General
The 1H-NMR and 13C-NMR data were recorded in CDCl3 solution with Bruker NMR spectrometers (DRX 500, AM 300) if not noted otherwise. The chemical shifts are measured relative to TMS (δ = 0) or chloroform (δ = 7.26) and the coupling J is expressed in Hertz. Mass spectra were recorded on a Thermo Scientific TSQ Quantum Access MAX mass spectrometer (ESI, positive or negative). Standard flash chromatography was employed to purify the crude reaction mixture using 200–300 mesh silica gel (Tsingdao Ocean Company, Tsingdao, China) under a positive nitrogen pressure. Tetrahydrofuran (THF) and diethyl ether were freshly distilled from lithium aluminium hydride under an argon atmosphere. Dichloromethane, hexane and toluene were freshly distilled from calcium hydride under argon.
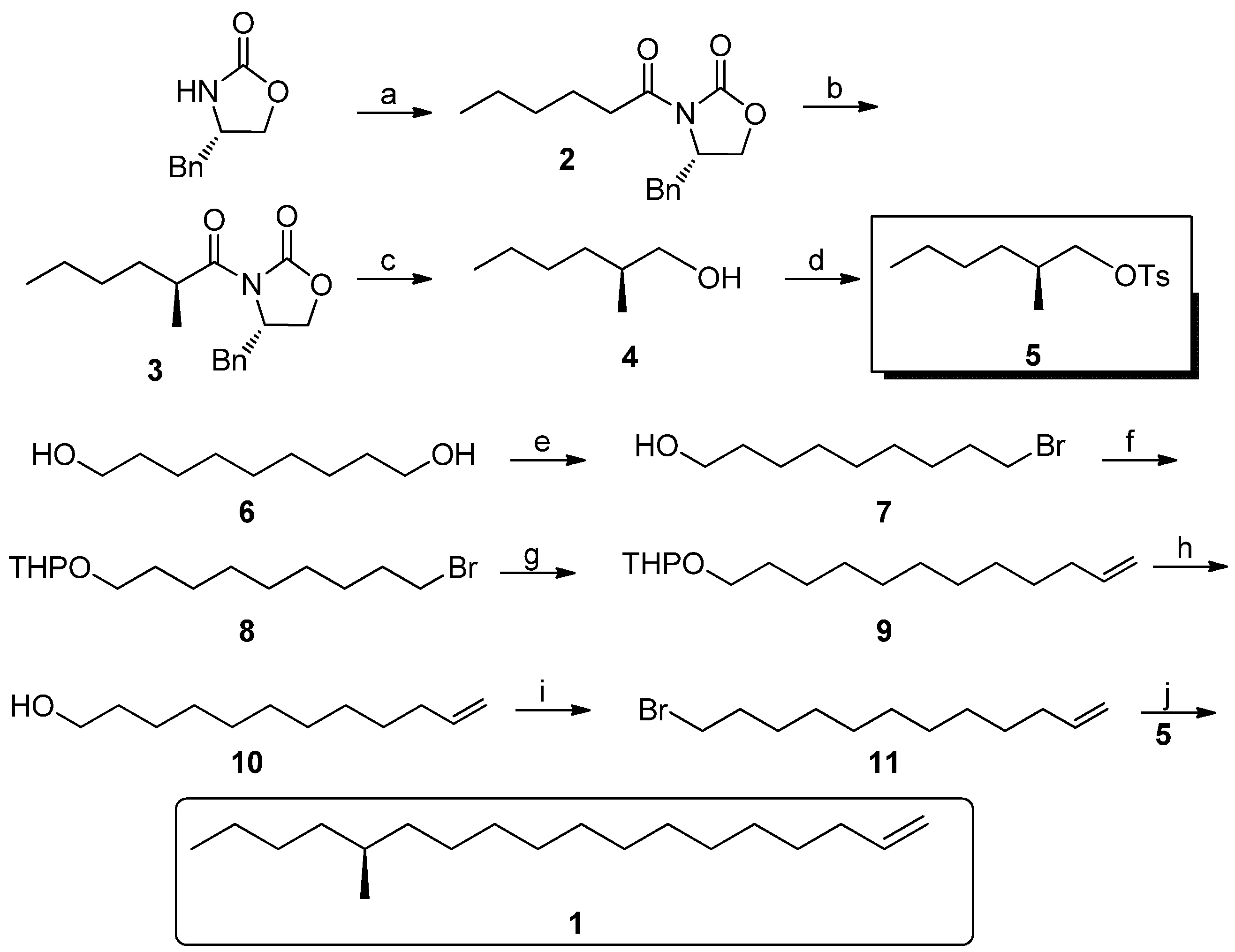
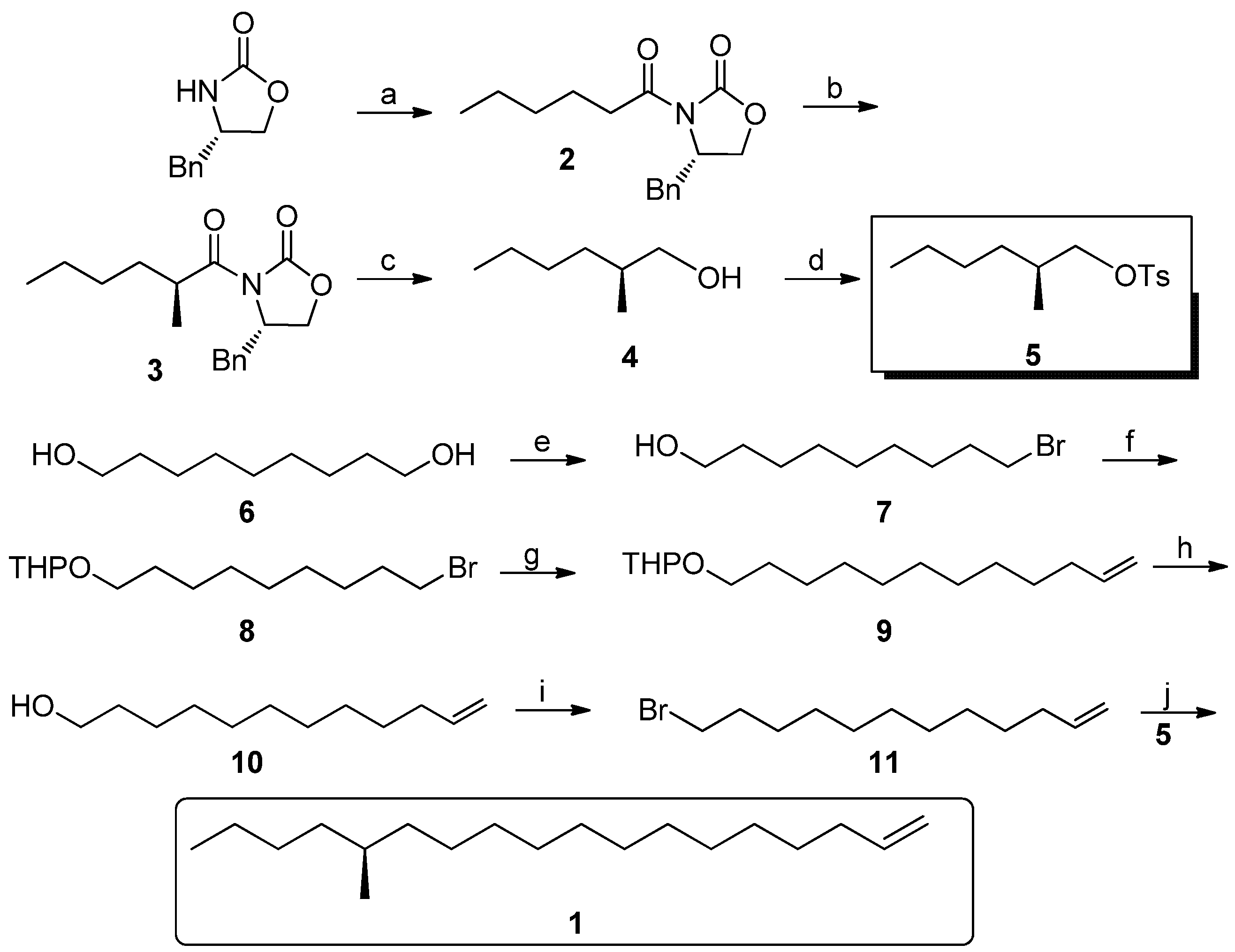
(S)-4-Benzyl-3-hexanoyloxazolidin-2-one (2). At −78 °C, to a solution of (S)-4-benzyloxazolidin-2-one (5.32 g, 0.03 mol) in anhydrous THF (50 mL) was added BuLi (0.036 