
QUBO Problem Formulation of Fragment-Based Protein–Ligand Flexible Docking




Abstract
1. Introduction
2. QUBO Problem Formulation of Fragment-Based Docking
2.1. Fundamental Factors of Fragment-Based Docking Calculation
- Fragment of and fragment of are different.
- and do not clash.
- If and are connected in the compound structure of interest, the placements , can be connected in the same manner as the compound.
- Decompose a compound into multiple fragments (chemical substructures with no internal degree of freedom) and regard chemical compound structure as a set of fragment placements.
- Enumerate candidate fragment placements by independent fragment docking.
- Choose a single placement for each fragment.
- Consider clashes between fragments.
- For fragments that have covalent bonds with each other, consider the bond distance between the placements.
2.2. Similarities and Differences between Polyomino Puzzle and Fragment-Based Docking
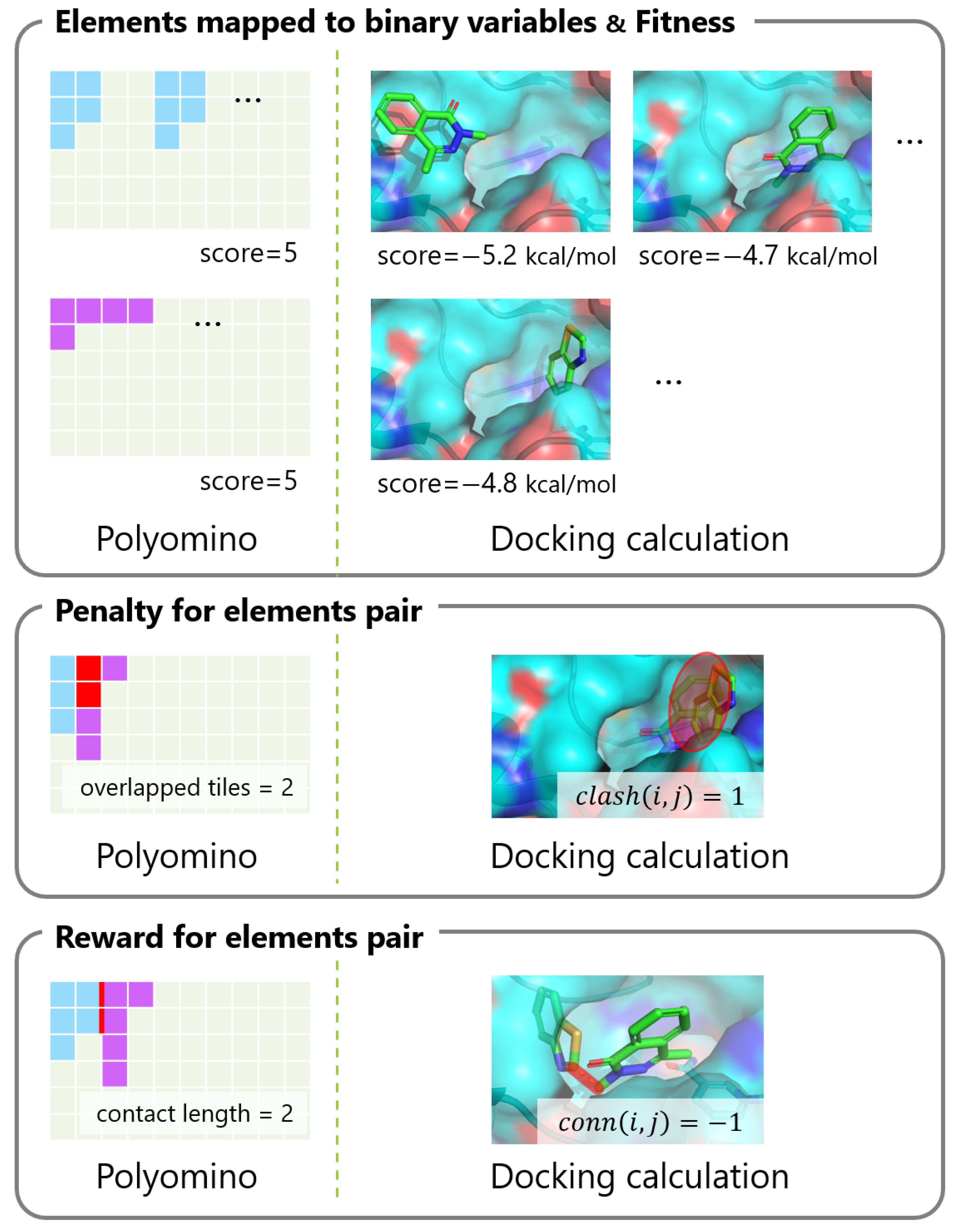
2.3. Elements to Be Mapped to Binary Variables
2.4. Fitness for Each Variable
2.5. Penalty for Element Pair
2.6. Reward for Element Pair
2.7. Constraints for the Number of Selected Placements
3. Materials and Methods
3.1. Method Overview
3.2. Pose Enumeration with Protein–Fragment Rigid Docking
3.3. QUBO Formulation

- Furthermore, since the term is a constraint term whose condition must be satisfied, the weight D should be larger. In this study, the weights of each term were determined as according to a prior experiment based on the aforementioned assumptions, with the constant fixed.
