General
All reactions were performed under an atmosphere of argon in oven dried glassware using anhydrous solvents on 4 Å molecular sieves obtained from Acros Organics/Thermo Fisher Scientific (Pittsburgh, PA, USA). Solvents were always handled under argon atmosphere. The ambient temperature was set at 21 °C.
Yields refer to chromatographically and spectroscopically homogenous materials, unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm E. Merck silica gel plates (60F-254), using UV light as a visualizing agent, and an aqueous solution of basic potassium permanganate and heat as developing agents. Purifications were done on a Biotage flash purification system using pre-packed Biotage SNAP columns.
Products were characterized by 1H-NMR/13C-NMR on either a Varian 500 MHz, or 400 MHz spectrometer in a deuterated solvent using tetramethylsilane (TMS) as an internal standard. Chemical shifts are given in δ (ppm) and J-values in Hz. High-pressure liquid chromatography (HPLC) was performed using Waters 1525 binary HPLC pump and Waters 2489 UV/Visible detector operating at 254 nm on a Waters symmetry C18 3.5 μm, 4.6/75 mm column. MeOH HPLC solvent was obtained from Sigma-Aldrich (St. Louis, MO, USA).
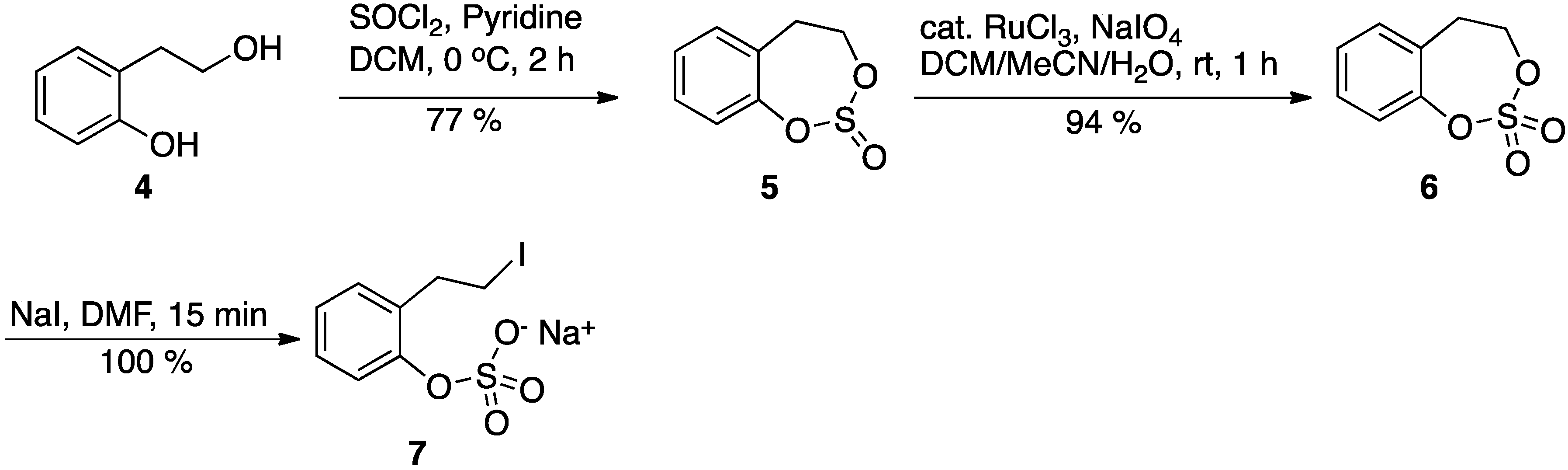
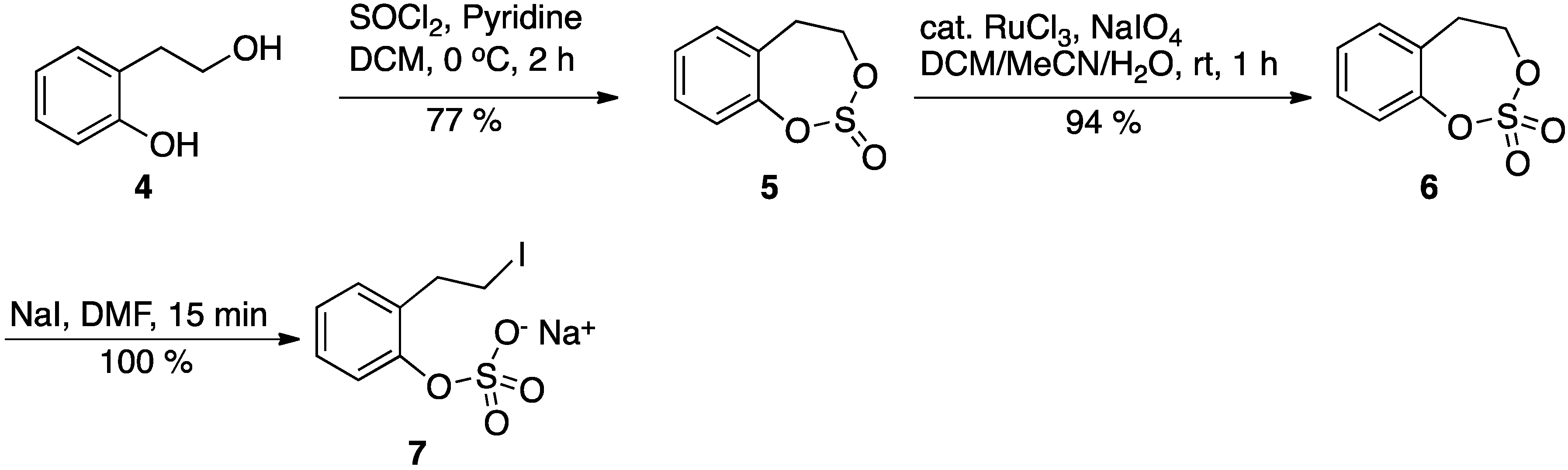
4,5-Dihydrobenzo[d][1,3,2]dioxathiepine 2,2-dioxide (6): Anhydrous pyridine (3.4 mL, 41.62 mmol) was added to a solution of the 2-phenethanol 4 (2.3 g, 16.65 mmol) in anhydrous DCM (70 mL), and the mixture was cooled to 0 °C in an ice bath. Then, freshly distilled thionyl chloride (1.22 mL, 16.65 mmol) was added to the above solution. The resultant reaction mixture was then stirred for 2 h with gradual warming to the ambient temperature. At this point, TLC of the reaction mixture indicated complete consumption of the starting material. The reaction mixture was then diluted with more DCM, transferred to a separatory funnel, and washed with cold dilute HCl twice. Then the DCM layer was washed with water (2×) and brine (1×), and, finally, the DCM layer was dried with anhydrous sodium sulfate and concentrated to give the cyclic sulfite 5, 2.8 g, as an oily liquid (Rf: 0.58; 20% EtOAc/hexane). The product was used in the subsequent step without purification.