mol), then freshly distilled hexanoyl chloride (4.63 g, 0.033 mol) in THF (25 mL) was added after half an hour. The mixture was gradually warmed to 25 °C and maintained for 12 h. After completion, the reaction was quenched by addition of saturated aqueous NH4Cl, and the volatiles were evaporated. The residue was extracted with ethyl acetate (100 mL × 3), and the combined organic layers were washed with dilute NaOH and brine, and dried over anhydrous MgSO4. Then the extract was evaporated, and purified by column chromatography (15–25% gradient, EtOAc-hexane) to give 2 as a yellowish oil (5.78 g, 70%). = +97.6° (c 0.36, MeOH); 1H-NMR (500 MHz, CDCl3): 0.90 (br s, 3H), 1.34 (br s, 4H), 1.67 (br s, 2H), 2.73–2.77 (m, 1H), 2.84–2.93 (m, 2H), 3.22 (d, J = 13.3, 1H), 4.10 (s, 2H), 4.62 (s, 1H), 7.17–7.28 (m,5H); 13C-NMR (125 MHz, CDCl3): 13.9, 22.4, 23.9, 31.3, 35.4, 37.8, 55.0, 66.1, 127.2, 128.8, 129.4, 135.5, 153.4, 173.2.
(S)-4-Benzyl-3-((S)-2-methylhexanoyl)oxazolidin-2-one (3). To a cooled (−78 °C) solution of LDA (0.051 mol) was added a solution of compound 2 (11.7 g, 43 mmol) in THF (90 mL). After 2 h, dry MeI (13.3 mL, 0.213 mol) was introduced through a dropping funnel at this temperature. The reaction mixture was allowed to stir at −78 °C for 3 h before being warmed to 25 °C and maintained overnight. The reaction was quenched with saturated aqueous NH4Cl (50 mL), and the aqueous layer was extracted with three 50 mL portions of EtOAc. The combined organic extracts were dried (MgSO4), concentrated in vacuo, and chromatographed (15–25% gradient, EtOAc-hexane) to provide of pure 3 (9.7 g, 78%): = +104.4° (c 0.28, MeOH); 1H-NMR (500 MHz, CDCl3) 0.89 (t, J = 6.8, 3H), 1.22 (d, J = 6.9, 3H), 1.27–1.40 (m, 4H), 1.40–1.44 (m, 1H), 1.72–1.76 (m, 1H), 2.77 (dd, J = 13.3, 9.6, 1H), 3.26 (dd, J = 13.3, 2.9, 1H), 3.38–3.73 (m, 1H), 4.15–4.21 (m, 2H), 4.65–4.69 (m, 1H), 7.21–7.34 (m, 5H); 13C-NMR (125 MHz, CDCl3): 14.0, 17.4, 22.7, 29.5, 33.2, 37.7, 37.9, 55.4, 66.0, 127.3, 128.9, 129.5, 135.4, 153.1, 177.4. ESI-MS: m/z: 290 (M+H), 276.
