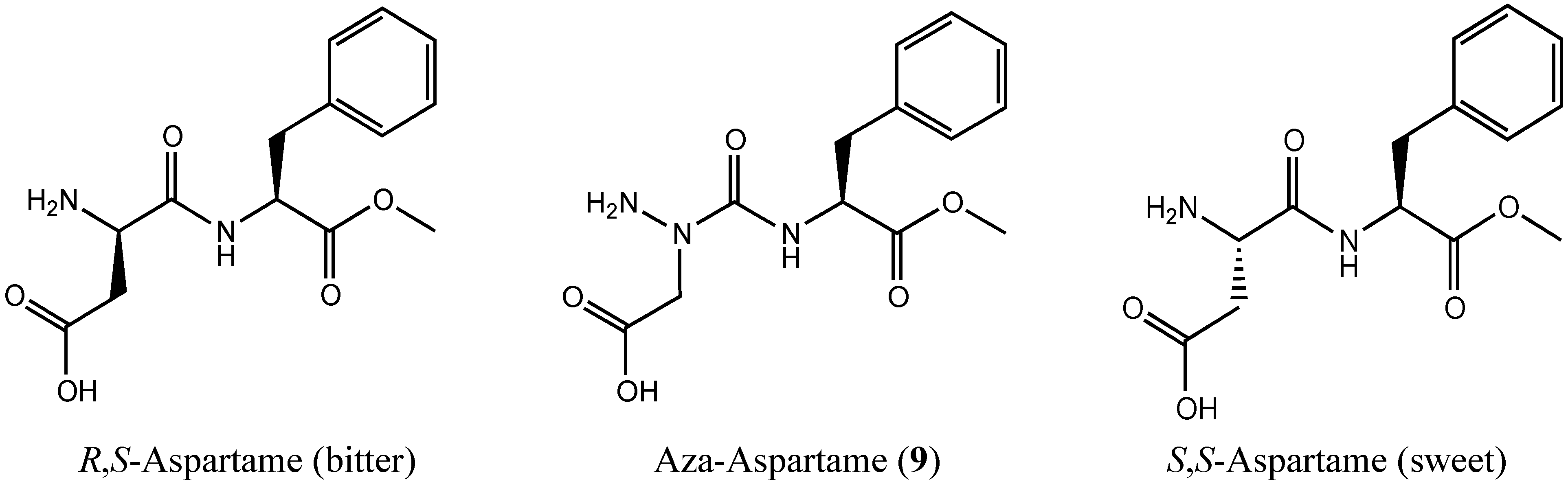
Examination of the Potential for Adaptive Chirality of the Nitrogen Chiral Center in Aza-Aspartame


Abstract
: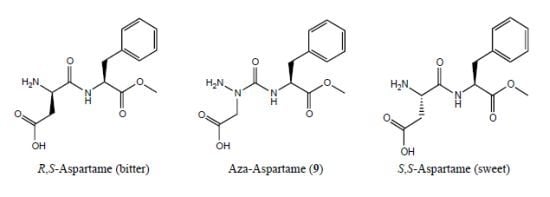
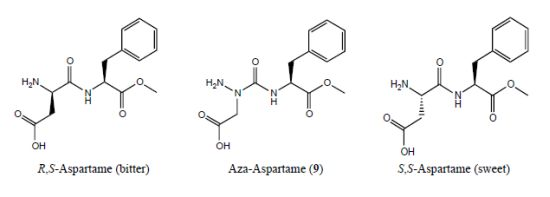
1. Introduction
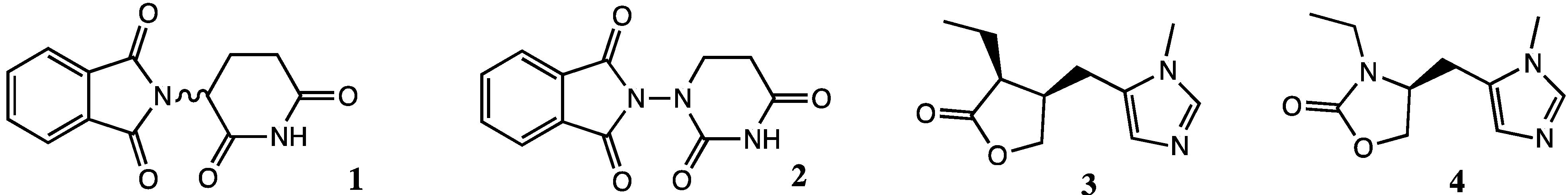
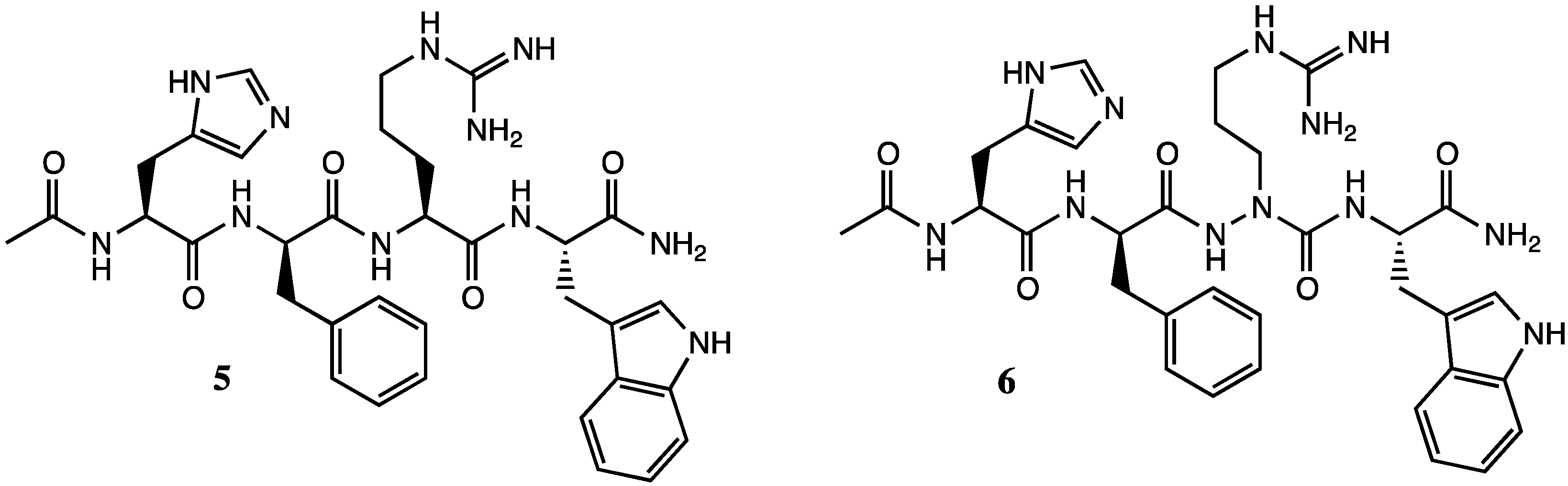
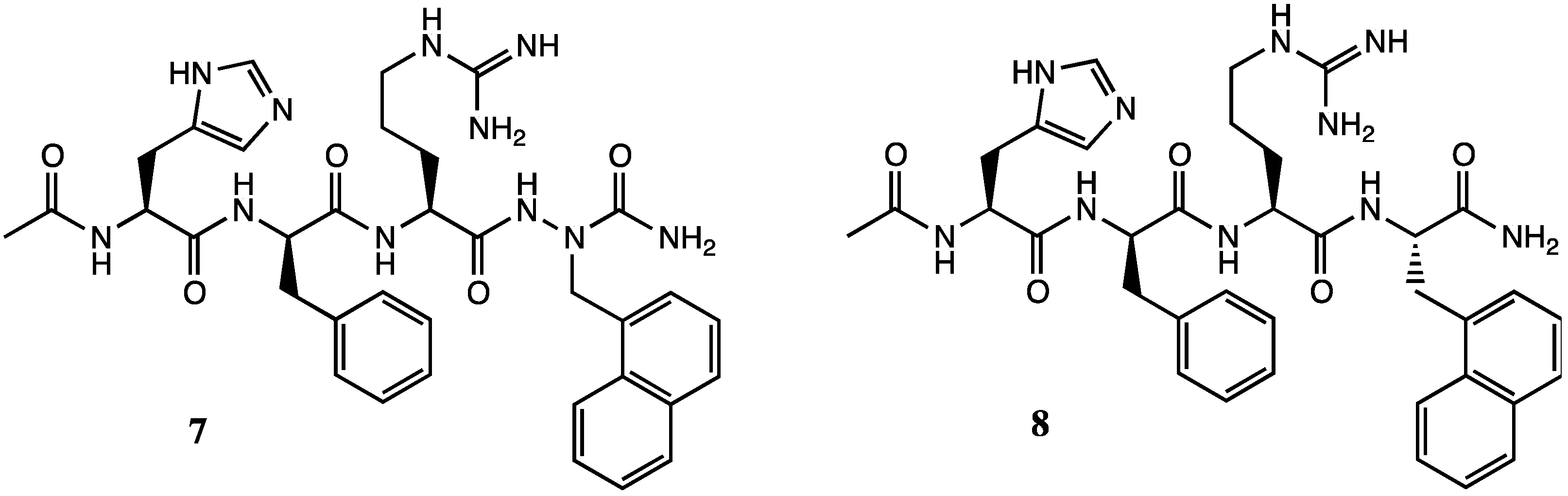
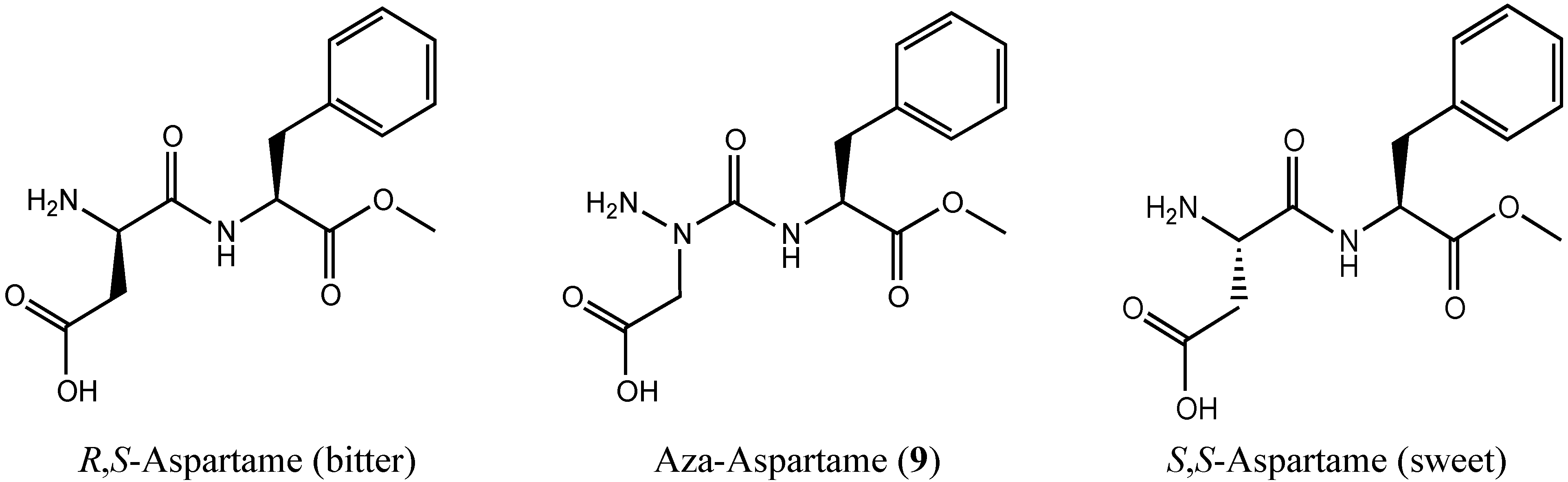
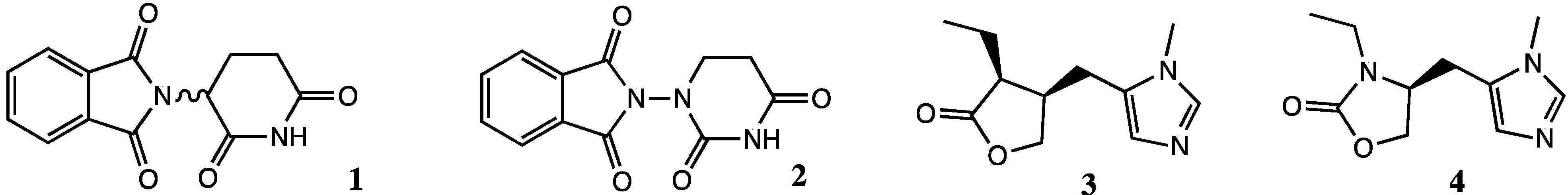
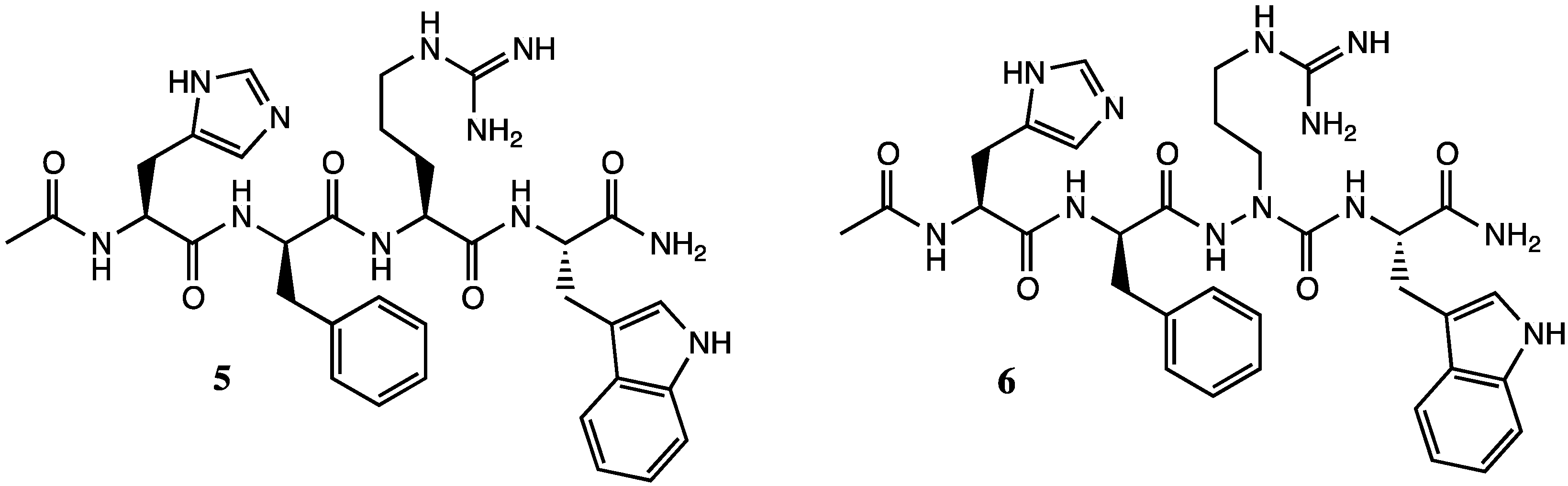
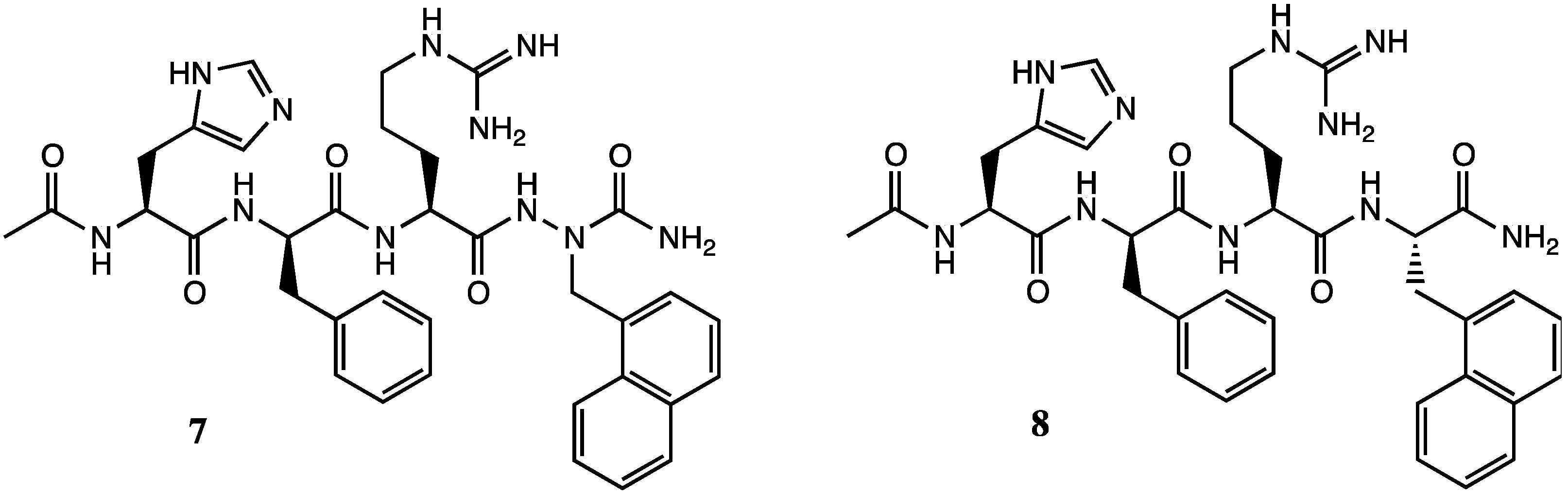
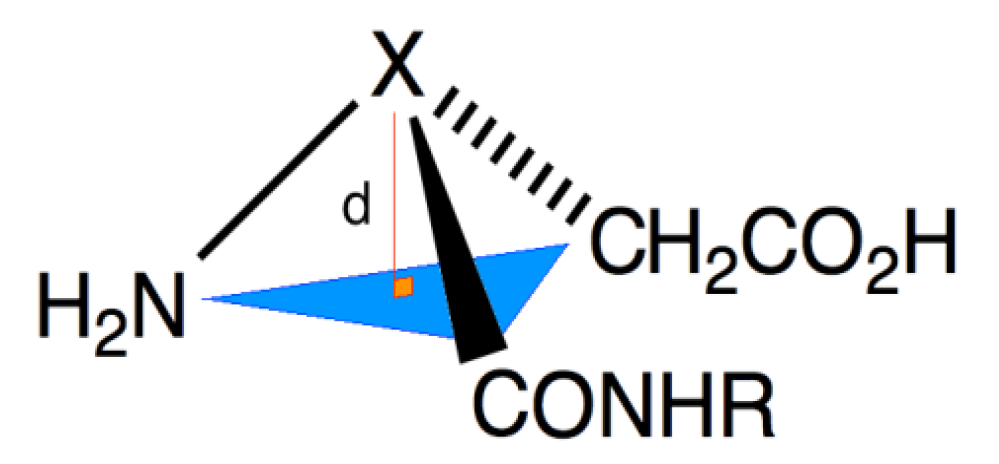
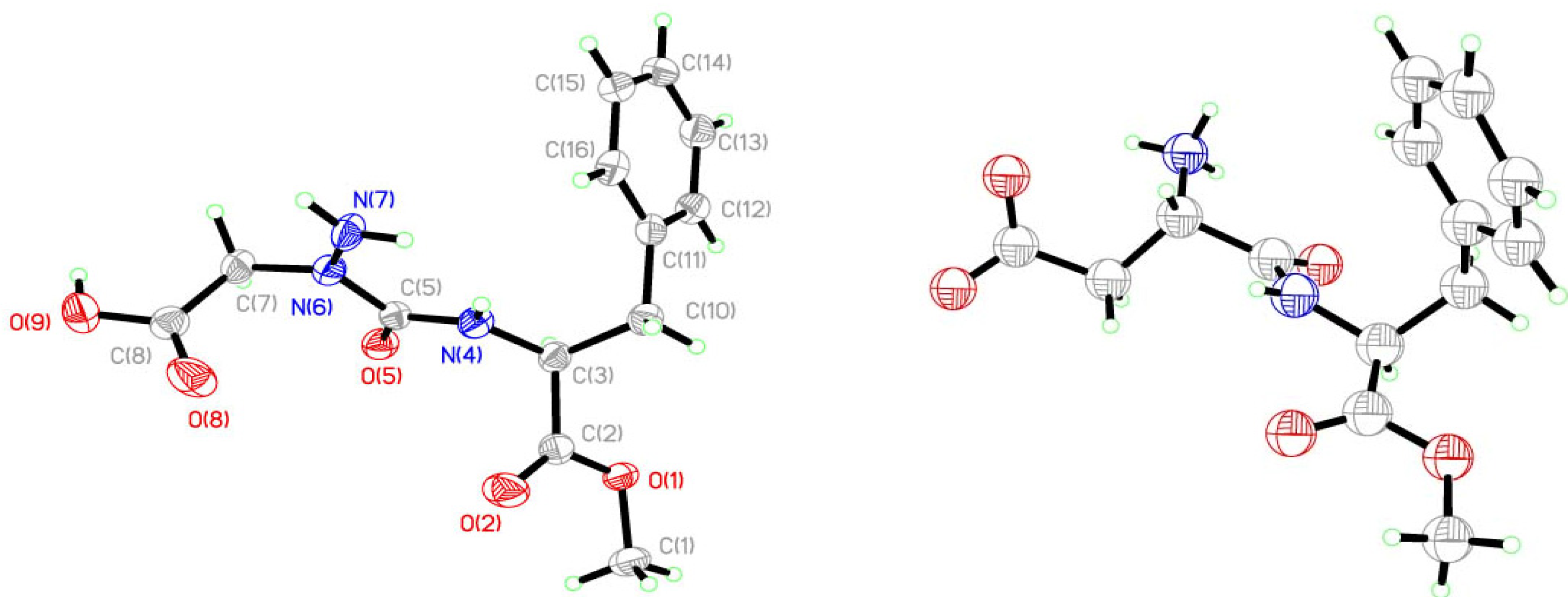
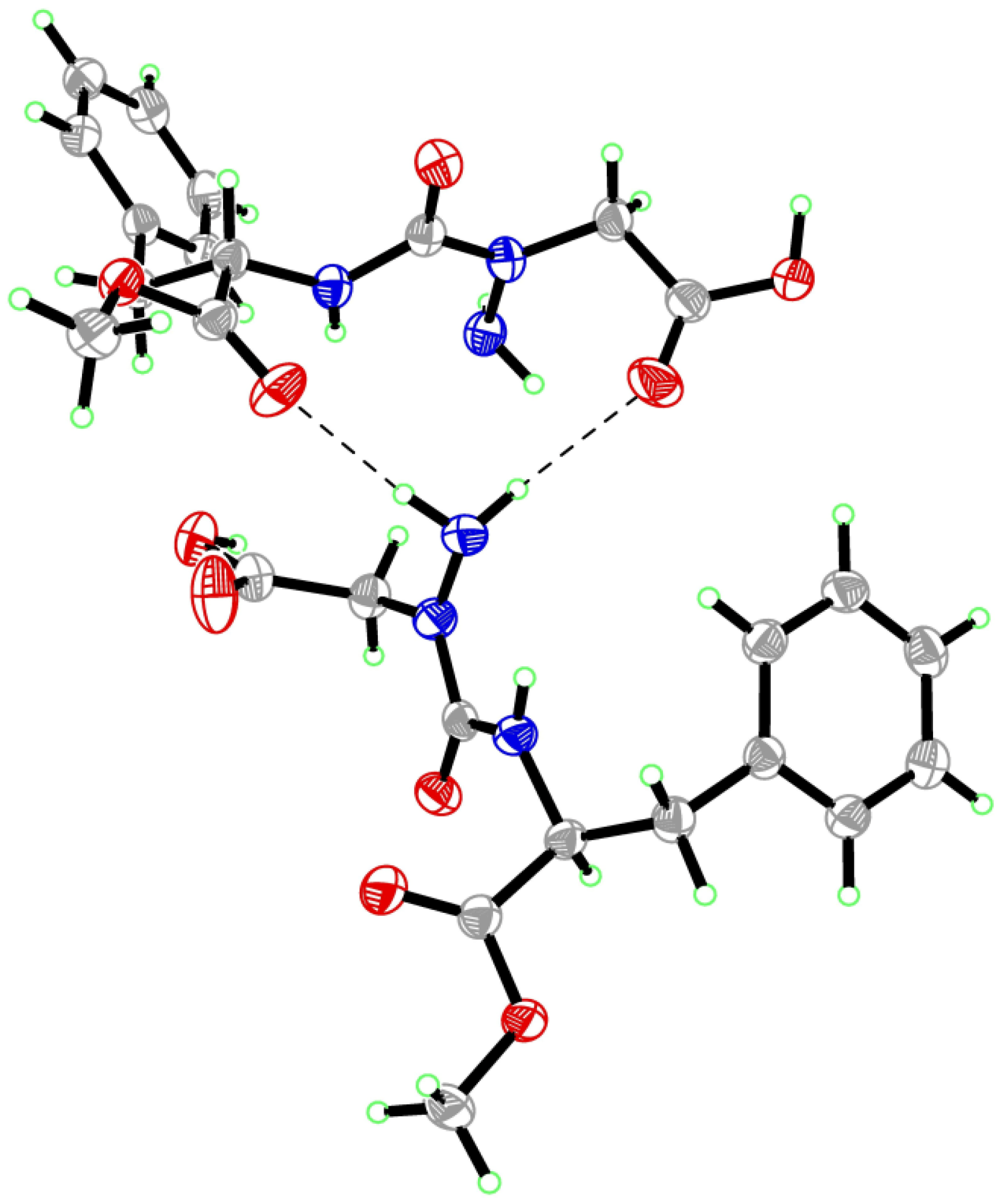
