
Bis-Pyrene Photo-Switch Open- and Closed-Form Differently Bind to ds-DNA, ds-RNA and Serum Albumin and Reveal Light-Induced Bioactivity




Abstract
1. Introduction
2. Results
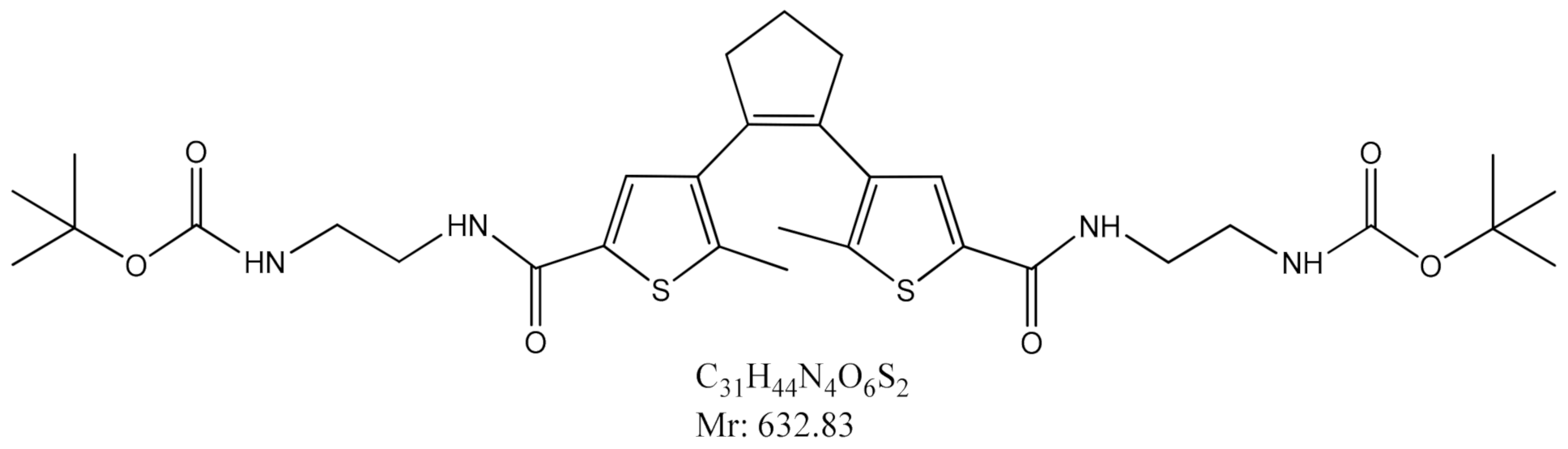
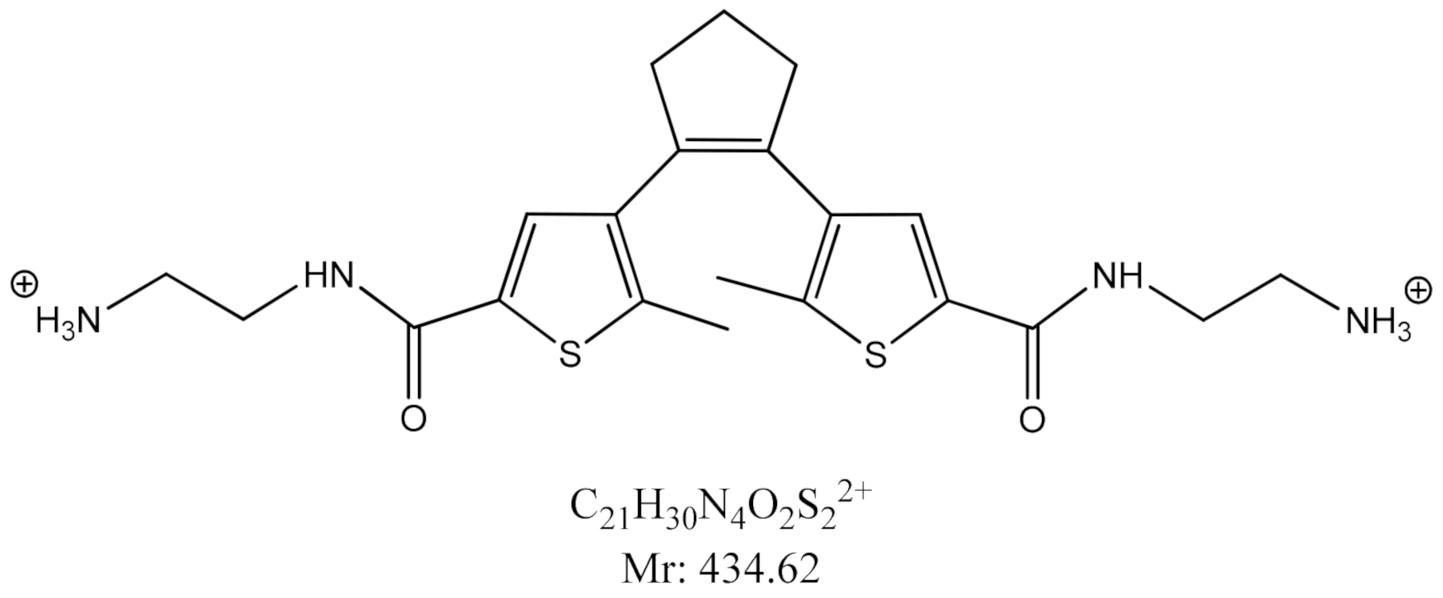