(S)-2-Methylhexyl 4-methylbenzenesulfonate (5). To a cooled (0 °C) suspension of LiAlH4 (606 mg, 16 mmol) in anhydrous THF (20 mL) was added a solution of the imide 3 (1.4 g, 5 mmol) in THF (30 mL) over a 15 min period. After an additional 30 min of stirring, the cold (0 °C) reaction was slowly quenched with water (0.6 mL), then 10% aqueous NaOH (1.2 mL) and water (1.8 mL) to precipitate the aluminum salts, which were then filtered. The filtrate was dried (MgSO4), and the solution was concentrated in vacuo. Because of the volatility of the product, the crude product was used directly for the next step. To the cooled (−10 °C) solution of the above alcohol 4 in pyridine (20 mL) was added 4-methylbenzene-1-sulfonyl chloride (1.0 g, 5.1 mmol). After the solution was stirred an additional 30 min at −10 °C, the reaction mixture was slowly warmed to 20 °C for an additional 30 min period. The reaction was then quenched with brine (30 mL), and the aqueous layer was extracted with three 30 mL portions of CH2Cl2. The combined organic extracts were washed with brine, diluted HCl and saturated aqueous CuSO4 and dried over MgSO4, concentrated in vacuo, and chromatographed (10% EtOAc-hexane) to provide pure 5 (0.75 g, 55% for two steps) = −2.6° (c 1.67, CH2Cl2); 1H-NMR (500 MHz, CDCl3) 0.85 (t, J = 7.0, 3H), 0.87 (d, J = 6.7, 3H), 1.09–1.13 (m, 2H), 1.20–1.25 (m, 4H), 1.73–1.79 (m, 1H), 2.45 (s, 3H), 3.80 (dd, J = 6.5, 6.4, 1H), 3.88 (dd, J = 5.6, 5.7, 1H), 7.34 (d, J = 8.1, 2H), 7.787(d, J = 8.1, 2H). ESI-MS: m/z: 270 (M+H), 173, 155, 91.
9-Bromononan-1-ol (7). A mixture of nonane-1,9-diol 6 (24 g, 0.15 mol), a catalytic amount of iodine (0.5 g, 2 mmol) and 40% HBr (33 mL) in toluene (240 mL) was heated to reflux and the water formed was separated continuously for 30 h. Then the mixture was washed successively with water, aqueous NaOH, HCl, water and brine. The organic phase was concentrated in vacuo, and chromatographed (10% EtOAc-hexane) to give 30 g of pure 7 as a colorless oil in 89% yield. 1H-NMR (500 MHz, CDCl3) 1.31–1.36 (m, 8H), 1.41–1.43 (m, 2H), 1.49 (br s, 1H), 1.53–1.59 (m, 2H), 1.82–1.88 (m, 2H), 3.41 (t, J = 6.85, 2H), 3.63(t, J = 6.62, 2H). ESI-MS: m/z: 223 (M+H), 207.
2-((9-Bromononyl)oxy)tetrahydro-2H-pyran (8). To a solution of 9-bromononan-1-ol (7, 25 g, 0.112 mol) in anhydrous CH2Cl2 (150 mL) was added 4-methylbenzenesulfonic acid (0.3 g) and freshly distilled 3, 4-dihydro-2H-pyran (11.24 mL, 0.123 mol) in CH2Cl2 (10 mL), at 0 °C. After addition, the mixture was allowed to stir for 5 h at room temperature and monitored by TLC. Then the solution was diluted by addition of another portion of CH2Cl2 (150 mL) and then the reaction mixture was washed with saturated aqueous NaHCO3 and brine, dried over anhydrous MgSO4 and then concentrated in vacuo and chromatographed to afford 31.6 g (92% yield) of pure 8 as a colorless oil. 1H-NMR (500 MHz, CDCl3): 1.36 (br s, 8H), 1.36–1.42 (m, 2H), 1.50–1.60 (m, 6H), 1.69–1.71 (m, 1H), 1.80–1.88 (m, 3H), 3.36–3.42 (m, 3H), 3.48–3.51 (m, 1H), 3.36–3.75 (m, 1H), 3.85–3.89 (m, 1H), 4.57 (t, J = 3.4, 1H); 13C-NMR (125 MHz, CDCl3) 19.7, 25.5, 26.2, 28.2, 28.7, 29.4, 29.4, 30.8, 32.8, 34.0, 62.3, 67.6, 98.8. EI-MS: m/z: 305 (M-H).