Catalytic ruthenium chloride (0.02 mol %, 63 mg) was added to the crude cyclic sulfite 5 (2.8 g, 15.2 mmol), in acetonitrile (40 mL) and DCM (40 mL). Then, a solution of sodium periodate (4.9 g, 22.8 mmol) in water (60 mL) was slowly added dropwise to the above solution at room ambient temperature. The reaction mixture was then stirred at room temperature until all of the starting material was consumed (about 1 h). All of the volatile solvents were removed in vacuo and the aqueous layer was extracted with EtOAc (3×). The combined ethyl acetate layer was washed with saturated sodium bicarbonate solution (2×), water (2×), and brine (1×). Finally, the organic layer was dried over anhydrous sodium sulfate, concentrated, and chromatographed (hexane/EtOAc 5–50%) over silica gel to give pure cyclic sulfate (6), 2.9 g, 87%, as white solid. Rf: 0.28 (EtOAc/hexane). 1H-NMR (CDCl3; 500 MHz) δ 7.35 (t, 7.5 Hz, 1 H), 7.26 (dd, 13.5 Hz, 7,5 Hz, 2 H), 7.19 (d, 7.5 Hz, 1 H), 4.64 (m, 2 H), 3.26 (m, 2 H); 13C-NMR (CDCl3, 125 MHz) δ 149.9, 131.1, 130.2, 129.5, 127.7, 121.9, 72.6, 33.6; HPLC: RT 9.1 min on Symmetry C18 column from Waters, flow 1 mL/min, linear gradient 5 to 95% MeOH/Water.
Sodium 2-(2-Iodoethyl)phenyl sulfate (7): Solid sodium iodide (13 mg, 0.088 mmol) was added to a solution of cyclic sulfate 6 (17.5 mg, 0.088 mmol) in the DMF (0.35 mL) at ambient temperature. The reaction was monitored by the TLC to document the consumption of the starting material. Within 15 min, all of the starting material was consumed. The DMF was removed in vacuo and the residue was dried thoroughly in vacuo at ambient temperature to give the pure product 7, yield 100%; 1H-NMR (DMF-d7, 400 MHz) δ 7.21 (m, 3 H), δ 7.01 (dt, 1 Hz, 8 Hz, 1 H), 3.55 (m, 2 H), 3.26 (m, 2 H); 13C-NMR (100 MHz, DMF-d7) δ 152.2, 133.2, 130.3, 127.7, 123.6, 122.3, 35.9, 6.5; HPLC: RT 6.1 min on Symmetry C18 column from Waters, flow 1 mL/min, linear gradient 5 to 95% MeOH/Water.
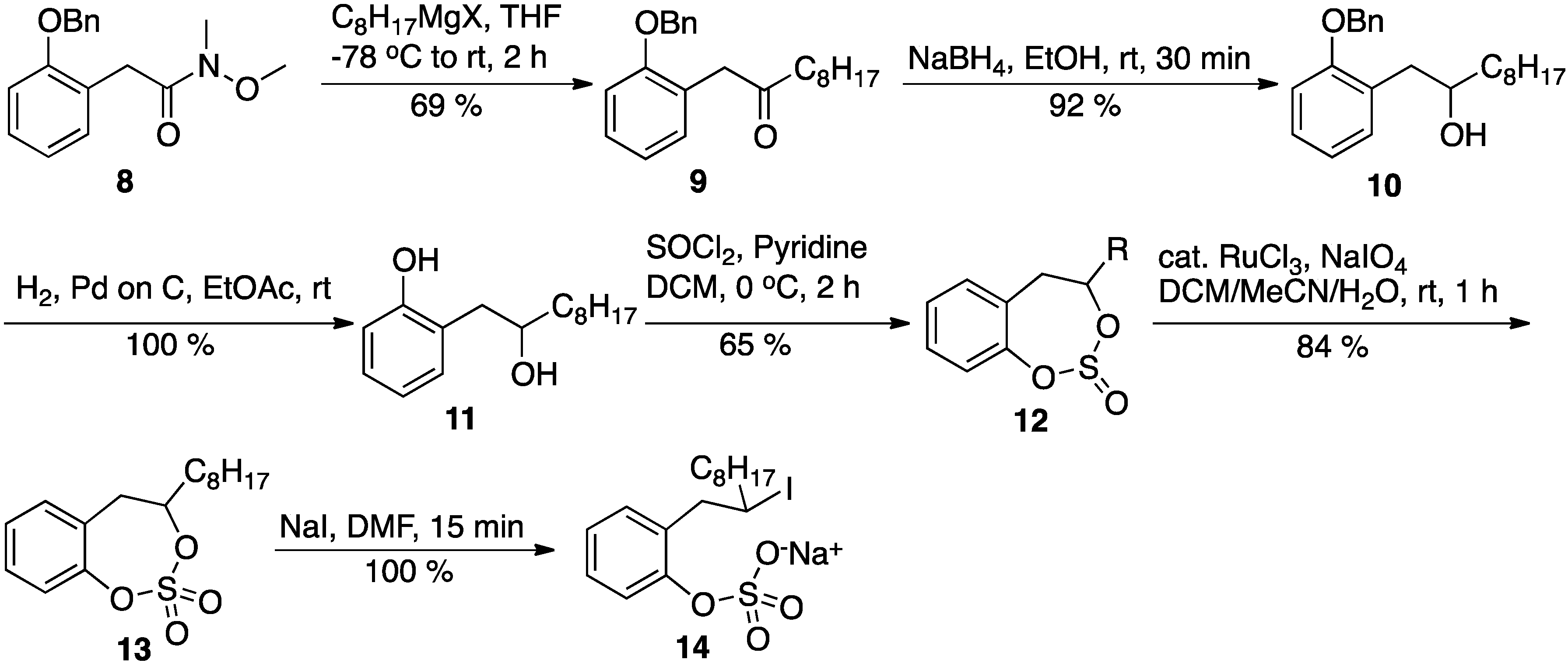