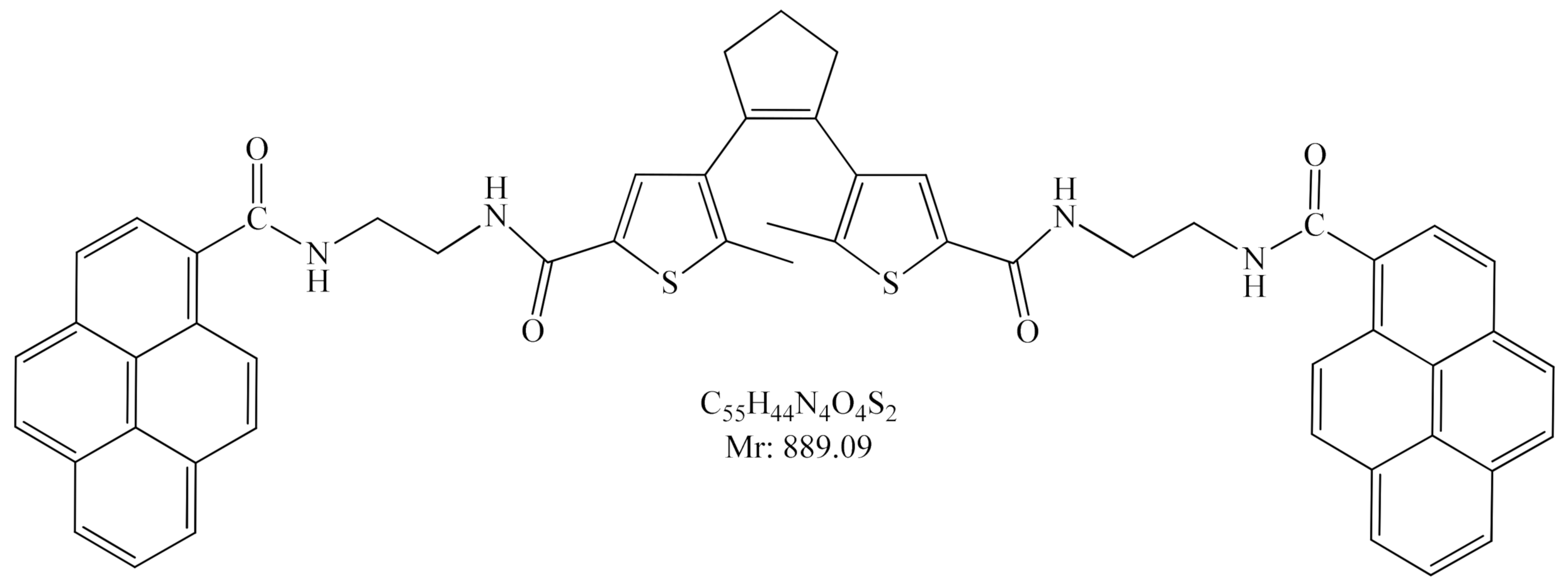
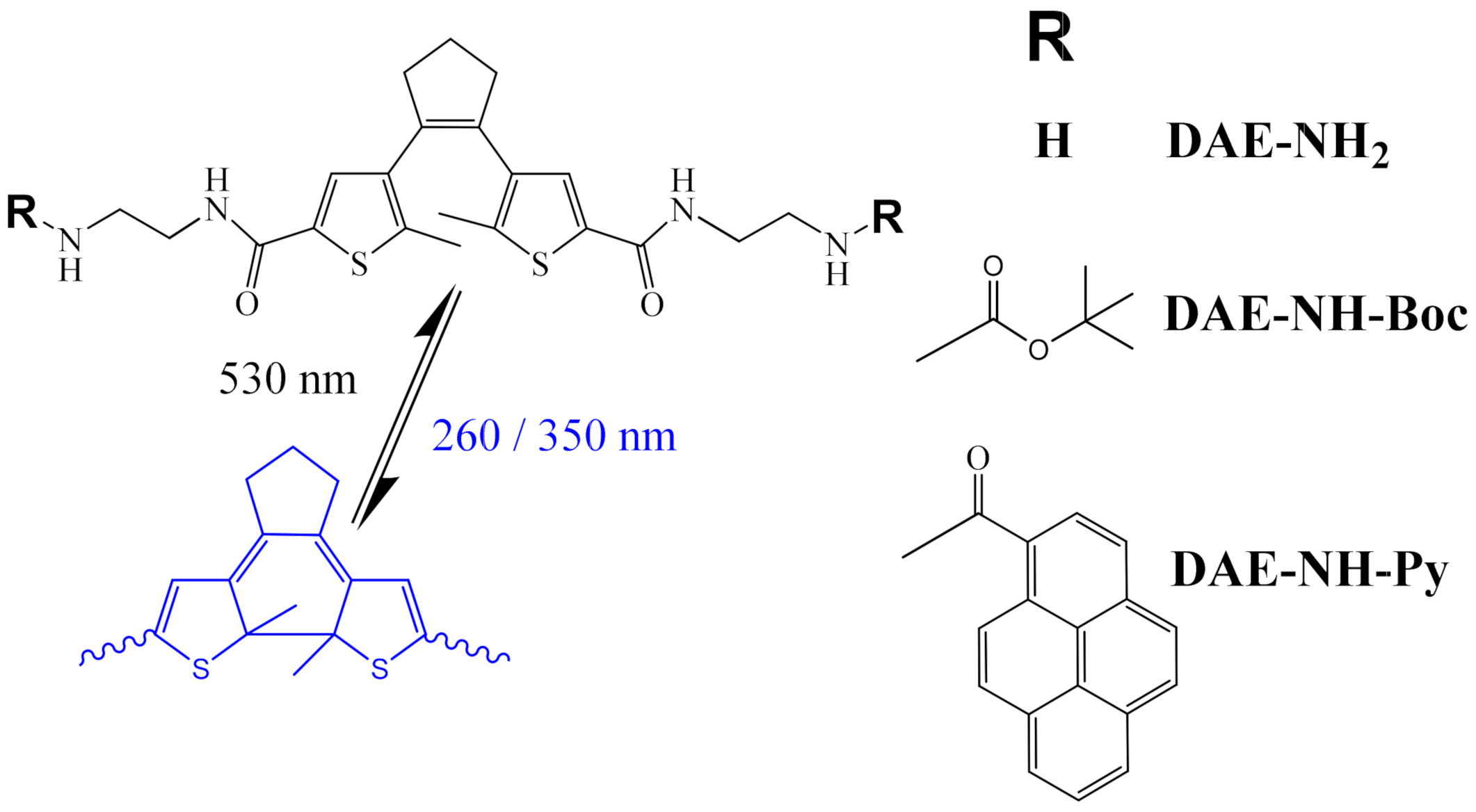