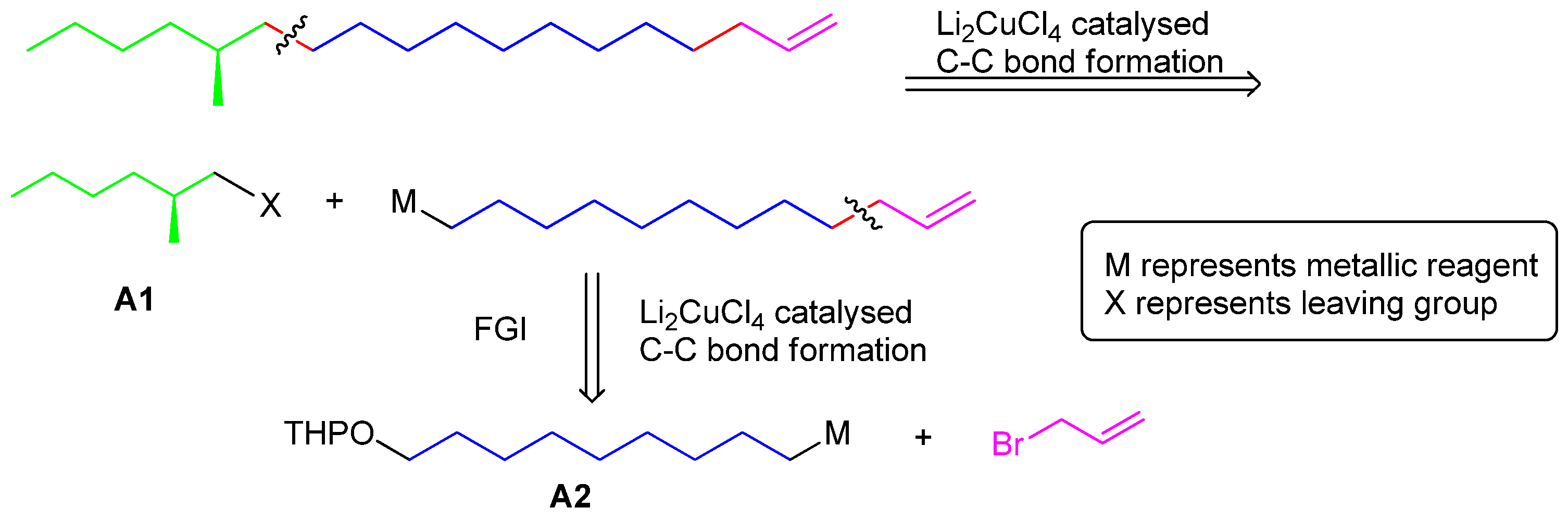
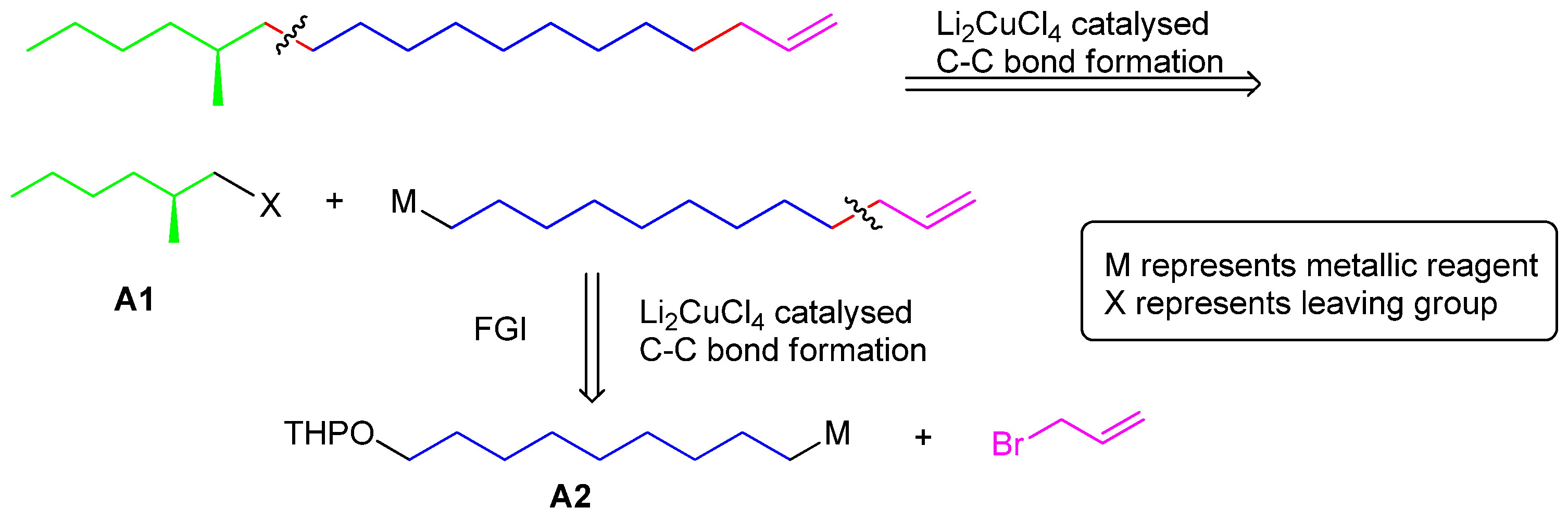
2-(Dodec-11-en-1-yloxy)tetrahydro-2H-pyran (9). Under a nitrogen atmosphere at 20 °C, Mg turnings (56 mmol, 1.37 g), a catalytic amount of iodine and anhydrous THF (25 mL) were placed in a dry 500 mL three-necked round-bottomed flask equipped with a condenser and a dropping funnel. A solution of compound 8 (13 g, 42 mmol) in THF (150 mL) was added dropwise to the above mixture (approx. 20 min). Then the mixture was allowed to reflux for another 5 h. Another dry 500 mL three-necked round-bottomed flask was charged with allyl bromide (84 mmol), NMP (4 mL) and Li2CuCl4 (0.3 M in THF, 4 mL). The above Grignard reagent was transferred to this flask through a double-ended needle with stirring. After transfer, the reaction mixture was warmed to 85 °C for 12 h. After cooling, the reaction mixture was quenched by dropwise addition of saturated aqueous NH4Cl solution. The THF was recovered on a rotavapor and the residue was extracted with ethyl acetate (100 mL × 3). The combined organic extracts were dried (MgSO4), concentrated in vacuo, and chromatographed (15–25% gradient, EtOAc-hexane) to provide 7.5 g (66%) of pure 9 as a colorless oil. 1H-NMR (500 MHz, CDCl3) 1.27–1.30 (m, 10 H), 1.40–1.57 (m, 4H), 1.58–1.94 (m, 8H), 1.95–2.13 (m, 2H), 3.37–3.38 (m, 2H), 3.68–3.73 (m, 2H), 4.57 (t, J = 6.8, 1H), 4.91–4.96 (m, 1H), 5.01–5.23 (m, 1H), 5.74–5.99 (m, 1H), 13C-NMR (125 MHz, CDCl3) some signals were overlapped. 139.2, 114.1, 98.8, 67.6, 62.2, 31.8, 30.8, 29.8, 29.6, 29.5, 29.3, 26.2, 25.5. EI-MS: m/z: 267 (M-H), 191.