1-(2-(Benzyloxy)phenyl)decan-2-one (9): A solution of amide 8 (570 mg, 2 mmol) in anhydrous THF (8 mL) was cooled to −78 °C in a dry ice-acetone bath under an argon atmosphere. To this cooled solution, a solution of octylmagnesium bromide (2 M, 1.25 mL, 2.5 mmol) in ether was added. After the addition was complete, the reaction was allowed to proceed for 2 h with gradual warming to the ambient temperature. The reaction was quenched by the addition of a saturated ammonium chloride solution and the THF was removed in vacuo. The reaction mixture was transferred to a separatory funnel and extracted with diethyl ether (3×). The combined organic layer was washed with water (1×) and brine (1×), dried over anhydrous sodium sulfate, concentrated and chromatographed on silica gel to give pure ketone 9, 415 mg, 61%, as viscous oil. Rf: 0.77 (15% EtOAc in hexane); 1H-NMR (CDCl3, 500 MHz) δ 7.39 (m, 5 H), 7.34 (m, 1 H), 7.25 (m, 1 H), 7.16 (m, 1 H), 6.96 (m, 1 H), 5.06 (s, 2 H), 3.71 (s, 2 H), 2.4 (t, 7 Hz, 2 H), 1.52 (m, 2 H), 1.23 (bm, 10 H), 0.89 (t, 7.5 Hz, 3 H); 13C-NMR (125 MHz, CDCl3) δ 206.8, 156.4, 136.9, 131.3, 128.5, 128.3, 127.8, 127.3, 124.1, 120.8, 111.6, 69.9, 44.7, 42.2, 31.8, 29.3, 29.1, 29.09, 23.7, 22.6, 14.0.
1-(2-(Benzyloxy)phenyl)decan-2-ol (10): To a solution of ketone 9 (415 mg, 1.23 mmol) in absolute ethanol (5 mL), solid sodium borohydride (93 mg, 2.46 mmol) was added at ambient temperature. The reaction mixture was stirred at ambient temperature for 30 min. At this point, the TLC indicated complete consumption of the starting material, and the reaction was quenched by the addition of a saturated ammonium chloride solution. All of the ethanol was removed in vacuo, and the residue was transferred to a separatory funnel and extracted with EtOAc (3×). The combined organic layer was washed with water (2×) and brine (1×). Finally, the organic layer was dried over anhydrous sodium sulfate, concentrated, and chromatographed on silica gel to give pure alcohol 10, 393 mg, 94%, as viscous oil. Rf: 0.48 (10% EtOAc in hexane); 1H-NMR (500 MHz, CDCl3) δ 7.41 (m, 5 H), 7.20 (m, 2 H), 6,94 (m, 2 H), 5.09 (s, 2 H), 3.89 (bs, 1 H), 3.00 (dd, 4 Hz, 14 Hz, 1 H), 2.68 (dd, 8 Hz, 14 Hz, 1 H), 1.93 (bs, 1 H), 1.50 (m, 2 H), 1.27 (bm, 12 H), 0.89 (t, 7 Hz, 3 H); 13C-NMR (125 MHz, CDCl3) δ 156.7, 136.9, 131.5, 128.5, 127.9, 127.7, 127.5, 127.2, 120.9, 111.7, 71.8, 70.0, 38.8, 37.2, 31.8, 29.7, 29.6, 29.2, 25.7, 22.6, 14.1.
