A New Cytotoxic Friedelane Acid – Pluricostatic Acid – and Other Compounds from the Leaves of Marila pluricostata

 , and
, and
Abstract
:Introduction
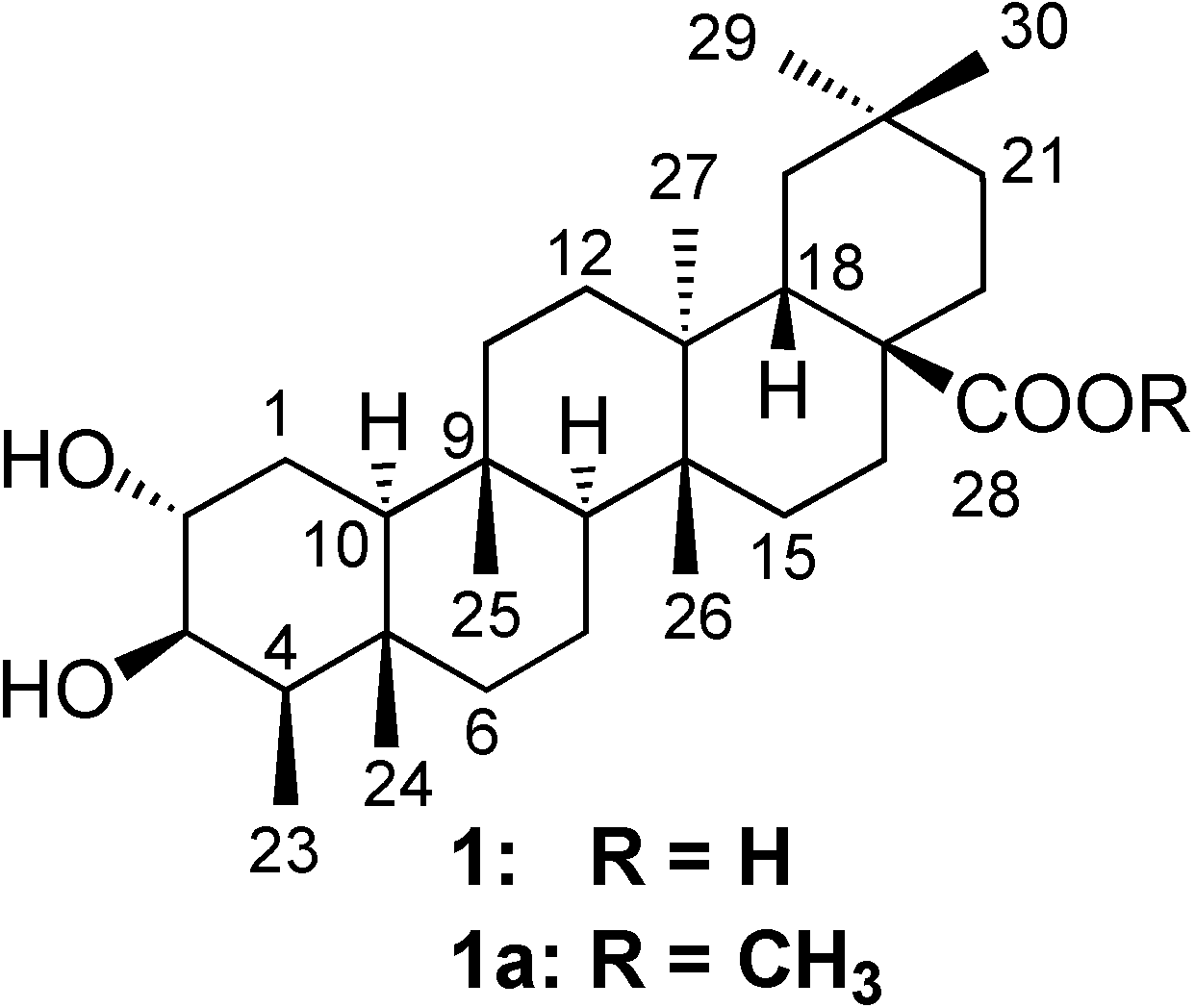
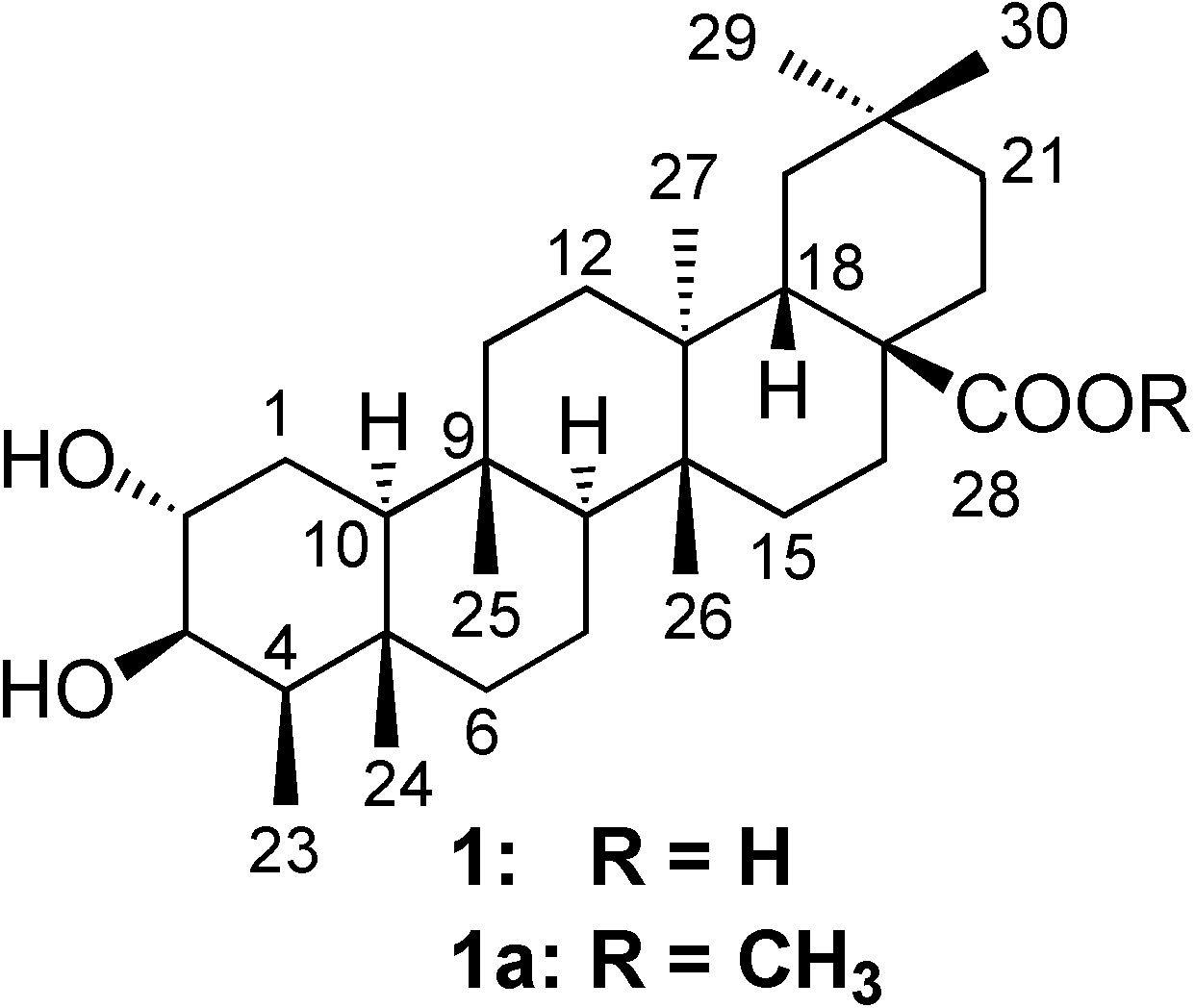
Results and Discussion
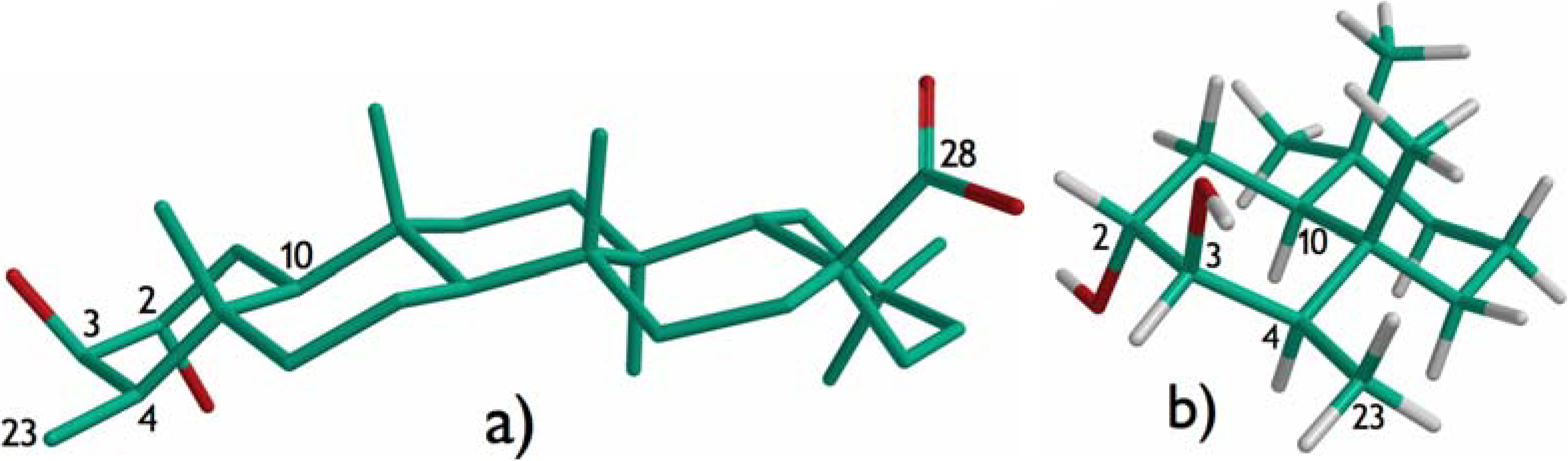
Biological activity

{kind=link}
{kind=link}
| GI50 (μg/mL) | |||
|---|---|---|---|
| MCF-7 | H-460 | SF-268 | |
| dichloromethane extract | 5.9 | 6.2 | 4.6 |
| methanolic fraction defatted | 8.3 | 7.8 | 5.5 |
| neutral fraction | 1.6 | 1.5 | 0.7 |
| acid fraction | 2.4 | 2.7 | 2.2 |
| 1 | 3.0 | 1.2 | 3.3 |
| 8 | 6.3 | 5.1 | 4.6 |
| 9 | 7.8 | 7.2 | > 10 |
| 10 | 7.2 | 5.3 | 6.5 |
| 11 | 5.0 | 3.6 | 5.1 |
| Adriamycin | 0.37 | 0.42 | 0.50 |
Experimental
General
Plant Material
Extraction, Isolation and Identification
| Components | Retention time (Rt) (min.) | Relative (%) | M+ m/z |
|---|---|---|---|
| fraction A | |||
| α-copaene | 17.33 | 2.6 | 220 |
| β-caryophyllene | 20.01 | 29.9 | 204 |
| zingiberene | 20.83 | 1.1 | 204 |
| germacrene-d | 21.84 | 0.6 | 204 |
| α-amorphene | 23.02 | 11.7 | 204 |
| β-selinene | 23.19 | 3.8 | 204 |
| calarene | 23.71 | 4.1 | 204 |
| α-muurolene | 24.17 | 3.6 | 204 |
| χ-cadinene | 24.87 | 7.7 | 204 |
| calamenene | 25.01 | 1.6 | 204 |
| δ-cadinene | 25.42 | 3.6 | 204 |
| α-cadinene | 26.09 | 3.0 | 204 |
| calacorene | 26.85 | 1.3 | 200 |
| α-caryophyllene oxide | 27.90 | 10.9 | 220 |
| Total compounds identified | 85.5 | ||
| fraction B | |||
| α-copaene | 17.37 | 36.0 | 220 |
| β-elemene | 18.41 | 0.9 | 204 |
| β-caryophyllene | 20.03 | 13.4 | 204 |
| β-cubenene | 20.25 | 3.0 | 204 |
| α-humulene | 21.18 | 0.8 | 204 |
