
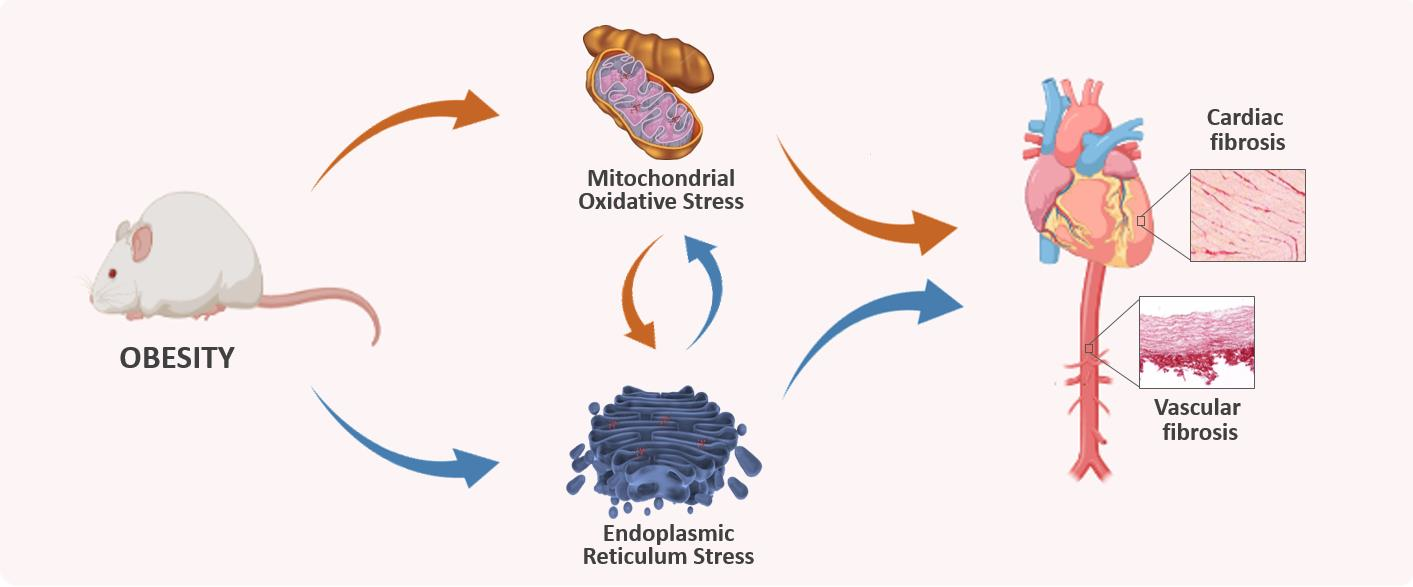
The Interplay of Mitochondrial Oxidative Stress and Endoplasmic Reticulum Stress in Cardiovascular Fibrosis in Obese Rats

 , , , ,
, , , ,  , ,
, ,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Histological Analysis
2.3. Cell Isolation and Cell Culture Conditions
2.4. Western Blot
2.5. Retrotranscription and Real-Time PCR
2.6. Measurement of Intracellular Superoxide Anion Production
2.7. Statistical Analysis
3. Results
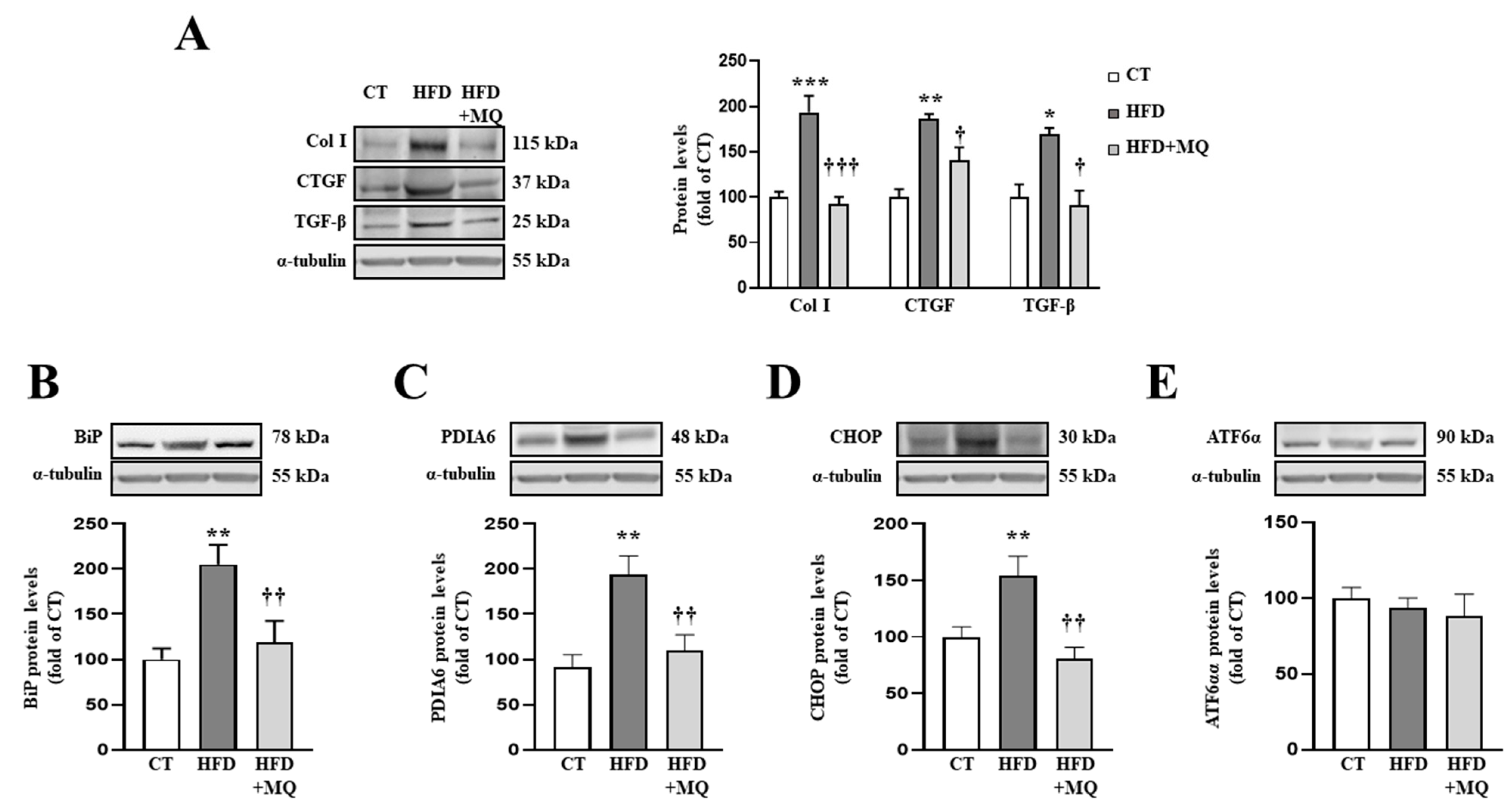
3.1. Mitochondrial Oxidative Stress Promotes ECM Protein Deposition and ER Stress Activation at Cardiac Level in Obesity
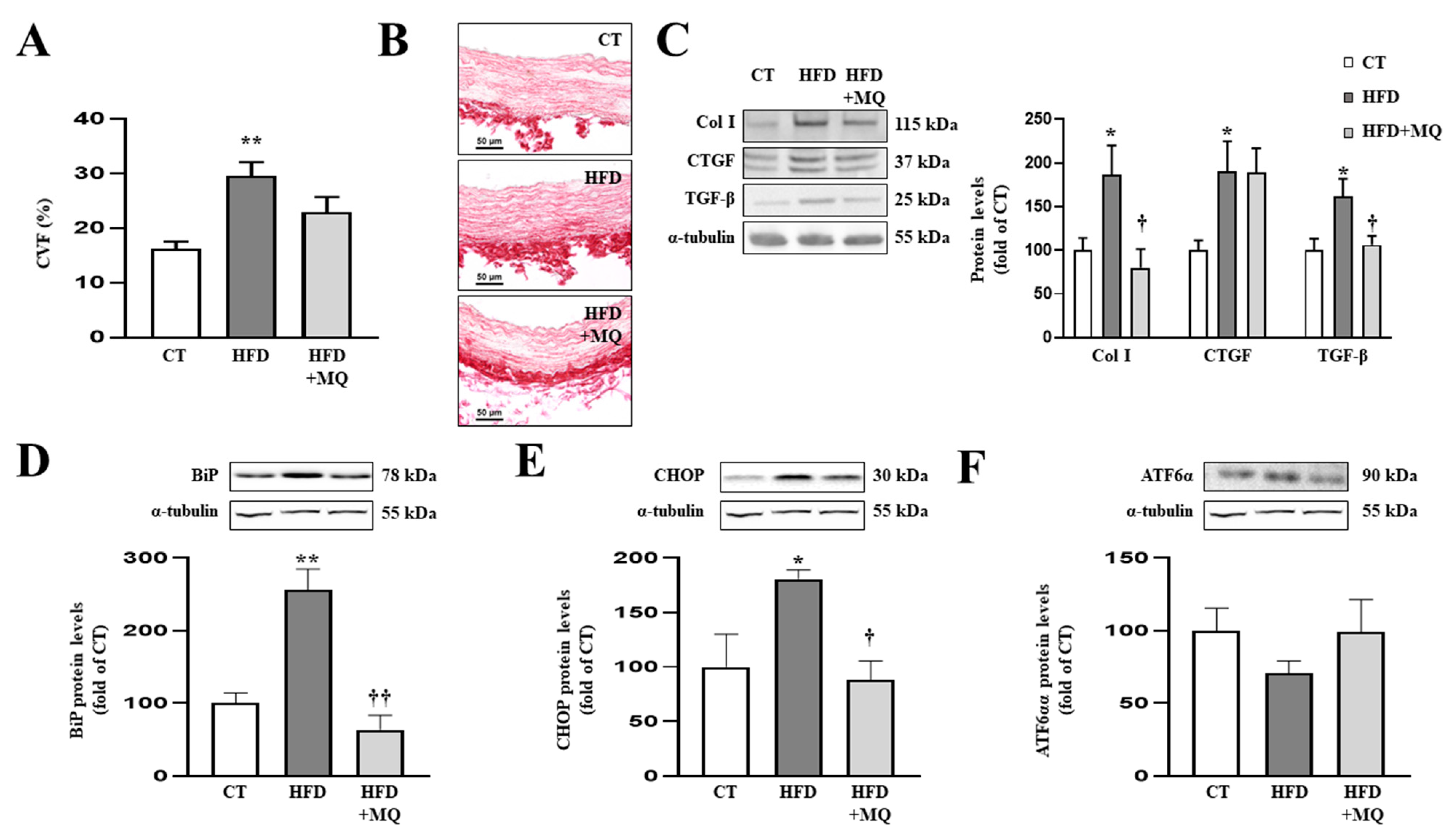
3.2. MitoQ Treatment Improves Vascular Fibrosis and ER Stress Activation in the Aorta of HFD Animals
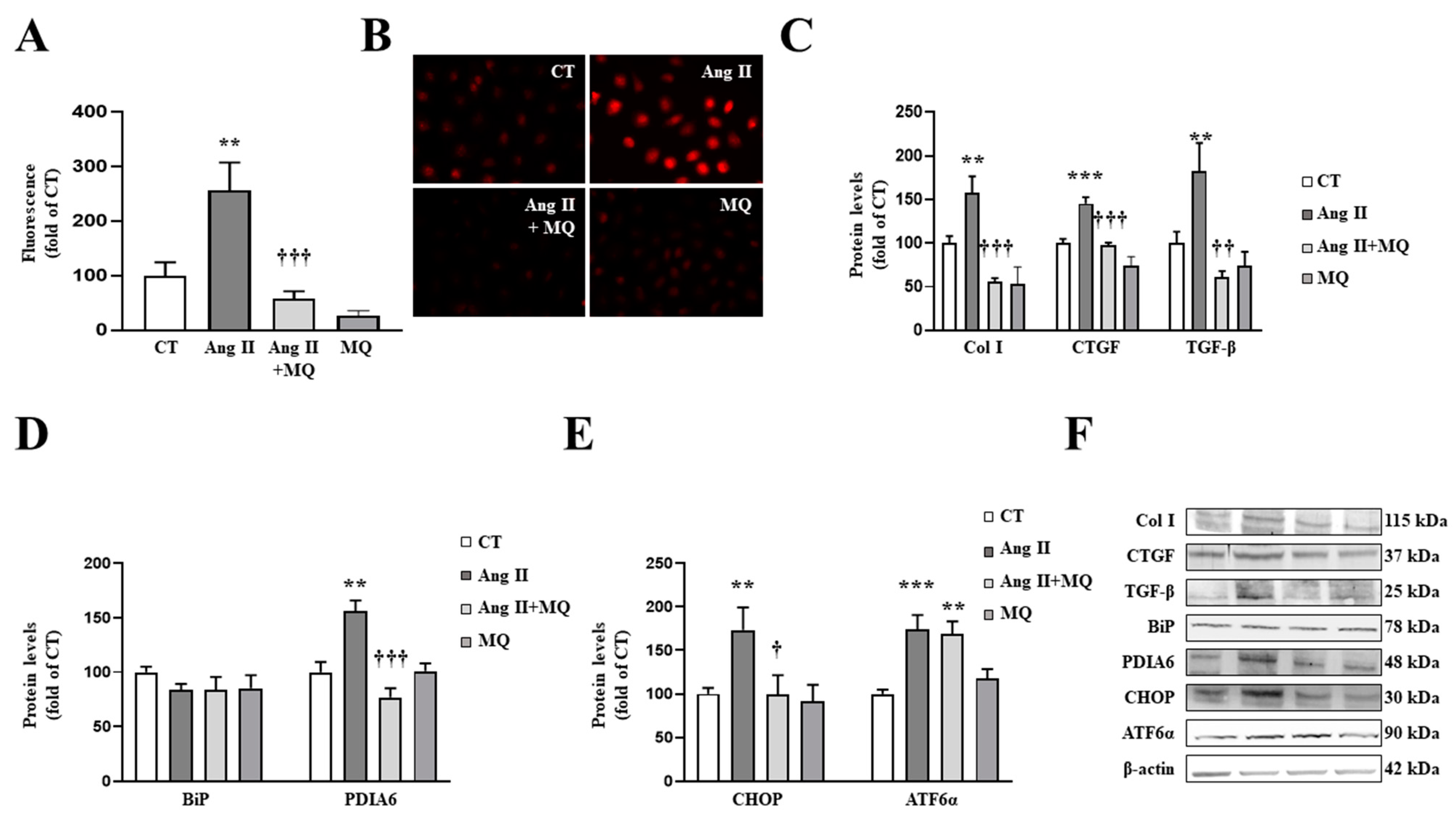
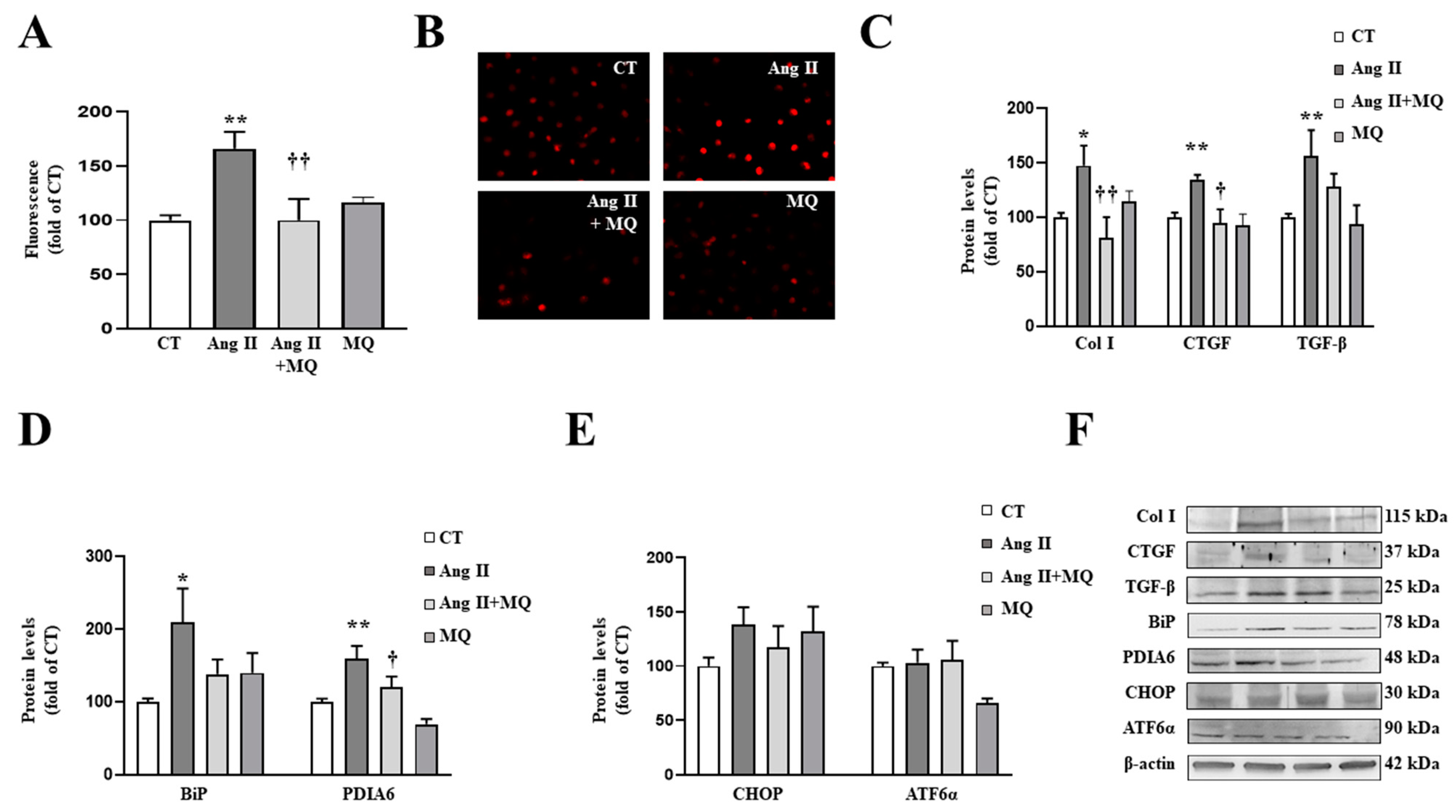
3.3. Mitochondrial Oxidative Stress Mediates the Effects of Ang II in Cardiac Fibroblasts
3.4. Mitochondrial Oxidative Stress Mediates the Effects of Ang II in Vascular Smooth Muscle Cells
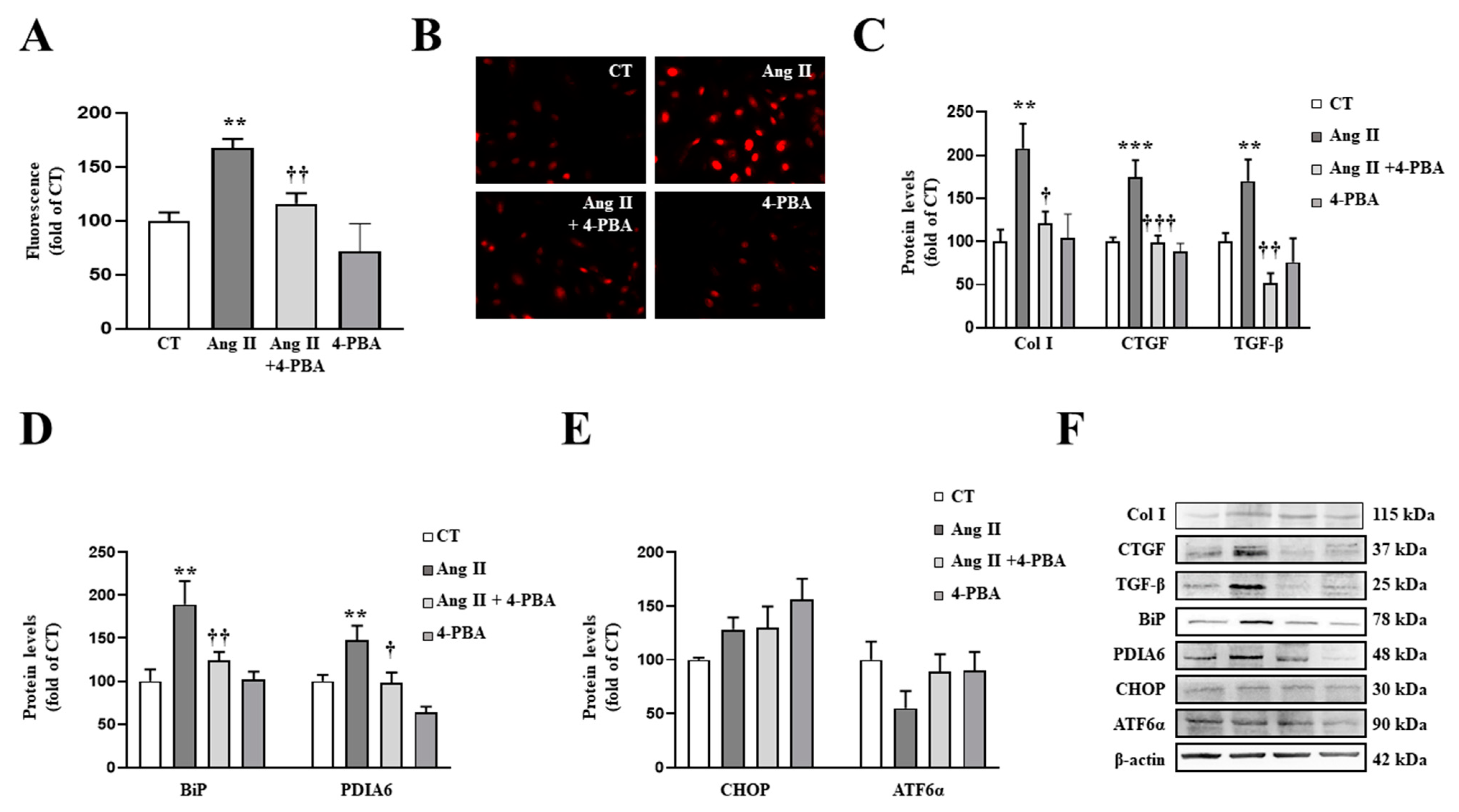
3.5. Inhibition of Endoplasmic Reticulum Stress Blocks the Prooxidant and Profibrotic Effects of Ang II in Cardiovascular Cells
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Czubryt, M.P. Cardiac Fibroblast to Myofibroblast Phenotype Conversion-An Unexploited Therapeutic Target. J. Cardiovasc. Dev. Dis. 2019, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, D.L.; Robinson, A.T.; Rossman, M.J.; Seals, D.R.; Edwards, D.G. Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2080–H2100. [Google Scholar] [CrossRef]
- Cavalera, M.; Wang, J.; Frangogiannis, N.G. Obesity, metabolic dysfunction, and cardiac fibrosis: Pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl. Res. 2014, 164, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Jurado-Lopez, R.; Valero-Munoz, M.; Bartolome, M.V.; Ballesteros, S.; Luaces, M.; Briones, A.M.; Lopez-Andres, N.; Miana, M.; Cachofeiro, V. Leptin induces cardiac fibrosis through galectin-3, mTOR and oxidative stress: Potential role in obesity. J. Hypertens. 2014, 32, 1104–1114; discussion 1114. