2.1. Materials
The aim of this study was to compare the predictive performances of three approaches for predicting drug pharmacokinetics over time of the antibiotic Dalbavancin, namely, compartment analysis, NLME and NODEs.
The dataset used in this study was collected from a cohort of 218 patients undergoing Dalbavancin treatment and who underwent therapeutic drug monitoring (TDM) of Dalbavancin at the IRCCS, Azienda Ospedaliero Universitaria di Bologna between April 2021 and December 2024. Dalbavancin is a long-acting antibiotic used for the treatment of skin and soft tissue infections and as a second-line agent in patients with staphylococcal infections, such as bone and joint infections, vascular prosthetic joint infections and endocarditis. At our center, Dalbavancin plasma exposure is optimized by means of TDM, which is the medical practice of measuring drug concentration in plasma in order to guide drug dosing for attaining a pre-defined target of efficacy. Dalbavancin is a semisynthetic lipoglycopeptide derived from the teicoplanin-like antibiotic A-40926, which is an agent naturally produced by the actinomycete Nonomuria spp. An important step in the synthesis of Dalbavancin is the amidation of the peptide carboxyl group in the teicoplanin structure with a 3,3 dimethylaminopropylamide group, resulting in the improved potency of Dalbavancin against staphylococci. Moreover, the removal of the acetylglucosamine group of Dalbavancin with respect to teicoplanin resulted in the improved activity against enterococci, even if Van-A enterococci remained resistant to Dalbavancin. The long lipophilic side chain in the Dalbavancin molecule serves a dual purpose by both extending the half-life, as well as anchoring the compound to the membrane, which enhances its antimicrobial activity [
8].
Targeting a Dalbavancin plasma concentration above an efficacy threshold, previously defined to be of
, may be challenged by pharmacokinetic variability. A Bayesian estimation approach aimed at forecasting the duration of concentration persisting over
was recently developed by means of standard population pharmacokinetics methods (NLME) [
9] and validated in a small cohort of patients.
The data acquisition process involved recording patient covariates, drug administration details and serial blood sampling for pharmacokinetic analysis. Dalbavancin doses ranged from to , with a total of 703 administrations recorded. Blood samples were obtained at various time points following drug administration, resulting in 669 concentration measurements. Standardized laboratory procedures were followed for quantifying Dalbavancin levels in plasma, ensuring accuracy and consistency across all samples.
All patients included in the study were adults, with 145 males and 73 females. All the dataset’s covariates collected were: age, height, weight, sex and serum creatine concentration. The values of these covariates are summarized in
Table 1.
To ensure robust model evaluation, we employed a cross-validation approach. Given the dataset size and the need for a balance between the training and validation data, we opted for a 6-fold cross-validation framework. This choice provides a good trade-off between statistical reliability and computational efficiency, allowing each model to be trained and validated on multiple subsets of the data while maintaining a sufficient amount of training data per fold.
2.2. Methods
The proposed model for fitting drug concentration curves is constructed using Neural Ordinary Differential Equations (NODEs) [
5,
10].
NODEs are a machine learning technique that extends traditional neural networks by formulating the transformation of hidden states of the neural network (NN) as a continuous-time dynamical system. The right-hand side of a differential equation is substituted with an NN, and then it is integrated through a traditional ODE solver (i.e., Runge–Kutta methods).
To model drug concentration over time, a nonlinear regression approach has been employed. The optimization is performed using the standard squared loss, given by:
where
M denotes the total number of patients,
represents the number of concentration measurements for patient
k,
corresponds to the observed drug concentration at time
and
is the predicted concentration obtained from the NODE model. The PyTorch V2.6.0 implementation of differentiable ODE solvers (torchdiffeq [
11]) has been used to perform gradient descent and optimize the NN parameter.
Since the compartment models are systems of differential equations, the replacement of these equations with an NN is direct:
The differential equation systems (
2a) represent a simple two-compartment model with the four parameters that need to be fitted:
and
are the volumes of the central and peripheral compartments,
the clearance,
Q the intercompartmental clearance and
and
the concentrations of the two compartments. Equation (2b) shows instead the substitution of all the right-hand side equations with the NN, which, in this case, became a one-dimensional expression;
represents the hidden state of the NODE that, in this work, represents the drug concentration, and
is the vector of the parameters of the NN.
The specific structure of NNs allows for the direct inclusion of patients’ covariates as model input. In this framework, during training, the NN learns how to integrate covariates into the prediction of concentration curves, inherently capturing all possible correlations. This approach is significantly more efficient than the traditional method of incorporating covariates into compartmental models and NLME. Moreover, it enables a fully data-driven approach, eliminating the need for prior assumptions about the system’s underlying relationships.