2. Results and Discussion
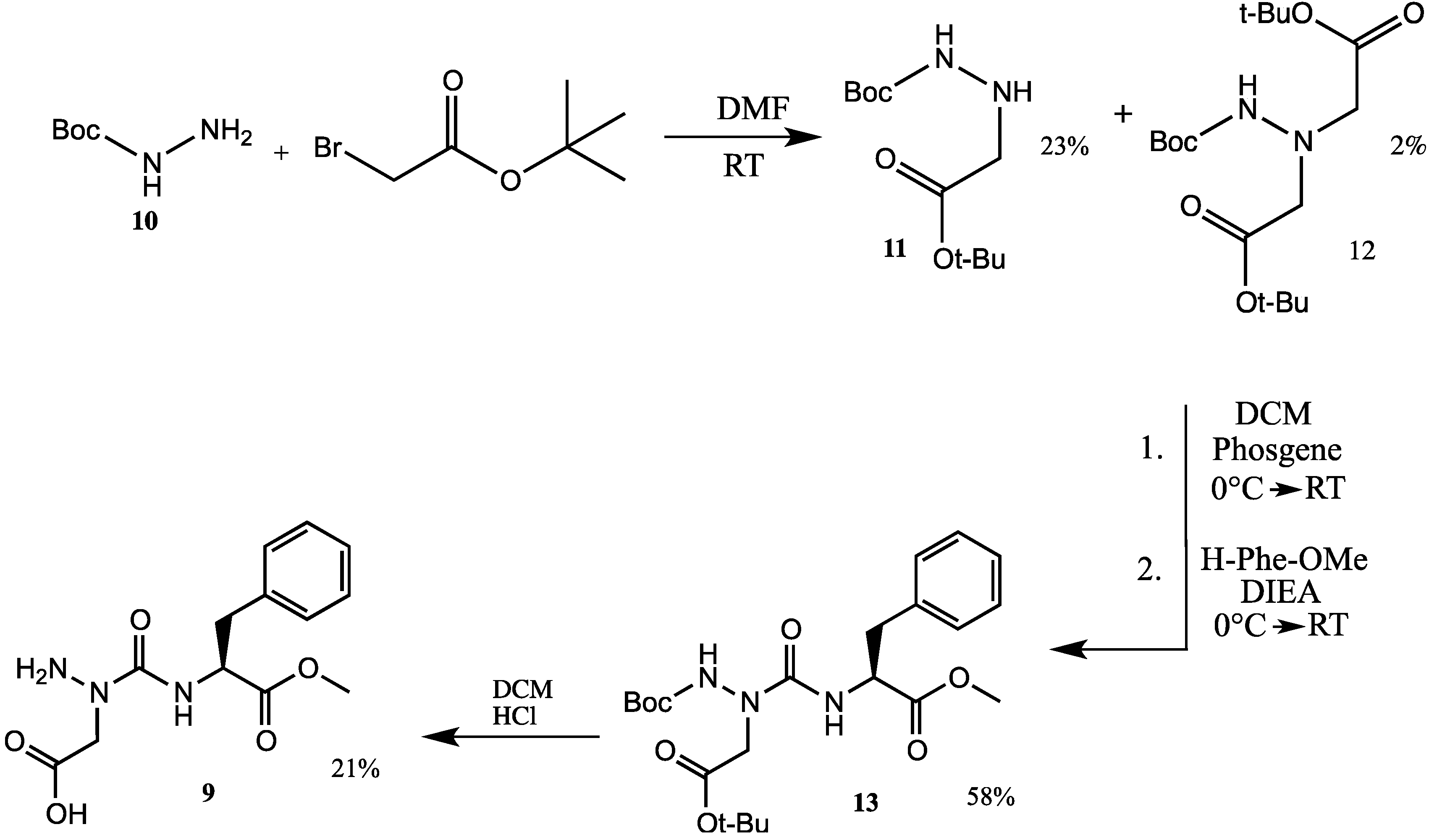
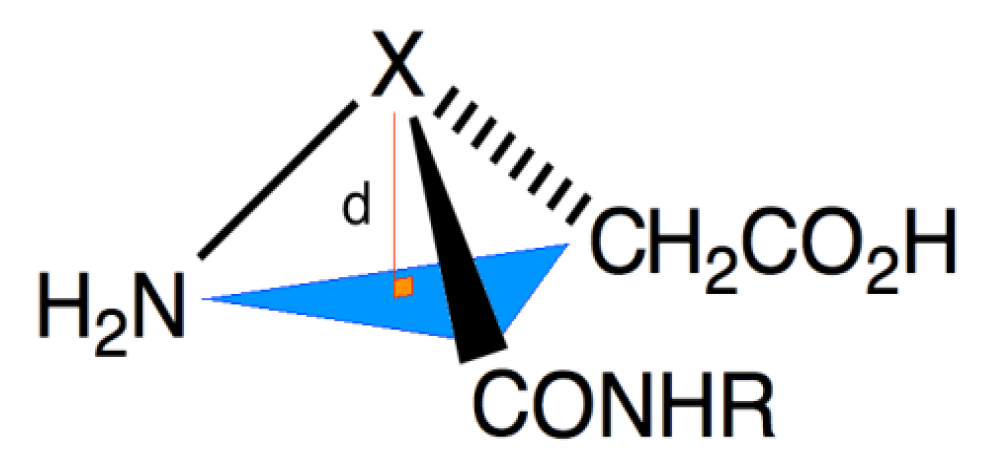
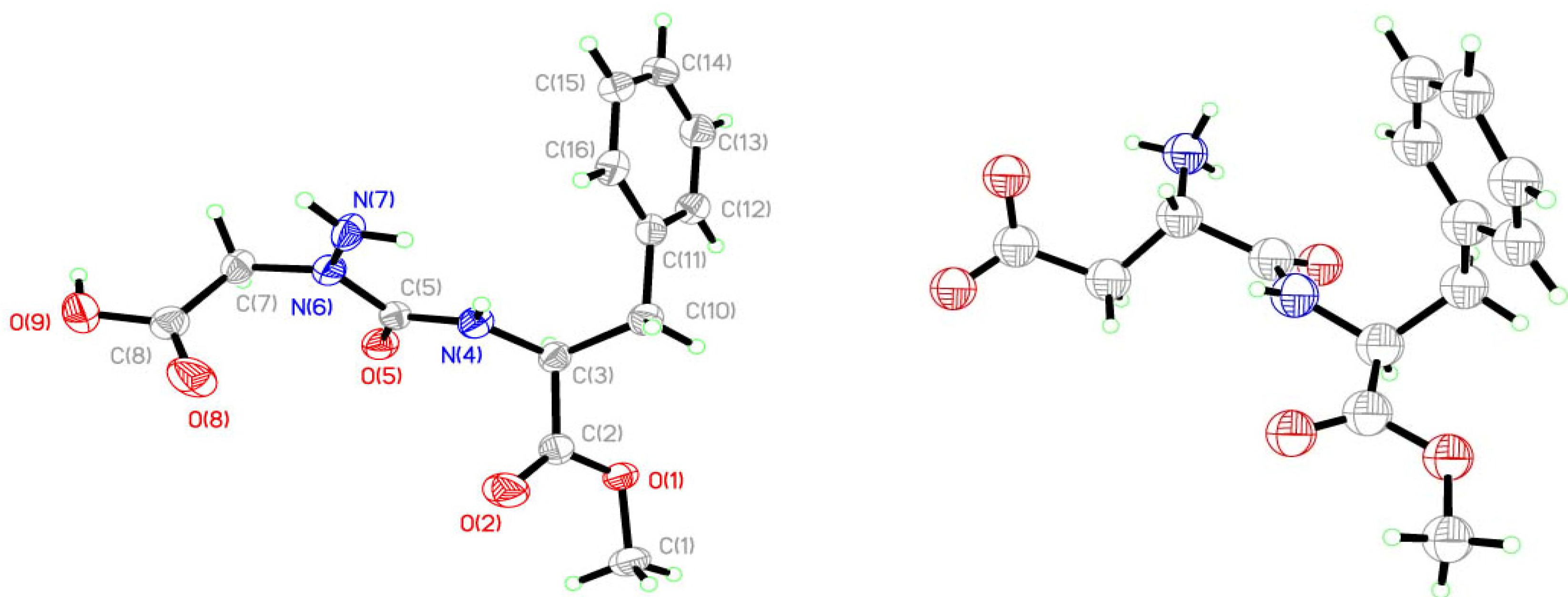

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
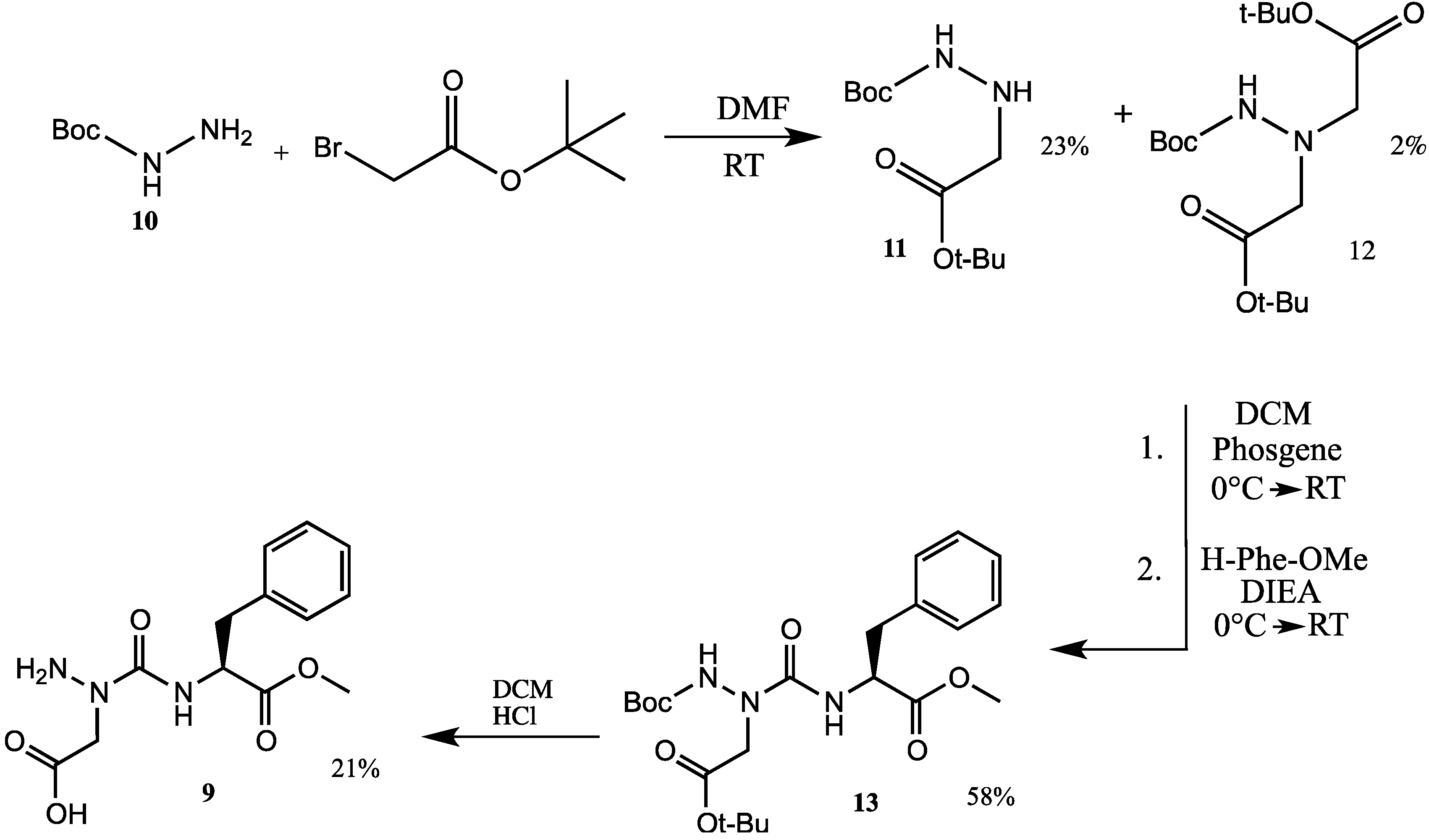{kind=link}
| Bonds | Lengths [Å] | Angle [°] | |
|---|---|---|---|
| C(1)-O(1) | 1.4441(14) | C(2)-O(1)-C(1) | 115.63(9) |
| O(1)-C(2) | 1.3275(14) | O(2)-C(2)-O(1) | 124.51(11) |
| C(2)-O(2) | 1.2030(14) | O(2)-C(2)-C(3) | 125.36(11) |
| C(2)-C(3) | 1.5291(16) | O(1)-C(2)-C(3) | 110.13(9) |
| C(3)-N(3) | 1.4407(15) | N(4)-C(3)-C(2) | 111.04(9) |
| C(3)-C(10) | 1.5465(16) | N(4)-C(3)-C(10) | 109.80(10) |
