrothiolation of unsaturated monosaccharide derivatives with exo- and endocyclic double bonds. Org. Biomol. Chem. 2013, 11, 5339–5350. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, J.; Vesely, P.; Netíková, I.; Matousková, E.; Petruzelka, L. The protective effect of pyrimidine nucleosides on human HaCaT keratinocytes treated with 5-FU. Anticancer Res. 2015, 35, 1303–1310. [Google Scholar] [PubMed]
- Nagy, G.; Király, G.; Veres, P.; Lázár, I.; Fábián, I.; Bánfalvi, G.; Juhász, I.; Kalmár, J. Controlled release of methotrexate from functionalized silica-gelatin aerogel microparticles applied against tumor cell growth. Int. J. Pharm. 2019, 558, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Schuler, P.J.; Trellakis, S.; Greve, J.; Bas, M.; Bergmann, C.; Bölke, E.; Lehnerdt, G.; Mattheis, S.; Albers, A.E.; Brandau, S.; et al. In vitro Chemosensitivity of Head and Neck Cancer Cell Lines. Eur. J. Med. Res. 2010, 15, 337–344. [Google Scholar] [CrossRef]
- Griffith, D.A.; Jarvis, S.M. Nucleoside and nucleobase transport systems of mammalian cells. Biochim. Biophys. Acta Rev. Biomembr. 1996, 1286, 153–181. [Google Scholar] [CrossRef]
- Kraupp, M.; Marz, R. Nucleobase and nucleoside transport in mammalian cells. Wien. Klin. Wochenschr. 1995, 107, 677–680. [Google Scholar]
- Stuurman, F.E.; Voest, E.E.; Awada, A.; Witteveen, P.O.; Bergeland, T.; Hals, P.A.; Rasch, W.; Schellens, J.H.; Hendlisz, A. Phase I study of oral CP-4126, a gemcitabine derivative, in patients with advanced solid tumors. Invest. New Drugs 2013, 31, 959–966. [Google Scholar] [CrossRef]
- Adema, A.D.; Smid, K.; Losekoot, N.; Honeywell, R.J.; Verheul, H.M.; Myhren, F.; Sandvold, M.L.; Peters, G.J. Metabolism and accumulation of the lipophilic deoxynucleoside analogs elacytarabine and CP-4126. Investig. New Drugs 2012, 30, 1908–1916. [Google Scholar] [CrossRef]
- Szilágyi, Á.; Fenyvesi, F.; Majercsik, O.; Pelyvás, I.F.; Bácskay, I.; Fehér, P.; Váradi, J.; Vecsernyés, M.; Herczegh, P. Synthesis and Cytotoxicity of Leinamycin Antibiotic Analogues. J. Med. Chem. 2006, 49, 5626–5630. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.A.; Oliveira, M.; Christiansen, M.A.; Cutler, C.E. Preliminary SAR analysis of novel antiproliferative N6,5’-bis-ureidoadenosine derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 6775–6779. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.R.; Cutler, C.E.; Browning, M.S.; Balzarini, J.; Peterson, M.A. Synthesis and SAR of 2’,3’-bis-O-substituted N6,5’-bis-ureidoadenosine derivatives: Implications for prodrug delivery and mechanism of action. Bioorg. Med. Chem. Lett. 2012, 22, 6067–6071. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, C.; Pérez-Pérez, M.J.; Rodríguez-Barrios, F.; Gago, F.; De Clercq, E.; Balzarini, J.; San-Félix, A.; Camarasa, M.J. Exploring the role of the 5’-position of TSAO-T. Synthesis and anti-HIV evaluation of novel TSAO-T derivatives. Antivir. Res. 2001, 50, 207–222. [Google Scholar] [CrossRef]