The NN represented as
in Equation (2b) is a feedforward neural network (FNN), for which two different architectures have been considered in this work. The first and simpler architecture takes four input: time, concentration, number of previous administrations and dose. It produces a single output, i.e., the derivative of the concentration curve. The network includes two hidden layers with 20 neurons each, selected via elbow rule. Different numbers of hidden layers were tested, and two was the smallest number at which the model’s performance reached a plateau; see
Appendix A. The activation function used is softplus. This architecture, without covariates, was employed to first compare NODEs with NLMEs and the traditional two-compartment model when provided with the same information. The covariates were not included in the NLME and two-compartment models because, when tested, their inclusion did not improve the results; see
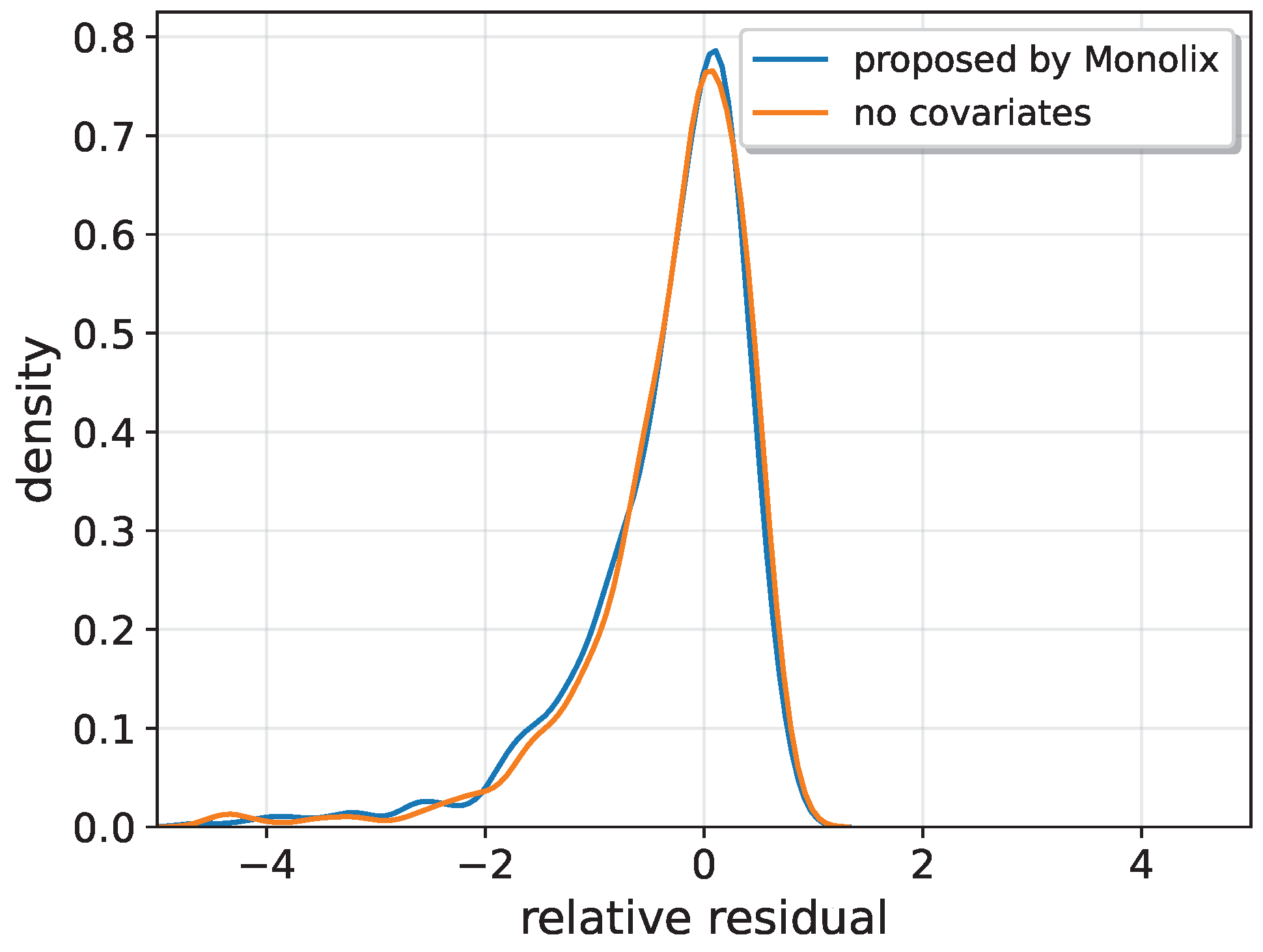
Appendix B. Therefore, no covariates have been included in these models. In this way, the comparison between the NODE (both with and without covariates) and the other models was always conducted using the most accurate and well-optimized versions of each model.
The second architecture follows a similar FNN structure but takes nine input variables: the original four together with five covariates. Additionally, an extra hidden layer with 20 neurons was added to take into account the increasing complexity of the model.