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Kim, D.H.; Park, G.; Park, S.; Kim, H.S. Clinical significance of anti-dense fine speckled 70 antibody in patients with fibromyalgia. Korean J. Intern. Med. 2019, 34, 426–433. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Gonzalez, S.; Marin-Royo, G.; Jurado-Lopez, R.; Bartolome, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martinez-Martinez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef]
- Feillet-Coudray, C.; Fouret, G.; Ebabe Elle, R.; Rieusset, J.; Bonafos, B.; Chabi, B.; Crouzier, D.; Zarkovic, K.; Zarkovic, N.; Ramos, J.; et al. The mitochondrial-targeted antioxidant MitoQ ameliorates metabolic syndrome features in obesogenic diet-fed rats better than Apocynin or Allopurinol. Free Radic. Res. 2014, 48, 1232–1246. [Google Scholar] [CrossRef]
- Coudray, C.; Fouret, G.; Lambert, K.; Ferreri, C.; Rieusset, J.; Blachnio-Zabielska, A.; Lecomte, J.; Ebabe Elle, R.; Badia, E.; Murphy, M.P.; et al. A mitochondrial-targeted ubiquinone modulates muscle lipid profile and improves mitochondrial respiration in obesogenic diet-fed rats. Br. J. Nutr. 2016, 115, 1155–1166. [Google Scholar] [CrossRef]
- Marin-Royo, G.; Rodriguez, C.; Le Pape, A.; Jurado-Lopez, R.; Luaces, M.; Antequera, A.; Martinez-Gonzalez, J.; Souza-Neto, F.V.; Nieto, M.L.; Martinez-Martinez, E.; et al. The role of mitochondrial oxidative stress in the metabolic alterations in diet-induced obesity in rats. FASEB J. 2019, 33, 12060–12072. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Devaraj, S. Gut Microbiome in Obesity, Metabolic Syndrome, and Diabetes. Curr. Diab. Rep. 2018, 18, 129. [Google Scholar] [CrossRef]
- Dabke, K.; Hendrick, G.; Devkota, S. The gut microbiome and metabolic syndrome. J. Clin. Investig. 2019, 129, 4050–4057. [Google Scholar] [CrossRef]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef]
- Astudillo, A.A.; Mayrovitz, H.N. The Gut Microbiome and Cardiovascular Disease. Cureus 2021, 13, e14519. [Google Scholar] [PubMed]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Karbach, S.H.; Schonfelder, T.; Brandao, I.; Wilms, E.; Hormann, N.; Jackel, S.; Schuler, R.; Finger, S.; Knorr, M.; Lagrange, J.; et al. Gut Microbiota Promote Angiotensin II-Induced Arterial Hypertension and Vascular Dysfunction. J. Am. Heart Assoc. 2016, 5, e003698. [Google Scholar] [CrossRef]