3.4. Criteria of Covalent Bonding and Collision
- Calculate interaction energy for each fragment placements pair without adding a covalent bond.
- Calculate interaction energy for each fragment placement pair with the addition of a covalent bond if the fragments and have a covalent bond.
- Set if the fragments and have a covalent bond and ; otherwise, set . Note that is an energy tolerance level.
- Set if and ; otherwise, set .
3.5. Combinatorial Optimization by SQBM+
3.6. Postprocessing
3.7. Dataset Preparation
4. Results and Discussion
4.1. Fragment Docking
4.2. Local Solutions Enumerated by SQBM+
4.3. Energy Minimization of Reconstructed Compound Structure
4.4. Toward Virtual Screening Applications
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALDR | Aldose Reductase |
| FPGA | Field-Programmable Gate Array |
| GPU | Graphics Processing Unit |
| OPLS | Optimized Potentials for Liquid Simulations |
| PDB | Protein Data Bank |
| QUBO | Quadratic Unconstrained Binary Optimization |
| RMSD | Root Mean Square Deviation |
| SB | Simulated Bifurcation |
| SBM | Simulated Bifurcation Machine |
| SMILES | Simplified Molecular Input Line Entry System |
| UFF | Universal Force Field |
References
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A Fast Flexible Docking Method using an Incremental Construction Algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K.; Kubota, R.; Yoshikawa, Y.; Ohue, M.; Akiyama, Y. Effective Protein–Ligand Docking Strategy via Fragment Reuse and a Proof-of-Concept Implementation. ACS Omega 2022, 7, 30265–30274. [Google Scholar] [CrossRef]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. eHiTS: A new fast, exhaustive flexible ligand docking system. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Lenz, W. Beitrag zum Verständnis der magnetischen Erscheinungen in festen Körpern. Z. Phys. 1920, 21, 613–615. [Google Scholar]
- Eagle, A.; Kato, T.; Minato, Y. Solving tiling puzzles with quantum annealing. arXiv 2019, arXiv:1904.01770. [Google Scholar]
- Jain, S. Solving the Traveling Salesman Problem on the D-Wave Quantum Computer. Front. Phys. 2021, 9, 760783. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Leleu, T.; Ganguli, S.; Mabuchi, H. Coherent Ising machines-Quantum optics and neural network Perspectives. Appl. Phys. Lett. 2020, 117, 160501. [Google Scholar] [CrossRef]
- Aramon, M.; Rosenberg, G.; Valiante, E.; Miyazawa, T.; Tamura, H.; Katzgraber, H.G. Physics-Inspired Optimization for Quadratic Unconstrained Problems Using a Digital Annealer. Front. Phys. 2019, 7, 48. [Google Scholar] [CrossRef]
- Goto, H.; Tatsumura, K.; Dixon, A.R. Combinatorial optimization by simulating adiabatic bifurcations in nonlinear Hamiltonian systems. Sci. Adv. 2019, 5, eaav2372. [Google Scholar] [CrossRef]
- Kajiura, M.; Akiyama, Y.; Anzai, Y. Solving large scale puzzles with neural networks. In Proceedings of the IEEE International Workshop on Tools for Artificial Intelligence, Fairfax, VA, USA, 23–25 October 1989; pp. 562–569. [Google Scholar] [CrossRef]
- Manabe, S.; Asai, H. A Neuro-Based Optimization Algorithm for Tiling Problems with Rotation. Neural Process. Lett. 2001, 13, 267–275. [Google Scholar] [CrossRef]
- Takabatake, K.; Yanagisawa, K.; Akiyama, Y. Solving Generalized Polyomino Puzzles Using the Ising Model. Entropy 2022, 24, 354. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Ogata, K.; Isomura, T.; Utsunomiya, S.; Yamamoto, Y.; Aihara, K. Boltzmann Sampling by Degenerate Optical Parametric Oscillator Network for Structure-Based Virtual Screening. Entropy 2016, 18, 365. [Google Scholar] [CrossRef]
- Banchi, L.; Fingerhuth, M.; Babej, T.; Ing, C.; Arrazola, J.M. Molecular docking with Gaussian Boson Sampling. Sci. Adv. 2020, 6, eaax1950. [Google Scholar] [CrossRef]
- Zha, J.; Su, J.; Li, T.; Cao, C.; Ma, Y.; Wei, H.; Huang, Z.; Qian, L.; Wen, K.; Zhang, J. Encoding Molecular Docking for Quantum Computers. J. Chem. Theory Comput. 2023, 19, 9018–9024. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Toshiba Digital Solutions Corporation. About SQBM+. Available online: https://www.global.toshiba/ww/products-solutions/ai-iot/sbm/intro.html (accessed on 28 April 2024).