| α-amorphene | 23.05 | 11.7 | 204 |
| α-aromadendrene | 23.11 | 3.8 | 204 |
| Total compounds identified | 69.6 | ||
| fraction C | |||
| α-cubebene | 16.06 | 5.1 | 204 |
| α-copaene | 17.35 | 38.2 | 220 |
| β-bourbonene | 17.76 | 7.6 | 204 |
| β-cubenene | 20.25 | 11.3 | 204 |
| α-aromadendrene | 23.13 | 0.7 | 204 |
| β-sesquiphellandrene | 25.34 | 0.9 | 204 |
| Total compounds identified | 36.4 | ||
| fraction D | |||
| β-farnesene | 16.48 | 2.8 | 204 |
| α-curcumene | 16.92 | 1.8 | 202 |
| β-bisabolene | 17.72 | 1.9 | 204 |
| Squalene | 42.40 | 89.9 | 410 |
| Total compounds identified | 96.4 | ||
Test for in vitro Antineoplastic Cytotoxicity
Molecular Modeling
Acknowledgment
References
- Woodson, R.E.; Shery, R.W. Flora of Panama. Ann. Mo. Bot. Gard. 1980, 67, 969–1043. [Google Scholar] [CrossRef]
- D’Arcy, W.G. Flora of Panama, Checklist and Index. Part II. Ann. Mo. Bot. Gard. 1987, 1–670. [Google Scholar]
- Correa, M.D.; Foster, R.; Galdames, C.; Stapf, M.S. Catálogo de Plantas Vasculares de Panamá, 1st ed.; Novo Art: Bogotá, Colombia, 2004; pp. 1–600. [Google Scholar]
- Jorgensen, P.; León, Y. Catalogue of the Vascular Plants of Ecuador. Monographs in Systematic Botany. Ann. Mo. Bot. Gard 1999, 75, 1–1182. [Google Scholar]
- Schultes, R. De Plantis toxicariis e mundo novo tropicale comentationes. XXX. Biodynamic Gutiferous plants of Northwest Amazon. Bot. Mus. Leafl. Harv. Univ. 1983, 29, 49–57. [Google Scholar]
- López-Pérez, J.L.; Olmedo, D.A.; Del Olmo, E.; Vásquez, Y.; Solís, P.N.; Gupta, M.P.; San Feliciano, A. Cytotoxic 4-phenylcoumarins from the leaves of Marila pluricostata. J. Nat. Prod. 2005, 68, 369–373. [Google Scholar] [CrossRef]