- Martinez, B.K.; White, C.M. The Emerging Role of Inflammation in Cardiovascular Disease. Ann. Pharm. 2018, 52, 801–809. [Google Scholar] [CrossRef]
- Saad, M.J.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Hernandez, A.; Martinez-Martinez, E.; Gomez-Gordo, R.; Lopez-Andres, N.; Fernandez-Celis, A.; Gutierrez-Miranda, B.; Nieto, M.L.; Alarcon, T.; Alba, C.; Gomez-Garre, D.; et al. The Interaction between Mitochondrial Oxidative Stress and Gut Microbiota in the Cardiometabolic Consequences in Diet-Induced Obese Rats. Antioxidants 2020, 9, 640. [Google Scholar] [CrossRef]
- Blackwood, E.A.; Hofmann, C.; Santo Domingo, M.; Bilal, A.S.; Sarakki, A.; Stauffer, W.; Arrieta, A.; Thuerauf, D.J.; Kolkhorst, F.W.; Muller, O.J.; et al. ATF6 Regulates Cardiac Hypertrophy by Transcriptional Induction of the mTORC1 Activator, Rheb. Circ. Res. 2019, 124, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Chen, B.; Wang, X. 4-PBA prevents pressure overload-induced myocardial hypertrophy and interstitial fibrosis by attenuating endoplasmic reticulum stress. Chem. Biol. Interact. 2015, 242, 99–106. [Google Scholar] [CrossRef]
- Thuerauf, D.J.; Marcinko, M.; Gude, N.; Rubio, M.; Sussman, M.A.; Glembotski, C.C. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res. 2006, 99, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Myoishi, M.; Hao, H.; Minamino, T.; Watanabe, K.; Nishihira, K.; Hatakeyama, K.; Aasada, Y.; Okada, K.; Ishibashi-Ueda, H.; Gabbiani, G.; et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation 2007, 116, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, X.; Qin, F.; Huang, K. Selenium suppresses oxidative-stress-enhanced vascular smooth muscle cell calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress. J. Biol. Inorg. Chem. 2014, 19, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Kassan, M.; Galan, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arter. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef]