Dodec-11-en-1-ol (10). To a solution of compound 9 (7.0 g, 26 mmol) in MeOH (100 mL) was added a catalytic amount of PPTS with stirring. Then the reaction mixture was heated to 50 °C overnight and monitored by TLC. After completion, the methanol was removed on a rotavapor, And the residue was purified by column chromagraphy (15–25% gradient, EtOAc-hexane) to give dodec-11-en-1-ol (4.3 g, 96% yield) as a colorless oil. 1H-NMR (500 MHz, CDCl3) 1.27–1.30 (m, 14H) 1.52–1.59 (m, 2H), 2.01–2.06 (m, 2H), 2.10 (br s, 1H), 3.62 (t, J = 6.6, 2H), 4.92 (dd, J = 12.1, 1.6, 1H), 4.98 (dd, J = 17.1, 1.6, 1H), 5.75–5.83 (m, 1H); 13C-NMR (125 MHz, CDCl3) 139.2, 114.2, 62.90, 33.8, 32.7, 31.9, 29.7, 29.5, 29.4, 29.3, 29.1, 28.9. EI-MS: m/z: 184.
12-Bromododec-1-ene (11). A dry 250 mL three-necked round-bottomed flask was charged with PPh3 (14.7 g, 56 mmol), dichloromethane (80 mL) and CBr4 (22.2 g, 67 mmol) and the mixture was allowed to stir for 20 min. To the above solution was added compound 10 portionwise at −10 °C. After completion, the volatiles were removed on a rotavapor, and the residue was purified by column chromagraphy directly to give 12-bromododec-1-ene (11, 90% yield) as a colorless oil. 1H-NMR (500 MHz, CDCl3) 1.30–1.35 (m, 14H) 1.35–1.45 (m, 2H), 1.83–1.90 (m, 1H), 2.02–2.08 (m, 1H), 3.62 (t, J = 6.8, 2H), 4.93–4.96 (m, 1H), 5.00 (ddd, J = 21.4, 4.6, 1.6, 1H), 5.75–5.83 (m, 1H); 13C-NMR (125 MHz, CDCl3) 139.2, 114.1, 33.9, 33.8, 32.7, 31.9, 29.7, 29.5, 29.4, 29.3, 29.1, 28.9. EI-MS: m/z: 247 (M-H), 245, 191, 189.