2-(2-Hydroxydecyl)phenol (11): Catalytic 10% palladium on carbon (20 mg) was added to a solution of alcohol 10 (393 mg, 1.15 mmol) in EtOAc (4 mL). Then the reaction flask was purged with hydrogen (by applying vacuum and flushing hydrogen gas). This process was repeated three times, and then the reaction proceeded overnight under a balloon of hydrogen gas. The reaction mixture was then filtered through a pad of Celite, and the Celite pad was washed thoroughly with EtOAc. Finally, EtOAc was removed in vacuo to give the desired product 11—a pale yellow oil (285 mg, 99%). Thus, the product obtained was sufficiently pure for use in the next step; an analytically pure sample for NMR was obtained by chromatographing a small sample on silica gel. Rf: 0.45 (25% EtOAc in hexane); 1H-NMR (500 MHz, CDCl3) δ 8.19 (bs, 1 H), 7.15 (m, 1 H), 7.02 (m, 1 H), 6.91 (m, 1 H), 6.84 (m, 1 H), 4.00 (m, 1 H), 2.85 (dd, 2 Hz, 15 Hz, 1 H), 2.80 (dd, 7 Hz, 15 Hz, 1 H), 2.39 (bs, 1 H), 1.53 (m, 1 H), 1.28 (m, 12 H), 0.88 (t, 7 Hz, 3 H); 13C-NMR (125 MHz, CDCl3) δ 155.9, 131.5, 128.3, 125.9, 120.2, 117.2, 74.6, 40.3, 38.9, 36.9, 31.8, 29.5, 29.4, 25.6, 22.6, 14.1.
4-Octyl-4,5-dihydrobenzo[d][1,3,2]dioxathiepine 2,2-dioxide (13): Anhydrous pyridine (330 μL, 3.6 mmol) was added to a solution of the substituted 2-phenethanol 11 (340 mg, 1.44 mmol) in anhydrous DCM (6 mL), and the mixture was cooled to 0 °C in an ice bath. Then, freshly distilled thionyl chloride (115 μL, 1.58 mmol) was added to this solution. The reaction mixture was then removed from the ice bath and stirred for 2 h while warming up to the ambient temperature. At this point, the TLC of the reaction mixture indicated the complete consumption of the starting material. The reaction mixture was diluted with more DCM, transferred to separatory funnel, and washed with cold dilute HCl (2×). Then, the DCM layer was washed with water (2×) and brine (1×). Finally, the DCM layer was dried over anhydrous sodium sulfate and concentrated to give the cyclic sulfite 12 (360 mg, 87%) as an oily liquid. The product was then used in the next step without purification. The product formed two spots on the TLC (Rf: 0.80 & 0.72; 10% EtOAc/hexane) due to the formation of diastereomers.