- Li, S.J.; Liu, C.H.; Chu, H.P.; Mersmann, H.J.; Ding, S.T.; Chu, C.H.; Chen, C.Y. The high-fat diet induces myocardial fibrosis in the metabolically healthy obese minipigs-The role of ER stress and oxidative stress. Clin. Nutr. 2017, 36, 760–767. [Google Scholar] [CrossRef]
- Noyan-Ashraf, M.H.; Shikatani, E.A.; Schuiki, I.; Mukovozov, I.; Wu, J.; Li, R.K.; Volchuk, A.; Robinson, L.A.; Billia, F.; Drucker, D.J.; et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation 2013, 127, 74–85. [Google Scholar] [CrossRef]
- Wu, S.; Zou, M.H. Mitochondria-associated endoplasmic reticulum membranes in the heart. Arch. Biochem. Biophys. 2019, 662, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Barahona, A.; Alonso-Barroso, E.; Perez, B.; Murphy, M.P.; Richard, E.; Desviat, L.R. Treatment with antioxidants ameliorates oxidative damage in a mouse model of propionic acidemia. Mol. Genet. Metab. 2017, 122, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Miana, M.; Jurado-Lopez, R.; Bartolome, M.V.; Souza Neto, F.V.; Salaices, M.; Lopez-Andres, N.; Cachofeiro, V. The potential role of leptin in the vascular remodeling associated with obesity. Int. J. Obes. 2014, 38, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Murdoch, C.E.; Brewer, A.C.; Ivetic, A.; Evans, P.; Shah, A.M.; Zhang, M. Endothelial NADPH oxidase 4 protects against angiotensin II-induced cardiac fibrosis and inflammation. ESC Heart Fail. 2021, 8, 1427–1437. [Google Scholar] [CrossRef]
- Broekmans, K.; Giesen, J.; Menges, L.; Koesling, D.; Russwurm, M. Angiotensin II-Induced Cardiovascular Fibrosis Is Attenuated by NO-Sensitive Guanylyl Cyclase1. Cells 2020, 9, 2436. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Brugnolaro, C.; Ibarrola, J.; Ravassa, S.; Buonafine, M.; Lopez, B.; Fernandez-Celis, A.; Querejeta, R.; Satamaria, E.; Fernandez-Irigoyen, J.; et al. CT-1 (Cardiotrophin-1)-Gal-3 (Galectin-3) Axis in Cardiac Fibrosis and Inflammation. Hypertension 2019, 73, 602–611. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Buonafine, M.; Boukhalfa, I.; Ibarrola, J.; Fernandez-Celis, A.; Kolkhof, P.; Rossignol, P.; Girerd, N.; Mulder, P.; Lopez-Andres, N.; et al. Aldosterone Target NGAL (Neutrophil Gelatinase-Associated Lipocalin) Is Involved in Cardiac Remodeling after Myocardial Infarction through NFkappaB Pathway. Hypertension 2017, 70, 1148–1156. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Machu, J.L.; Girerd, N.; Jaisser, F.; Thum, T.; Butler, J.; Gonzalez, A.; Diez, J.; Heymans, S.; McDonald, K.; et al. Rationale of the FIBROTARGETS study designed to identify novel biomarkers of myocardial fibrosis. ESC Heart Fail. 2018, 5, 139–148. [Google Scholar] [CrossRef]
- Safar, M.E.; Czernichow, S.; Blacher, J. Obesity, arterial stiffness, and cardiovascular risk. J. Am. Soc. Nephrol. 2006, 17, S109–S111. [Google Scholar] [CrossRef]
- Chen, Z.W.; Qian, J.Y.; Ma, J.Y.; Chang, S.F.; Yun, J.; Jin, H.; Sun, A.J.; Zou, Y.Z.; Ge, J.B. TNF-alpha-induced cardiomyocyte apoptosis contributes to cardiac dysfunction after coronary microembolization in mini-pigs. J. Cell Mol. Med. 2014, 18, 1953–1963. [Google Scholar] [CrossRef]
- Su, Q.; Li, L.; Sun, Y.; Yang, H.; Ye, Z.; Zhao, J. Effects of the TLR4/Myd88/NF-kappaB Signaling Pathway on NLRP3 Inflammasome in Coronary Microembolization-Induced Myocardial Injury. Cell Physiol. Biochem. 2018, 47, 1497–1508. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Lopez-Andres, N.; Jurado-Lopez, R.; Rousseau, E.; Bartolome, M.V.; Fernandez-Celis, A.; Rossignol, P.; Islas, F.; Antequera, A.; Prieto, S.; et al. Galectin-3 Participates in Cardiovascular Remodeling Associated with Obesity. Hypertension 2015, 66, 961–969. [Google Scholar] [CrossRef]
- Zhou, Y.; Long, M.Y.; Chen, Z.Q.; Huang, J.W.; Qin, Z.B.; Li, L. Downregulation of miR-181a-5p alleviates oxidative stress and inflammation in coronary microembolization-induced myocardial damage by directly targeting XIAP. J. Geriatr. Cardiol. 2021, 18, 426–439. [Google Scholar] [PubMed]
- Orlandi, M.; Masi, S.; Bhowruth, D.; Leira, Y.; Georgiopoulos, G.; Yellon, D.; Hingorani, A.; Chiesa, S.T.; Hausenloy, D.J.; Deanfield, J.; et al. Remote Ischemic Preconditioning Protects against Endothelial Dysfunction in a Human Model of Systemic Inflammation: A Randomized Clinical Trial. Arter. Thromb. Vasc. Biol. 2021, 41, e417–e426. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Rosa, G.D.; Gitto, E.; Arrigo, T. Oxidative stress in obesity: A critical component in human diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef] [PubMed]