- Harmse, L.; Dahan-Farkas, N.; Panayides, J.L.; van Otterlo, W.; Penny, C. Aberrant Apoptotic Response of Colorectal Cancer Cells to Novel Nucleoside Analogues. PLoS ONE 2015, 10, e0138607. [Google Scholar] [CrossRef] [PubMed]
- Panayides, J.L.; Mathieu, V.; Banuls, L.M.Y.; Apostolellis, H.; Dahan-Farkas, N.; Davids de Leonie Harmse, H.; Rey, M.E.C.; Green, I.R.; Pelly, S.C.; Kiss, R.; et al. Synthesis and in vitro growth inhibitory activity of novel silyl- and trityl-modified nucleosides. Bioorg. Med. Chem. 2016, 24, 2716–2724. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Boukamp, P.; Stanbridge, E.J.; Foo, D.Y.; Cerutti, P.A.; Fusenig, N.E. c-Ha-ras oncogene expression in immortalized human keratinocytes (HaCaT) alters growth potential in vivo but lacks correlation with malignancy. Cancer Res. 1990, 50, 2840–2847. [Google Scholar]
- Boukamp, P.; Popp, S.; Altmeyer, S.; Hülsen, A.; Fasching, C.; Cremer, T.; Fusenig, N.E. Sustained nontumorigenic phenotype correlates with a largely stable chromosome content during long-term culture of the human keratinocyte line HaCaT. Genes Chromosom. Cancer 1997, 19, 201–214. [Google Scholar] [CrossRef]
- Sominski, D.D.; Rafferty, P.; Brosnan, K.; Volk, A.; Walker, M.; Capaldi, D.; Emmell, E.; Johnson, K.; Weinstock, D. Development of a squamous cell carcinoma mouse model for immunotoxicity testing. J. Immunotoxicol. 2016, 13, 226–234. [Google Scholar] [CrossRef]
- De Abreu, L.C.; Fernandes, M.O.H.; Dos Santos, M.G.; Meireles, A.B.; De Ameida, V.G.; De Fátima, W.G.; De Avelar-Freitas, B.A.; Brito-Melo, G.E.A. Dimethyl Sulfoxide (DMSO) Decreases Cell Proliferation and TNF-α, IFN-γ, and IL-2 Cytokines Production in Cultures of Peripheral Blood Lymphocytes. Molecules 2017, 22, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.; Nagy, M.; Kiss, A.; Rácz, D.; Barna, B.; Könczöl, P.; Bankó, C.; Bacsó, Z.; Kéki, S.; Bánfalvi, G.; et al. MICAN, a new fluorophore for vital and non-vital staining of human cells. Toxicol. In Vitro 2018, 48, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Hennig, W.; Petrenyi, K.; Kovács, L.; Pócsi, I.; Dombrádi, V.; Bánfalvi, G. Time-lapse video microscopy and image analysis of adherence and growth patterns of Candida albicans strains. Appl. Microbiol. Biotechnol. 2014, 98, 5185–5194. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Újvárosi, K.; Nagy, G.; Posta, J.; Bánfalvi, G. Apoptogenic and necrogenic effects of mercuric acetate on the chromatin structure of K562 human erythroleukemia cells. Toxicol. In Vitro 2010, 24, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Fiji. Available online: https://fiji.sc (accessed on 8 June 2019).
- Stack Deflicker. Available online: http://www.phage.dk/plugins/deflicker.htmL (accessed on 8 June 2019).
- ImageJ User Guide. Available online: https://imagej.nih.gov/ij/docs/guide/146-29.htmL (accessed on 8 June 2019).
- Adjust-ImageJ User Guide. Available online: https://imagej.nih.gov/ij/docs/guide/146-28.htmL#toc-Subsection-28.2 (accessed on 8 June 2019).