To account for multiple administrations, the integration of the NODE is performed piece-wise over the interval between two consecutive doses. The initial condition for each subsequent administration incorporates the residual concentration at that time point, ensuring continuity in the system’s state evolution. This approach effectively treats the problem as a sequence of Initial Value Problems (IVP), each solved independently over its respective subinterval while maintaining consistency across transitions. By adopting this framework, the model captures the cumulative effects of repeated dosing, addressing nonlinearity in drug accumulation and clearance dynamics. The piece-wise integration scheme also facilitates efficient numerical implementation and execution time both for the forward and backward pass. In this way, it is not necessary to integrate the NODE in each administration for a single patient up to the required time to then apply the superposition principle. This results in a more linear and stable gradient computation.
In addition to the main FNN, the model includes an additional parameter, the distribution volume (), which is learned through gradient descent with the whole FNN. This parameter is essential for converting the administered dose into a concentration, analogous to the compartment volume in a two-compartment model. However, unlike in the compartmental approach, where two distinct compartments correspond to two separate volumes, the neural ordinary differential equation (NODE) framework employs a single global volume, making direct comparisons between the models unsuitable.
In the case where covariates are included, the distribution volume is estimated using a simple FNN constituted by five input nodes corresponding to the five covariates, a single output and one hidden layer with 10 neurons with a softplus activation function.
A schematic view of the working principle of the algorithm is summarized in
Figure 1.
The dataset employed in this study is characterized by a sparse and irregular sampling of drug concentration measurements across patients. Frequently, patients undergo multiple drug administrations before any pharmacokinetic measurement is recorded, with some measurements occurring only after several doses. This discontinuity and sparsity introduce significant challenges for model training, particularly in maintaining numerical stability and robustness.
To mitigate these issues, a data augmentation strategy was adopted. During each fold of the cross-validation process, a two-compartment pharmacokinetic model was fitted to the available data using nonlinear least squares estimation. The fitted parameters were then used to simulate drug concentration-time profiles under various dosing regimens. From each simulated profile, 50 time points were sampled on a logarithmic time scale to generate synthetic training data. While the use of synthetic data for machine learning is known to down-perform in prediction accuracy and precision with respect to real data trainings [
12], in this study, an ablation analysis comparing training with and without synthetic pre-training was attempted. However, training without synthetic data proved largely unstable in this study due to the discontinuous and sparse nature of the original dataset. In most cases, gradient divergence prevented full convergence of the models, making a fair performance comparison unfeasible. Nevertheless, the results suggest that biases introduced by the parametric model during pre-training are effectively mitigated during subsequent fine-tuning on real data, which does not carry the same parametric model biases.
The synthetic data were used to perform an initial pre-training phase for the NODE, aimed at guiding the model to approximate the expected trend of the concentration curve. Following this step, the model underwent fine-tuning using real-world data. The pre-training phase was carried out over 100 epochs using the Adam optimizer [
13], with a learning rate of
and a weight decay of
. The fine-tuning process began with an additional 100 epochs using the same optimizer and hyper-parameter settings. Subsequently, the model was trained for another 100 epochs, with a reduced learning rate of
.
The performance of the resulting NODE has been compared to two of the most widely used models in pharmacokinetics: the compartment models and the NLME models.
In the case of Dalbavancin, multiple compartment models with varying numbers of compartments were evaluated, and the two-compartment model, outlined in
Figure 2, has been chosen for the comparison since it provided the best performances [
14,
15]. For the two-compartment model, the following parameters need to be estimated: the volumes of the two compartments (
and
), the intercompartmental clearance (
Q) and the clearance (
).
In this work, the fitting procedures of the two compartment models have been performed via nonlinear least squares (NLLS).
For what concerns the NLME model, the Monolix software [
16] has been used to perform the fitting and compute the predictions. The popPK model settings used in Monolix are: two-compartment distribution, infusion, no delay and linear elimination, where the drug is eliminated from the central compartment at a rate proportional to its concentration. This is represented by a linear dependence on the concentration of the central compartment in the ODE that represents the system (Equation (
2a)). Covariates can also be incorporated into the model for each parameter. Typically, when a covariate is included in the model through a specific parameter (let
be that parameter), an additional population parameter,
, is introduced. This parameter is modeled exponentially as
and is estimated along with the other parameters. To estimate the model parameters, Maximum Likelihood Estimation has been used.
Finally, in many fields, particularly in clinical applications, model explainability can be just as crucial as prediction accuracy. Over the years, several methods have been developed to improve explainability. Many of these algorithms rely on similar underlying assumptions and can be integrated into a unified framework: Shapley Additive Explanations (SHAP) [
17]. This framework aims to quantify the contribution of each input feature through the computation of Shapley values [
18]. In this work, these values were calculated for both NODE configurations, with and without covariates. The Shapley values provide insights into the model’s behavior and enable an analysis of the models personalization capabilities.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