- McNulty, M.; Mahmud, A.; Spiers, P.; Feely, J. Collagen type-I degradation is related to arterial stiffness in hypertensive and normotensive subjects. J. Hum. Hypertens. 2006, 20, 867–873. [Google Scholar] [CrossRef]
- Johnston, E.F.; Gillis, T.E. Transforming growth factor beta-1 (TGF-beta1) stimulates collagen synthesis in cultured rainbow trout cardiac fibroblasts. J. Exp. Biol. 2017, 220, 2645–2653. [Google Scholar] [PubMed]
- Duncan, M.R.; Frazier, K.S.; Abramson, S.; Williams, S.; Klapper, H.; Huang, X.; Grotendorst, G.R. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: Down-regulation by cAMP. FASEB J. 1999, 13, 1774–1786. [Google Scholar] [CrossRef]
- Roy Sarkar, S.; Banerjee, S. Gut microbiota in neurodegenerative disorders. J. Neuroimmunol. 2019, 328, 98–104. [Google Scholar] [CrossRef]
- Wu, S.; Liu, X.; Jiang, R.; Yan, X.; Ling, Z. Roles and Mechanisms of Gut Microbiota in Patients with Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 650047. [Google Scholar] [CrossRef]
- Muscogiuri, G.; Cantone, E.; Cassarano, S.; Tuccinardi, D.; Barrea, L.; Savastano, S.; Colao, A.; on behalf of the Obesity Programs of nutrition, Education Research Assessment Group. Gut microbiota: A new path to treat obesity. Int. J. Obes. Suppl. 2019, 9, 10–19. [Google Scholar] [CrossRef]
- Leocadio, P.C.L.; Oria, R.B.; Crespo-Lopez, M.E.; Alvarez-Leite, J.I. Obesity: More Than an Inflammatory, an Infectious Disease? Front. Immunol. 2019, 10, 3092. [Google Scholar] [CrossRef]
- Boulange, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Homma, T.; Kobayashi, S.; Seo, H.G. Mutual interaction between oxidative stress and endoplasmic reticulum stress in the pathogenesis of diseases specifically focusing on non-alcoholic fatty liver disease. World J. Biol. Chem. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 2012, 14, 434–456. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Escribano-Lopez, I.; Banuls, C.; Diaz-Morales, N.; Iannantuoni, F.; Rovira-Llopis, S.; Gomis, R.; Rocha, M.; Hernandez-Mijares, A.; Murphy, M.P.; Victor, V.M. The Mitochondria-Targeted Antioxidant MitoQ Modulates Mitochondrial Function and Endoplasmic Reticulum Stress in Pancreatic beta Cells Exposed to Hyperglycaemia. Cell Physiol. Biochem. 2019, 52, 186–197. [Google Scholar]