Sample Availability: Samples of the compounds 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 and 19 are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R’ | Temperature | Product | d.r. a | R | Yield |
|---|---|---|---|---|---|---|
| 1 | Ethyl | −80 °C | 8 | 9:1 | Me | 86% |
| 2 | n-Propyl | −80 °C | 5 | 13:1 | Me | 49% |
| 3 | i-Propyl | −40 to 0 °C | 9 | 33:1 | Me | 34% |
| 4 | n-Butyl | −80 to −40 °C | 10 | 20:1 | Me | 62% |
| 5 | n-Butyl | 0 °C | 10 | 10:1 | Me | 65% |
| 6 | i-Butyl | 0 °C | 11 | 22:1 | Me | 36% |
| 7 | t-Butyl | −80 to 0 °C | 12 | 14:1 | Me | 54% |
| 8 | n-Hexyl | −80 to −40 °C | 13 | 30:1 | Me | 46% |
| 9 | n-Octyl | 0 °C | 14 | 24:1 | Me | 29% |
| 10 | n-Dodecyl | 0 °C | 15 | 22:1 | Me | 29% |
| 11 | Phenyl | −80 °C −r.t. | - | - | Me | no reaction |
| 12 | Benzyl | −40 °C | 16 | 10:1 | Me | 82% |
| 13 | Hydroxyethyl | −80 °C | 17 | 3:1 | Me | 75% |
| 14 | Hydroxyethyl | −40 °C | 17 | 4:1 | Me | 72% |
| 15 | Hydroxyethyl | 0 °C | 17 | 4:1 | Me | 74% |
| 16 | n-Propyl | −80 °C | 2 | 50:1 | H | 75% |
| 17 | n-Butyl | −40 °C | 18 | 60:1 | H | 59% |
| 18 | n-Butyl | 0 °C | 18 | 12:1 | H | 66% |

| Compound | R | NB | H-1’ | H-4’ | H-2’ | H-3’ | C-1’ | C-3’ |
|---|---|---|---|---|---|---|---|---|
| 3-d-ribo a | GlcPerAc (D-ribo) | T | 5.81, d | 4.22–4.17 | 4.42, dd | 2.45–2.38 | 90.7 | 41.6 |
| J = 1.9 Hz | m b | J = 1.9, 4.9 Hz | m | |||||
| 3 a | GlcPerAc (D-xylo) | T | 5.92, d | 4.33, d | 4.10, dd | 2.80–2.76 | 87.1 | 47.5 |
| J = 6.9 Hz | J = 8.2 Hz | J = 7.0, 9.1 Hz | m | |||||
| 8 | Ethyl | T | 6.12, d | N. A. | N. A. | 3.01–2.94 | 87.4 | 46.5 |
| J = 6.9 Hz | m | |||||||
| 9 | i-Propyl | T | 6.10, d | 4.44, d | 4.28, dd | 2.82, td | 87.2 | 46.9 |
| J = 6.8 Hz | J = 8.2 Hz | J = 8.9, 7.3 Hz | J = 16.3, 18.8 Hz | |||||
| 10 | n-Butyl | T | 6.11, d | 4.46, d | 4.28, dd | 2.94–2.98 | 87.2 | 46.5 |
| J = 6.9 Hz | J = 8.0 Hz | J = 8.8, 7.2 Hz | m | |||||
| 11 | i-Butyl | T | 6.12, d | 4.48, d | 4.29, dd | 3.02–2.79 | 87.3 | 46.7 |
| J = 6.9 Hz | J = 8.0 Hz | J = 9.0, 7.0 Hz | m | |||||
| 12 | t-Butyl | T | 6.10, d | 4.41, d | 4.29, dd | 2.87–2.76 | 87.2 | 48 |
| J = 6.9 Hz | J = 8.3 Hz | J = 9.2, 7.1 Hz | m | |||||
| 13 | n-Hexyl | T | 6.12, d | 4.46, d | 4.29, dd | 2.89–2.79 | 87.3 | 46.5 |
| J = 6.9 Hz | J = 8.1 Hz | J = 8.9, 7.1 Hz | m | |||||
| 14 | n-Octyl | T | 6.12, d | 4.46, d | 4.29, dd | 2.73–2.63 | 87.3 | 46.5 |
| J = 6.9 Hz | J = 8.1 Hz | J = 8.9, 7.1 Hz | m | |||||
| 15 | n-Dodecyl | T | 6.12, d | 4.47, d | 4.29, dd | N. A. | 87.3 | 46.5 |
| J = 6.9 Hz | J = 8.1 Hz | J = 9.1, 7.0 Hz | ||||||
| 16 | Benzyl | T | 6.10, d | N. A. | N. A. | 2.88–2.75 b | 87.1 | 45.8 |
| J = 6.9 Hz | m | |||||||
| 17 | Hydroxyethyl | T | 6.09, d | 4.48, d | N. A. | N. A. | 87.4 | 46.7 |
| J = 6.9 Hz | J = 8.3 Hz | |||||||
| 18 | n-Butyl | U | 6.13, d | N. A. | N. A. | 2.95–2.79 | 87.7 | 46.6 |
| J = 6.6 Hz | m |
| Compound | R | HaCaT IC50 a | SCC IC50 a | logP b | SI c |