| N(4)-C(5) | 1.3426(16) | C(2)-C(3)-C(10) | 110.45(9) |
| C(5)-O(5) | 1.2514(15) | C(5)-N(4)-C(3) | 123.47(10) |
| C(5)-N(6) | 1.3513(15) | O(5)-C(5)-N(4) | 123.55(11) |
| N(6)-N(7) | 1.4089(15) | O(5)-C(5)-N(6) | 120.53(11) |
| N(6)-C(7) | 1.4434(15) | N(4)-C(5)-N(6) | 115.88(10) |
| C(7)-C(8) | 1.5225(16) | C(5)-N(6)-N(7) | 117.97(10) |
| C(8)-O(8) | 1.2029(16) | C(5)-N(6)-C(7) | 121.27(10) |
| C(8)-O(9) | 1.3237(15) | N(7)-N(6)-C(7) | 120.31(10) |
| C(10)-C(11) | 1.5084(16) | N(6)-C(7)-C(8) | 111.34(10) |
| C(11)-C(16) | 1.3951(17) | O(8)-C(8)-O(9) | 121.34(12) |
| C(11)-C(12) | 1.3976(17) | O(8)-C(8)-C(7) | 123.01(11) |
| C(12)-C(13) | 1.3885(18) | O(9)-C(8)-C(7) | 115.64(10) |
| C(13)-C(14) | 1.3832(19) | C(11)-C(10)-C(3) | 112.66(9) |
| C(14)-C(15) | 1.3895(19) | C(16)-C(11)-C(12) | 118.28(11) |
| C(15)-C(16) | 1.3892(18) | C(16)-C(11)-C(10) | 122.30(10) |
| C(12)-C(11)-C(10) | 119.42(10) | ||
| C(13)-C(12)-C(11) | 120.94(12) | ||
| C(14)-C(13)-C(12) | 120.15(12) | ||
| C(13)-C(14)-C(15) | 119.66(12) | ||
| C(16)-C(15)-C(14) | 120.18(12) | ||
| C(15)-C(16)-C(11) | 120.77(11) |
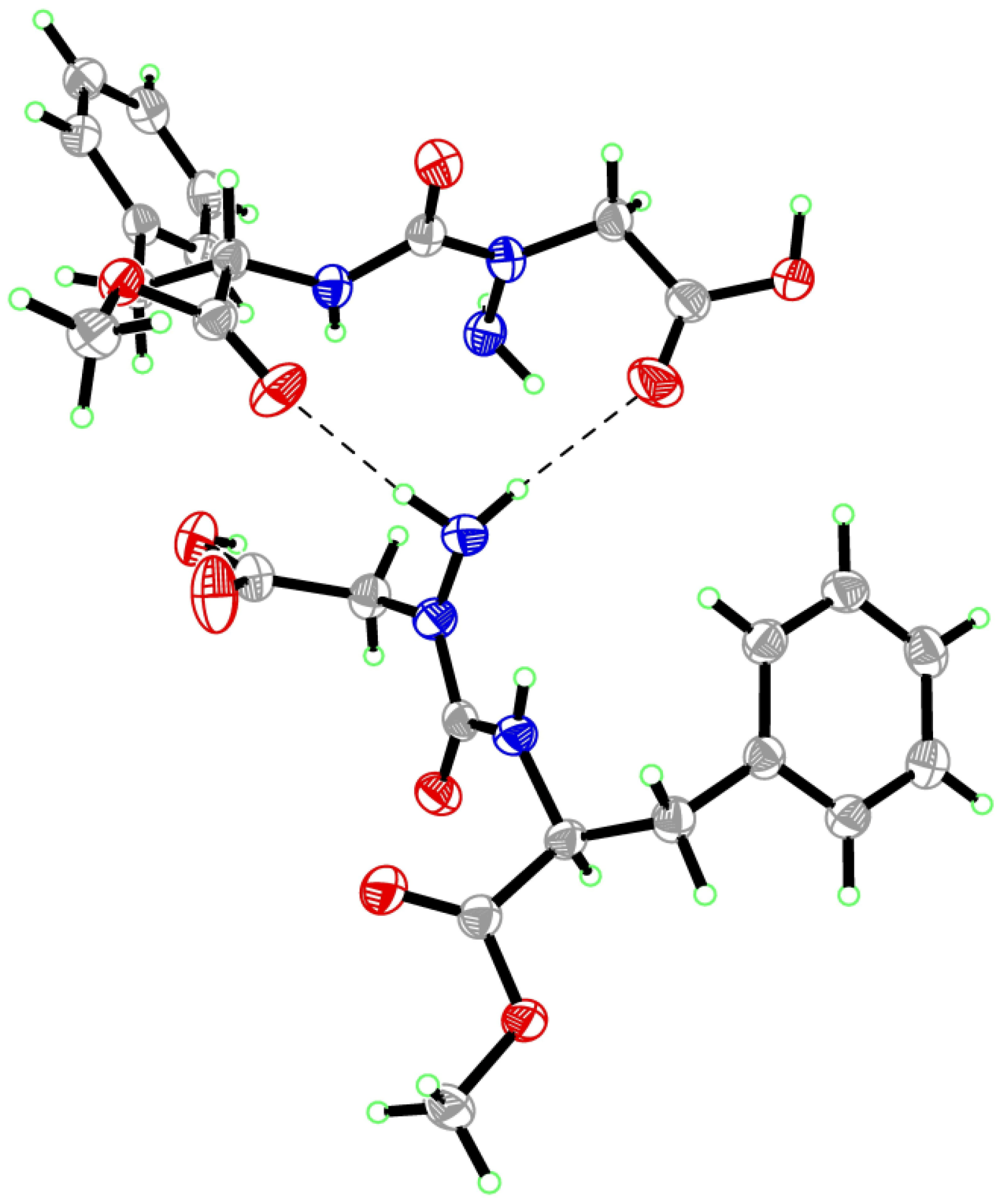
3. Experimental
General
4. Conclusions
Acknowledgments
Conflicts of Interest
Reference