- Fu, H.Y.; Okada, K.; Liao, Y.; Tsukamoto, O.; Isomura, T.; Asai, M.; Okuda, K.; Asano, Y.; Sanada, S.; Asanuma, H.; et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010, 122, 361–369. [Google Scholar] [CrossRef]
- Gregor, M.F.; Yang, L.; Fabbrini, E.; Mohammed, B.S.; Eagon, J.C.; Hotamisligil, G.H.; Klein, S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 2009, 58, 693–700. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.; Zazueta, C.; Aguilera-Aguirre, L. Oxidative Stress and Inflammation in Cardiovascular Disease. Oxid. Med. Cell Longev. 2017, 2017, 5853238. [Google Scholar] [CrossRef]
- Martin, R.; Miana, M.; Jurado-Lopez, R.; Martinez-Martinez, E.; Gomez-Hurtado, N.; Delgado, C.; Bartolome, M.V.; San Roman, J.A.; Cordoba, C.; Lahera, V.; et al. DIOL triterpenes block profibrotic effects of angiotensin II and protect from cardiac hypertrophy. PLoS ONE 2012, 7, e41545. [Google Scholar] [CrossRef]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Cabandugama, P.K.; Gardner, M.J.; Sowers, J.R. The Renin Angiotensin Aldosterone System in Obesity and Hypertension: Roles in the Cardiorenal Metabolic Syndrome. Med. Clin. N. Am. 2017, 101, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Menikdiwela, K.R.; Ramalingam, L.; Allen, L.; Scoggin, S.; Kalupahana, N.S.; Moustaid Moussa, N. Angiotensin II Increases Endoplasmic Reticulum Stress in Adipose Tissue and Adipocytes. Sci. Rep. 2019, 9, 8481. [Google Scholar] [CrossRef]
- Ha, T.S.; Park, H.Y.; Seong, S.B.; Ahn, H.Y. Angiotensin II induces endoplasmic reticulum stress in podocyte, which would be further augmented by PI3-kinase inhibition. Clin. Hypertens 2015, 21, 13. [Google Scholar] [CrossRef]
- Ramalingam, L.; Sopontammarak, B.; Menikdiwela, K.R.; Moustaid-Moussa, N. Endoplasmic Reticulum (ER) Stress in Part Mediates Effects of Angiotensin II in Pancreatic Beta Cells. Diabetes Metab. Syndr. Obes. 2020, 13, 2843–2853. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef]