- Bedoya, L.M.; Beltrán, M.; Sancho, R.; Olmedo, D.A.; Sánchez-Palomino, S.; Del Olmo, E.; López-Pérez, J.L.; Muñoz, E.; San Feliciano, A. Alcamí. 4-Phenylcoumarins as HIV transcription inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 4447–4450. [Google Scholar] [CrossRef]
- López-Pérez, J.L.; Therón, R.; Del Olmo, E.; Díaz, D. NAPROC-13: a database for the dereplication of natural product mixtures in bioassay-guided protocols. Bioinformatics 2007, 23, 3256–3257. [Google Scholar] [CrossRef]
- Kumar, N.S.; Muthukuda, P.M.; MacLeod, J.K. New D: A-friedooleananes from Euonymus revolutus (Celastraceae). J. Chem. Soc. Perkin Trans. I. 1985, 685–689. [Google Scholar]
- Patra, A.; Chaudhuri, S.K.; Acharya, A.K. Applications of two-dimensional NMR in spectral assignments of some friedelanes and secofriedelanes. Magn. Reson. Chem. 1990, 28, 85–89. [Google Scholar] [CrossRef]
- Akihisa, T.; Yamamoto, K.; Tamura, T.; Kumura, Y.; Iida, T.; Nambara, T.; Chang, F.C. Triterpenoid ketones from Lingnania chungii McClure: arborinone, friedelin and glutinone. Chem. Pharm. Bull. 1992, 40, 789–791. [Google Scholar] [CrossRef]
- Chang, W-C.; Wu, T-S.; Hsieh, Y-S.; Kuo, S-C.; Cha, P-D. Terpenoids of Syzygium formosanum. J. Nat. Prod. 1999, 62, 327–328. [Google Scholar] [CrossRef]
- Nozaki, H.; Suzuki, H.; Hirayama, T.; Kasai, R.; Wu, R.-Y.; Lee, K.H. Antitumor triterpenes of Maytenus diversifolia. Phytochemistry 1986, 25, 479–485. [Google Scholar] [CrossRef]
- Salazar, G.C.M.; Silva, G.D.F.; Duarte, L.P.; Filho, S.A.V.; Lula, I.S. Two epimeric friedelane triterpenes isolated from Maytenus truncata Reiss: 1H and 13C chemical shift assignments. Magn. Reson. Chem. 2000, 38, 977–980. [Google Scholar] [CrossRef]