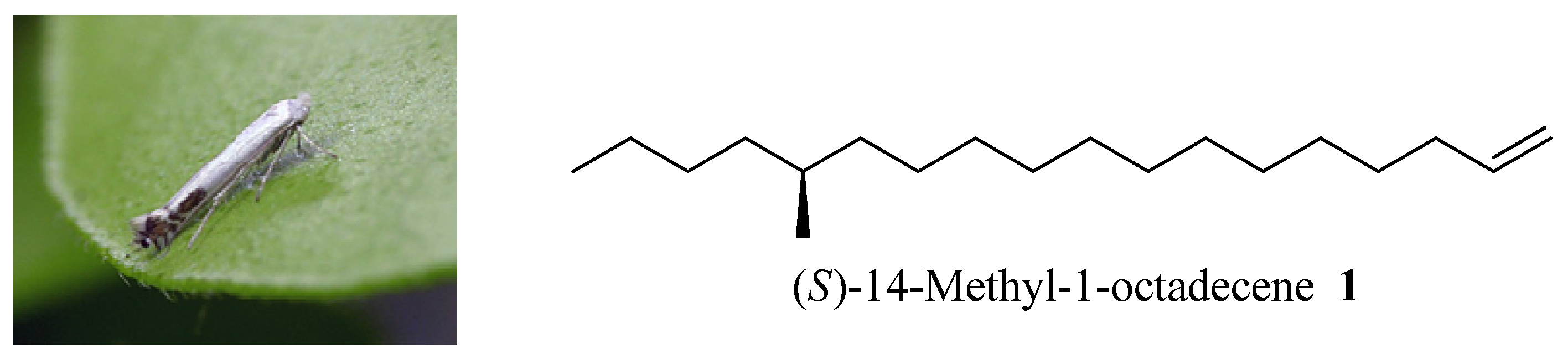
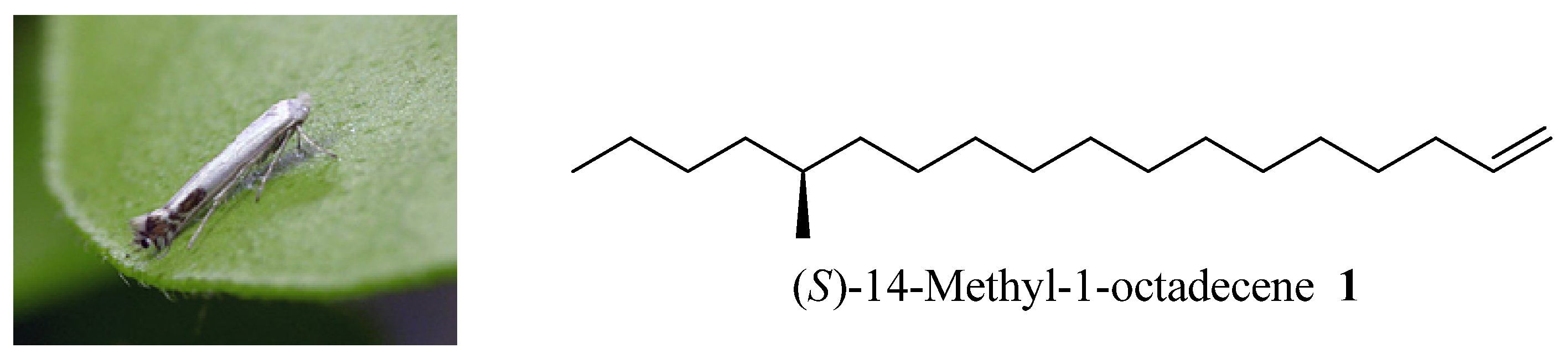
(S)-14-Methyloctadec-1-ene (1). Under nitrogen atmosphere at 20 °C, Mg turnings (0.2 g, 8.4 mmol), a catalytic amount of iodine, and anhydrous THF (5 mL) were placed in a dry 250 mL three-necked round-bottomed flask equipped with a condenser and a dropping funnel. A solution of compound 11 (1.6 g, 6.5 mmol) in THF (50 mL) was added dropwise to the above mixture (approx. 20 min). Then the mixture was allowed to reflux for another 5 h. Another dry 500 mL three-necked round-bottomed flask was charged with the chiral intermediate 5 (1.8 g, 6.5 mmol), NMP (2 mL) and Li2CuCl4 (0.3 M in THF, 2 mL). The above Grignard reagent was transferred to this flask through a double-ended needle with stirring. After transfer, the reaction mixture was warmed to 85 °C for 3 days. After cooling, the reaction mixture was quenched by dropwise addition of saturated aqueous NH4Cl solution. The THF was recovered on a rotavapor and the residue was extracted with ethyl acetate (100 mL × 3). The combined organic extracts were dried (MgSO4), concentrated in vacuo, and chromatographed to provide 0.8 g (65%) of pure 1 as a colorless oil. Colorless liquid, = +1.17° (c 1.82, n-hexane). 1H-NMR (300 MHz, CDCl3): 6.05–5.83 (1H, m), 5.03–4.90 (2H, m), 2.08–2.00 (2H, m), 1.57–1.09 (27H, m), 0.93 (6H, m). 13C-NMR (75 MHz, CDCl3): 139.3, 114.1, 41.6, 35.2, 34.6, 33.8, 31.9, 29.7, 29.6, 29.5, 29.3, 29.17, 29.0, 22.7, 14.1. EI-MS: m/z: 266 (M+, 3%), 43 (100%).

{kind=link}
{kind=link}
{kind=link}