- Cox, C.; Lectka, T. Solvent effect on the barrier to rotation in carbamates. J. Org. Chem. 1998, 63, 2426–2427. [Google Scholar] [CrossRef]
- Noguchi, T.; Shimazawa, R.; Nagasawa, K.; Hashimoto, Y. Thalidomide and its analogues as cyclooxygenase inhibitors. Bioorg. Med. Chem. Lett. 2002, 12, 1043–1046. [Google Scholar] [CrossRef]
- Sauerberg, P.; Chen, J.; WoldeMussie, E.; Rapoport, H. Cyclic carbamate analogues of pilocarpine. J. Med. Chem. 1989, 32, 1322–1326. [Google Scholar] [CrossRef]
- Drake, M.V.; O’Donnell, J.J.; Polansky, J.R. Isopilocarpine binding to muscarinic cholinergic receptors. J. Pharm. Sci. 1986, 75, 278–279. [Google Scholar] [CrossRef]
- Proulx, C.; Sabatino, D.; Hopewell, R.; Spiegel, J.; García-Ramos, Y.; Lubell, W.D. Azapeptides and their therapeutic potential. Future Med. Chem. 2011, 3, 1139–1164. [Google Scholar] [CrossRef]
- Boeglin, D.; Xiang, Z.; Sorenson, N.B.; Wood, M.S.; Haskell-Luevano, C.; Lubell, W.D. Aza-scanning of the potent melanocortin receptor agonist Ac-His-D-Phe-Arg-Trp-NH2. Chem. Biol. Drug Des. 2006, 67, 275–283. [Google Scholar] [CrossRef]
- Temussi, P.A. The good taste of peptides. J. Pept. Sci. 2012, 18, 73–82. [Google Scholar] [CrossRef]
- Still, W.C.; Kahn, M.; Mitra, A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978, 43, 2923–2925. [Google Scholar] [CrossRef]