|---|---|---|---|---|---|
| 1 | GlcPerAc | - d | - d | 3.83 | |
| 2 | n-Propyl | 12.6 ± 0.22 | 11.8 ± 0.22 | 5.82 | 1.07 |
| 3 | GlcPerAc | 22.4±2.91 | - d | 4.11 | |
| 4 | MannPerAc | - d | - d | 4.11 | |
| 5 | n-Propyl | 22.2 ± 1.70 | 15.2 ± 1.00 | 6.10 | 1.46 |
| 8 | Ethyl | 16.9 ± 0.55 | 15.5 ± 0.59 | 5.63 | 1.09 |
| 9 | i-Propyl | 22.7 ± 1.34 | 15.2 ± 0.48 | 5.93 | 1.49 |
| 10 | n-Butyl | 27.9 ± 1.85 | 17.0 ± 1.55 | 6.49 | 1.64 |
| 11 | i-Butyl | 15.5 ± 2.14 | 23.5 ± 0.12 | 6.50 | 0.66 |
| 12 | t-Butyl | >34.9 | 31.1 ± 0.79 | 5.90 | 1.12 |
| 13 | n-Hexyl | 22.9 ± 0.57 | 15.9 ± 0.32 | 7.28 | 1.44 |
| 14 | n-Octyl | 26.4 ± 0.37 | 26.0 ± 0.76 | 8.08 | 1.02 |
| 15 | n-Dodecyl | - d | - d | 9.66 | |
| 16 | Benzyl | 14.3 ± 0.36 | 14.6 ± 0.19 | 7.06 | 0.98 |
| 17 | Hydroxyethyl | 14.6 ± 0.82 | 16.0 ± 0.17 | 4.40 | 0.91 |
| 18 | n-Butyl | 14.4 ± 1.59 | 10.9 ± 0.18 | 6.21 | 1.32 |
| 19 | Ethyl | - d | - d | −0.10 | |
| 6T | - | 15.6 ± 4.45 | 18.8 ± 0.74 | 3.81 | 0.83 |
| Methotrexate e | - | 290.4 ± 2.76 | 215.6 ± 2.33 | ||
| 5-FU f | - | 60.7 ± 8.45 |
| Size of Control Cells (µm2) | DMSO Control Cells Size (µm2) | Treated Cells Size (µm2) a | Treated/DMSO Difference (%) b | |
|---|---|---|---|---|
| HaCaT (n = 15) | 22.81 ± 1.13 | 23.48 ± 1.87 | 21.60 ± 1.93 | −9.00 |
| SCC (n = 15) | 17.75 ± 1.09 | 19.00 ± 1.09 | 21.44 ± 1.42 | 12.80 |
| Control Gen. Time (h) | DMSO Control Gen. Time (h) | Treated Cells a Gen. Time (h) | Treated/DMSO Difference (%) | |
|---|---|---|---|---|
| HaCaT (n = 15) | 16.37 ± 0.092 | 17.11 ± 1.10 | 21.67 ± 1.01 | 20.80 |
| SCC (n = 15) | 11.04 ± 0.56 | 13.57 ± 0.91 | >48.00 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bege, M.; Kiss, A.; Kicsák, M.; Bereczki, I.; Baksa, V.; Király, G.; Szemán-Nagy, G.; Szigeti, M.Z.; Herczegh, P.; Borbás, A. Synthesis and Cytostatic Effect of 3’-deoxy-3’-C-Sulfanylmethyl Nucleoside Derivatives with d-xylo Configuration. Molecules 2019, 24, 2173. https://doi.org/10.3390/molecules24112173
Bege M, Kiss A, Kicsák M, Bereczki I, Baksa V, Király G, Szemán-Nagy G, Szigeti MZ, Herczegh P, Borbás A. Synthesis and Cytostatic Effect of 3’-deoxy-3’-C-Sulfanylmethyl Nucleoside Derivatives with d-xylo Configuration. Molecules. 2019; 24(11):2173. https://doi.org/10.3390/molecules24112173
Chicago/Turabian StyleBege, Miklós, Alexandra Kiss, Máté Kicsák, Ilona Bereczki, Viktória Baksa, Gábor Király, Gábor Szemán-Nagy, M. Zsuzsa Szigeti, Pál Herczegh, and Anikó Borbás. 2019. "Synthesis and Cytostatic Effect of 3’-deoxy-3’-C-Sulfanylmethyl Nucleoside Derivatives with d-xylo Configuration" Molecules 24, no. 11: 2173. https://doi.org/10.3390/molecules24112173
APA StyleBege, M., Kiss, A., Kicsák, M., Bereczki, I., Baksa, V., Király, G., Szemán-Nagy, G., Szigeti, M. Z., Herczegh, P., & Borbás, A. (2019). Synthesis and Cytostatic Effect of 3’-deoxy-3’-C-Sulfanylmethyl Nucleoside Derivatives with d-xylo Configuration. Molecules, 24(11), 2173. https://doi.org/10.3390/molecules24112173