- Tatsumura, K.; Hidaka, R.; Nakayama, J.; Kashimata, T.; Yamasaki, M. Pairs-Trading System Using Quantum-Inspired Combinatorial Optimization Accelerator for Optimal Path Search in Market Graphs. IEEE Access 2023, 11, 104406–104416. [Google Scholar] [CrossRef]
- Oya, K.; Fujimoto, H.; Hamakawa, Y.; Yamasaki, M.; Tatsumura, K. Proposal and Prototyping of Automotive Computing Platform with Quantum inspired Processing Unit. Trans. Soc. Automot. Eng. Jpn. 2023, 54, 1216–1221. [Google Scholar]
- Mpamhanga, C.P.; Chen, B.; McLay, I.M.; Willett, P. Knowledge-Based Interaction Fingerprint Scoring: A Simple Method for Improving the Effectiveness of Fast Scoring Functions. J. Chem. Inf. Model. 2006, 46, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Westbrook, J.D.; Woo, J.; et al. The RCSB Protein Data Bank: Views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015, 43, D345–D356. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Komine, S.; Suzuki, S.D.; Ohue, M.; Ishida, T.; Akiyama, Y. Spresso: An ultrafast compound pre-screening method based on compound decomposition. Bioinformatics 2017, 33, 3836–3843. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyomino Puzzle | Docking Calculation | |
|---|---|---|
| Elements mapped to binary variables | placements of polyominos on the board | placements of fragments |
| in the protein pocket | ||
| Fitness for each element | sizes of polyominos | binding free energy scores |
| to the protein | ||
| Penalty for elements pair | overlaps between polyomino placements | clashes between fragment placements |
| Reward for elements pair | length of touching borders | chemical bond between |
| between placements | fragment placements | |
| Constraints for the number | a single placement per one polyomino | a single placement per one fragment |
| of selected elements |
| Target | Aldose reductase (ALDR) |
| PDB ID | 2HV5 |
| Box center | ( Å, Å, Å) |
| Volume of the docking region | 14 Å × 14 Å × 14 Å |
| The number of subregions | subregions of 2 Å × 2 Å × 2 Å |
| Fragment SMILES | Number of Poses | Binding Energy Range (kcal/mol) |
|---|---|---|
| FC(F)F | 982 | – |
| OC=O | 884 | – |
| Cc1nc2c(s1)cccc2 | 641 | – |
| O=c1[nH]nc(c2c1cccc2)C | 498 | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanagisawa, K.; Fujie, T.; Takabatake, K.; Akiyama, Y. QUBO Problem Formulation of Fragment-Based Protein–Ligand Flexible Docking. Entropy 2024, 26, 397. https://doi.org/10.3390/e26050397
Yanagisawa K, Fujie T, Takabatake K, Akiyama Y. QUBO Problem Formulation of Fragment-Based Protein–Ligand Flexible Docking. Entropy. 2024; 26(5):397. https://doi.org/10.3390/e26050397
Chicago/Turabian StyleYanagisawa, Keisuke, Takuya Fujie, Kazuki Takabatake, and Yutaka Akiyama. 2024. "QUBO Problem Formulation of Fragment-Based Protein–Ligand Flexible Docking" Entropy 26, no. 5: 397. https://doi.org/10.3390/e26050397
APA StyleYanagisawa, K., Fujie, T., Takabatake, K., & Akiyama, Y. (2024). QUBO Problem Formulation of Fragment-Based Protein–Ligand Flexible Docking. Entropy, 26(5), 397. https://doi.org/10.3390/e26050397