- Ngouamegne, E.T.; Fongang, R.S.; Ngouela, S.; Boyom, F.F.; Rohmer, M.; Tsamo, E.; Gut, J.; Rosenthal, P.J. Endodesmiadiol, a friedelane triterpenoid, and other antiplasmodial compounds from Endodesmia calophylloides. Chem. Pharm. Bull. 2008, 56, 374–377. [Google Scholar] [CrossRef]
- Dharmaratne, H.R.W.; Sotheeswaran, S.; Balasubramaniam, S. Triterpene and neoflavonoids of Calophyllum lankaensis and Calophyllum thwaitesii. Phytochemisty 1984, 23, 2601–2603. [Google Scholar] [CrossRef]
- Katakawa, J.; Tetsumi, T.; Hasegawa, H.; Iida, T.; Katai, M. A new triterpene from leaves and stems of Euonymus fortunei. Nat. Med. 2000, 54, 18–21. [Google Scholar]
- Kitajima, J.; Shindo, M.; Tanaka, Y. Two new triterpenoid sulfates from the leaves of Schefflera octophylla. Chem. Pharm. Bull. 1990, 38, 714–716. [Google Scholar] [CrossRef]
- Siddiqui, S.; Hafeez, F.; Begum, S.; Siddiqui, B.S. Oleanderol, a new pentacyclic triterpene from the leaves of Nerium oleander. J. Nat. Prod. 1988, 51, 229–233. [Google Scholar] [CrossRef]
- Queiroz de Assis Galotta, A.L.; Boaventura, M.D.A. Constituintes químicos da raiz e do tallo da folha do acaí (Euterpe precatoria Mart., Arecaceae). Quím. Nova 2005, 28, 610–613. [Google Scholar]
- Amaral, M.D.C.E.; Faria, A.D.; Magalhaes, A.F.; Magalhaes, E.G.; Ruiz, A.L.T.G. Steroids and triterpenes from Eleocharis acutangula and E. sellowiana (Cyperaceae). Phytochem. Anal. 2004, 15, 125–129. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.A.; Johnson, G.S.; Paull, K.D.; Sausville, E.A. The NCI anti-cancer drug screen: A smart screen to identify effectors of novel drug targets. Anticancer Drug Des. 1997, 12, 533–541. [Google Scholar]