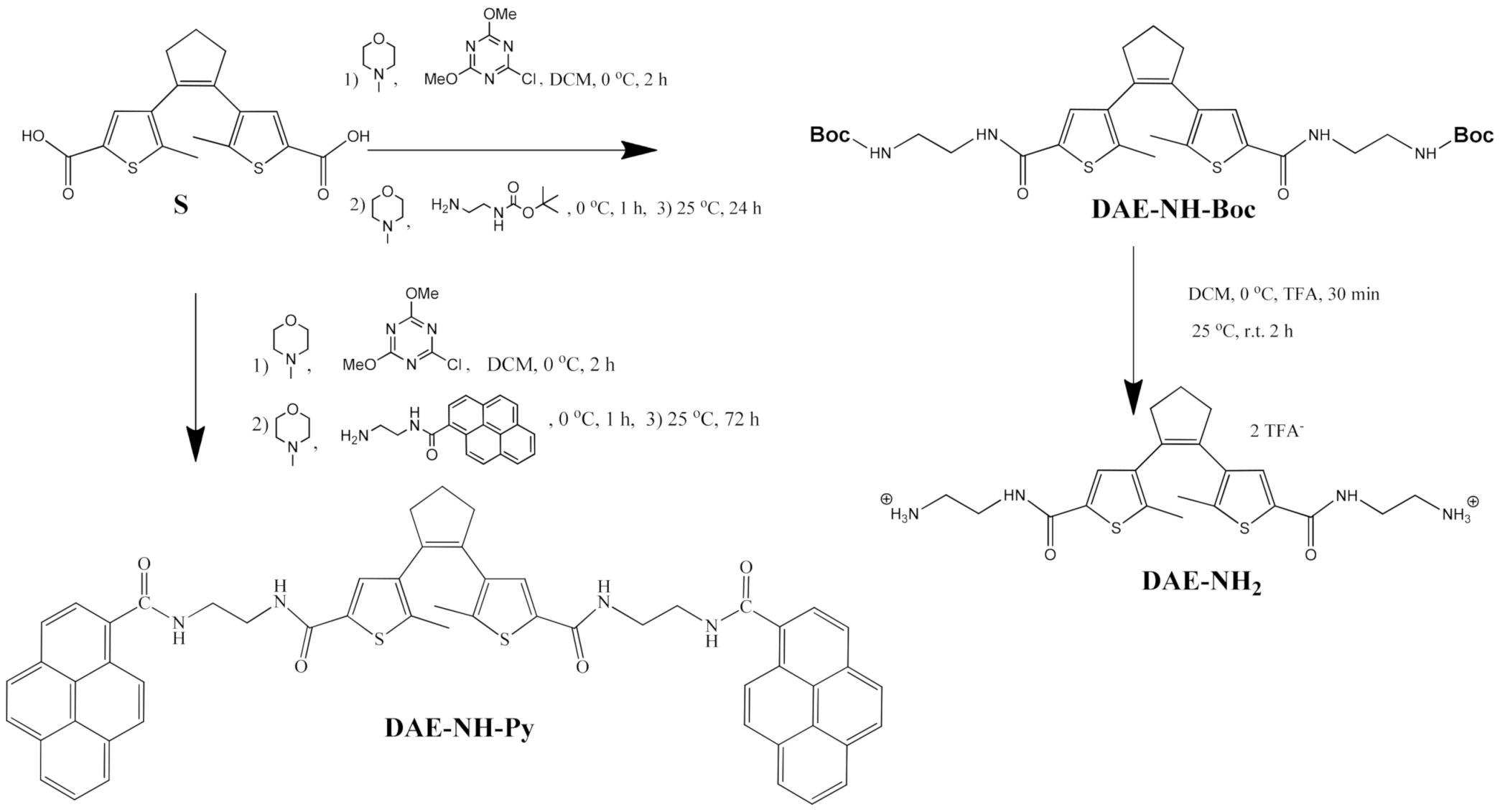
2.1. Synthesis
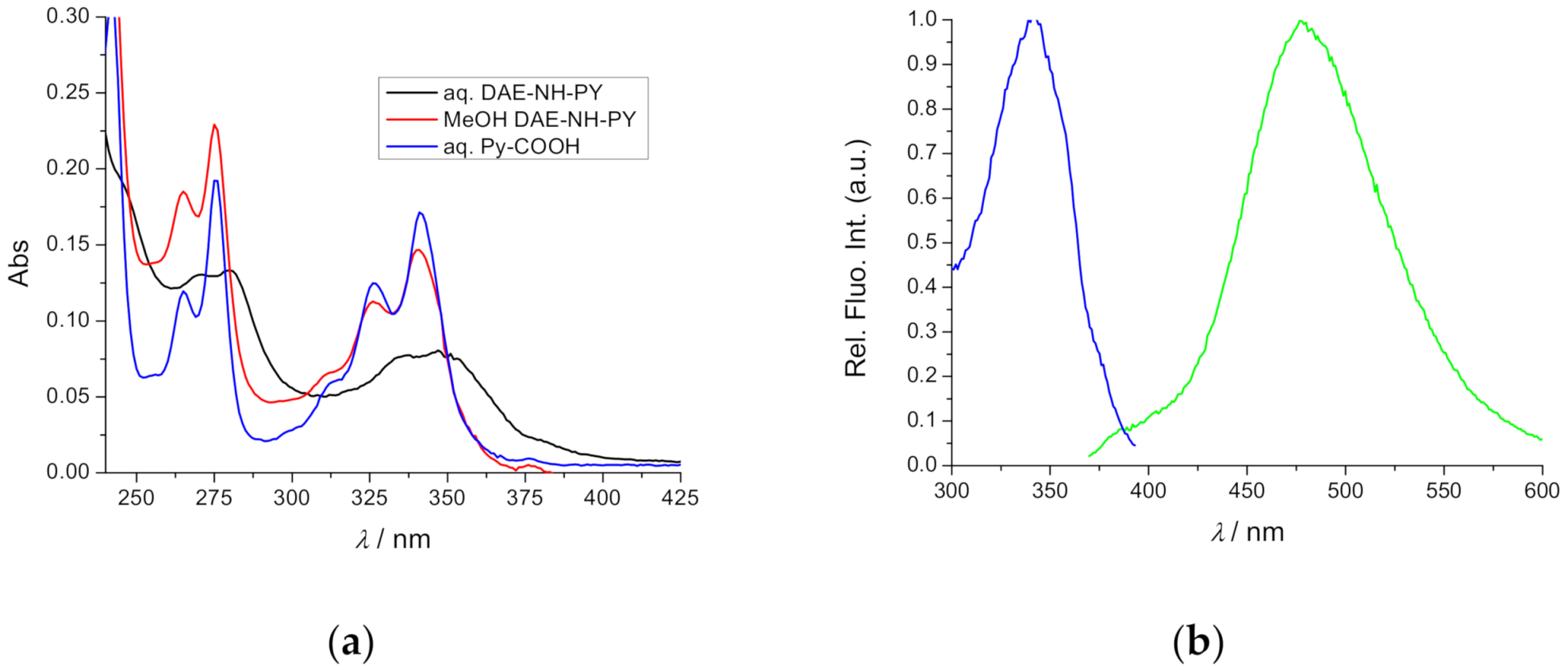