- Guguta, C.; Meekes, H.; de Gelder, R. Crystal structure of aspartame anhydrate from powder diffraction data. Cryst. Growth Des. 2006, 6, 2686–2692. [Google Scholar] [CrossRef]
- The Cabridg (CCDC 972051). Available online: www.ccdc.cam.ac.uk (accessed on 26 November 2013).
- Sample Availability: miligram samples of compounds 9 may be available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bouayad-Gervais, S.H.; Lubell, W.D. Examination of the Potential for Adaptive Chirality of the Nitrogen Chiral Center in Aza-Aspartame. Molecules 2013, 18, 14739-14746. https://doi.org/10.3390/molecules181214739
Bouayad-Gervais SH, Lubell WD. Examination of the Potential for Adaptive Chirality of the Nitrogen Chiral Center in Aza-Aspartame. Molecules. 2013; 18(12):14739-14746. https://doi.org/10.3390/molecules181214739
Chicago/Turabian StyleBouayad-Gervais, Samir H., and William D. Lubell. 2013. "Examination of the Potential for Adaptive Chirality of the Nitrogen Chiral Center in Aza-Aspartame" Molecules 18, no. 12: 14739-14746. https://doi.org/10.3390/molecules181214739
APA StyleBouayad-Gervais, S. H., & Lubell, W. D. (2013). Examination of the Potential for Adaptive Chirality of the Nitrogen Chiral Center in Aza-Aspartame. Molecules, 18(12), 14739-14746. https://doi.org/10.3390/molecules181214739