- Chen, Q.; Thompson, J.; Hu, Y.; Das, A.; Lesnefsky, E.J. Metformin attenuates ER stress-induced mitochondrial dysfunction. Transl. Res. 2017, 190, 40–50. [Google Scholar] [CrossRef]
- da Silva-Bertani, D.C.T.; Vileigas, D.F.; Mota, G.A.F.; de Souza, S.L.B.; Sant’Ana, P.G.; Freire, P.P.; de Tomasi, L.C.; Correa, C.R.; Padovani, C.R.; Fernandes, T.; et al. Increased angiotensin II from adipose tissue modulates myocardial collagen I and III in obese rats. Life Sci. 2020, 252, 117650. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souza-Neto, F.V.; Jiménez-González, S.; Delgado-Valero, B.; Jurado-López, R.; Genty, M.; Romero-Miranda, A.; Rodríguez, C.; Nieto, M.L.; Martínez-Martínez, E.; Cachofeiro, V. The Interplay of Mitochondrial Oxidative Stress and Endoplasmic Reticulum Stress in Cardiovascular Fibrosis in Obese Rats. Antioxidants 2021, 10, 1274. https://doi.org/10.3390/antiox10081274
Souza-Neto FV, Jiménez-González S, Delgado-Valero B, Jurado-López R, Genty M, Romero-Miranda A, Rodríguez C, Nieto ML, Martínez-Martínez E, Cachofeiro V. The Interplay of Mitochondrial Oxidative Stress and Endoplasmic Reticulum Stress in Cardiovascular Fibrosis in Obese Rats. Antioxidants. 2021; 10(8):1274. https://doi.org/10.3390/antiox10081274
Chicago/Turabian StyleSouza-Neto, Francisco V., Sara Jiménez-González, Beatriz Delgado-Valero, Raquel Jurado-López, Marie Genty, Ana Romero-Miranda, Cristina Rodríguez, María Luisa Nieto, Ernesto Martínez-Martínez, and Victoria Cachofeiro. 2021. "The Interplay of Mitochondrial Oxidative Stress and Endoplasmic Reticulum Stress in Cardiovascular Fibrosis in Obese Rats" Antioxidants 10, no. 8: 1274. https://doi.org/10.3390/antiox10081274
APA StyleSouza-Neto, F. V., Jiménez-González, S., Delgado-Valero, B., Jurado-López, R., Genty, M., Romero-Miranda, A., Rodríguez, C., Nieto, M. L., Martínez-Martínez, E., & Cachofeiro, V. (2021). The Interplay of Mitochondrial Oxidative Stress and Endoplasmic Reticulum Stress in Cardiovascular Fibrosis in Obese Rats. Antioxidants, 10(8), 1274. https://doi.org/10.3390/antiox10081274