Catalytic ruthenium chloride (0.02 mol %; 5 mg) was added to the solution of the crude cyclic sulfite 12 (350 mg, 1.20 mmol) in acetonitrile (4 mL) and DCM (4 mL). Then, a solution of sodium periodate (380 mg, 1.77 mmol) in water (6 mL) was added to this solution dropwise at ambient temperature. The reaction mixture was then stirred at ambient temperature until the starting material was consumed (about 1 h). All of the volatile solvents were removed in vacuo and the aqueous layer was extracted with EtOAc (3×). The combined ethyl acetate layer was washed with saturated sodium bicarbonate solution (2×), water (2×), and brine (1×). Finally, the organic layer was dried over anhydrous sodium sulfate, concentrated, and chromatographed (hexane/EtOAc 5–15%) on silica gel to give pure cyclic sulfate 13 (288 mg, 78%), as a pale yellow, viscous oil. Rf: 0.43 (10% EtOAc in hexane); 1H-NMR (500 MHz, CDCl3) δ 7.34 (m, 1 H), 7.25 (m, 2 H), 7.16 (m, 1 H), 4.80 (m, 1 H), 3.41 (dd, 9.5 Hz, 16 Hz, 1 H), 2.90 (d, 16 Hz, 1 H), 1.89 (m, 1 H), 1.73 (m, 1 H), 1.29 (m, 12 H), 0.88 (t, 7 Hz, 3 H); 13C-NMR (125 MHz, CDCl3) δ 150. 0, 131.2, 129.4, 127.6, 126.5, 86.2, 40.3, 38.9, 35.0, 31.8, 29.1, 29.0, 24.9, 22.6, 14.1; HPLC: RT 9.4 min, flow 1 mL/min, gradient 10 to 100% MeOH/Water.
Sodium 2-(2-iododecyl)phenyl sulfate: Solid sodium iodide (15 mg, 0.1 mmol) was added to a solution of cyclic sulfate (13, 26 mg, 0.083 mmol) in the DMF (0.5 mL) at ambient temperature. The reaction was monitored by TLC to document the consumption of the starting material. Within 15 min, the latter was consumed, and the DMF was removed in vacuo. The residue was dried thoroughly in vacuo at ambient temperature to give a pure product (yield 100%); 1H-NMR (DMF-d7, 400 MHz) δ 7.56 (dd, 1.2 Hz, 8 Hz, 1 H), 7.20 (m, 2 H), 7.00 (dt, 1.2 Hz, 8 Hz, 1 H), 3.79 (bs, 1 H), 3.40 (dd, 7 Hz, 14 Hz, 1 H), 3.33 (dd, 8 Hz, 14 Hz, 1 H), 1.73 (m, 2 H), 1.26 (m, 12 H), 0.86 (t, 6.8 Hz, 3 H); 13C-NMR (DMF-d7, 100 MHz) δ 153.1, 132.5, 131.0, 127.7, 123.3, 122.1, 43.4, 40.9, 40.2, 32.2, 30.0, 29.8, 29.6, 29.1, 22.9, 14.2; HPLC: RT 8.1 min, flow 1 mL/min, gradient 10 to 100% MeOH/Water.
Procedure for Labeling 6 with 124I: 200 μL of anhydrous MeCN were added to a solution of [124I]NaI (500 μCi) in 15 μL 0.02 M NaOH, and the solvents were evaporated under steady stream of argon. After the solvent was completely removed, an extra 100 μL of MeCN was added, and the solvent was removed again, as above. This procedure was repeated two more times to remove most of the water from [124I]NaI. Then, a solution of cyclic sulfate (100 μL, ~1 mg/mL) in DMF was added to the residue, and the iodination reaction was allowed to proceed for 30 min. Then, the reaction mixture was analyzed by HPLC using dual detection (UV and gamma) to distinguish the desired product from the unreacted [124I]iodide. Under the HPLC conditions (see above), the [124I]iodide had an RT of 1 min and the desired compound eluted at 7.3 min, as observed on gamma-detector. Retention times for the labeled compound on gamma-detector (7.3 min), and unlabeled compound on UV detector (6.1 min), are different because the gamma-detector was externally connected.

{kind=link}
{kind=link}
{kind=link}