- Adams, R.P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectroscopy; Allured Publishing Corporation: Illinois, USA, 1995; pp. 1–469. [Google Scholar]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel - an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Sample Availability: Milligram quantities of compounds 3-7 and 9-14 are available from the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Olmedo, D.A.; López-Pérez, J.L.; Del Olmo, E.; Vásquez, Y.; San Feliciano, A.; Gupta, M.P. A New Cytotoxic Friedelane Acid – Pluricostatic Acid – and Other Compounds from the Leaves of Marila pluricostata. Molecules 2008, 13, 2915-2924. https://doi.org/10.3390/molecules13112915
Olmedo DA, López-Pérez JL, Del Olmo E, Vásquez Y, San Feliciano A, Gupta MP. A New Cytotoxic Friedelane Acid – Pluricostatic Acid – and Other Compounds from the Leaves of Marila pluricostata. Molecules. 2008; 13(11):2915-2924. https://doi.org/10.3390/molecules13112915
Chicago/Turabian StyleOlmedo, Dionisio A., José L. López-Pérez, Esther Del Olmo, Yelkaira Vásquez, Arturo San Feliciano, and Mahabir P. Gupta. 2008. "A New Cytotoxic Friedelane Acid – Pluricostatic Acid – and Other Compounds from the Leaves of Marila pluricostata" Molecules 13, no. 11: 2915-2924. https://doi.org/10.3390/molecules13112915
APA StyleOlmedo, D. A., López-Pérez, J. L., Del Olmo, E., Vásquez, Y., San Feliciano, A., & Gupta, M. P. (2008). A New Cytotoxic Friedelane Acid – Pluricostatic Acid – and Other Compounds from the Leaves of Marila pluricostata. Molecules, 13(11), 2915-2924. https://doi.org/10.3390/molecules13112915