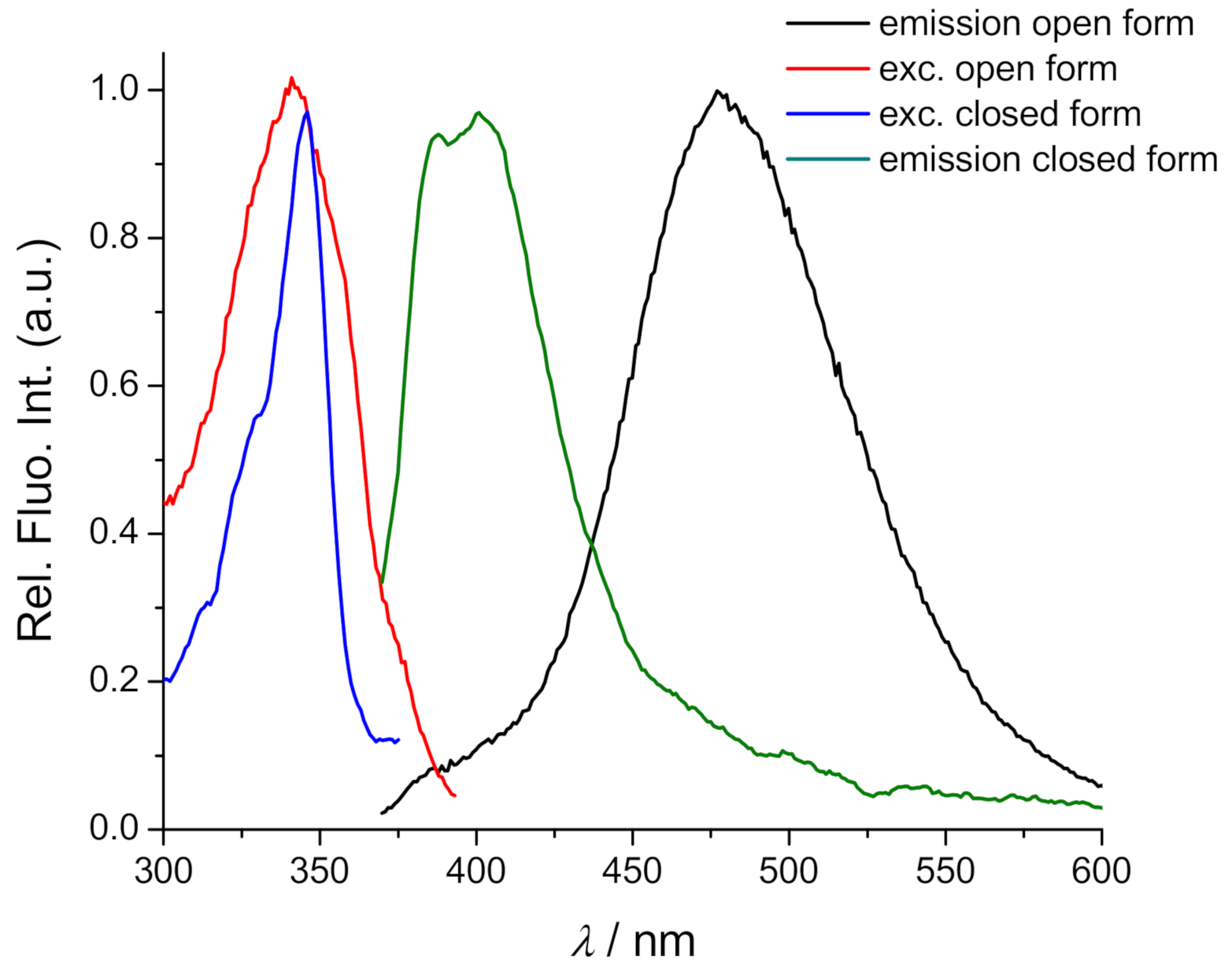
2.2. Spectrophotometric Characterisation
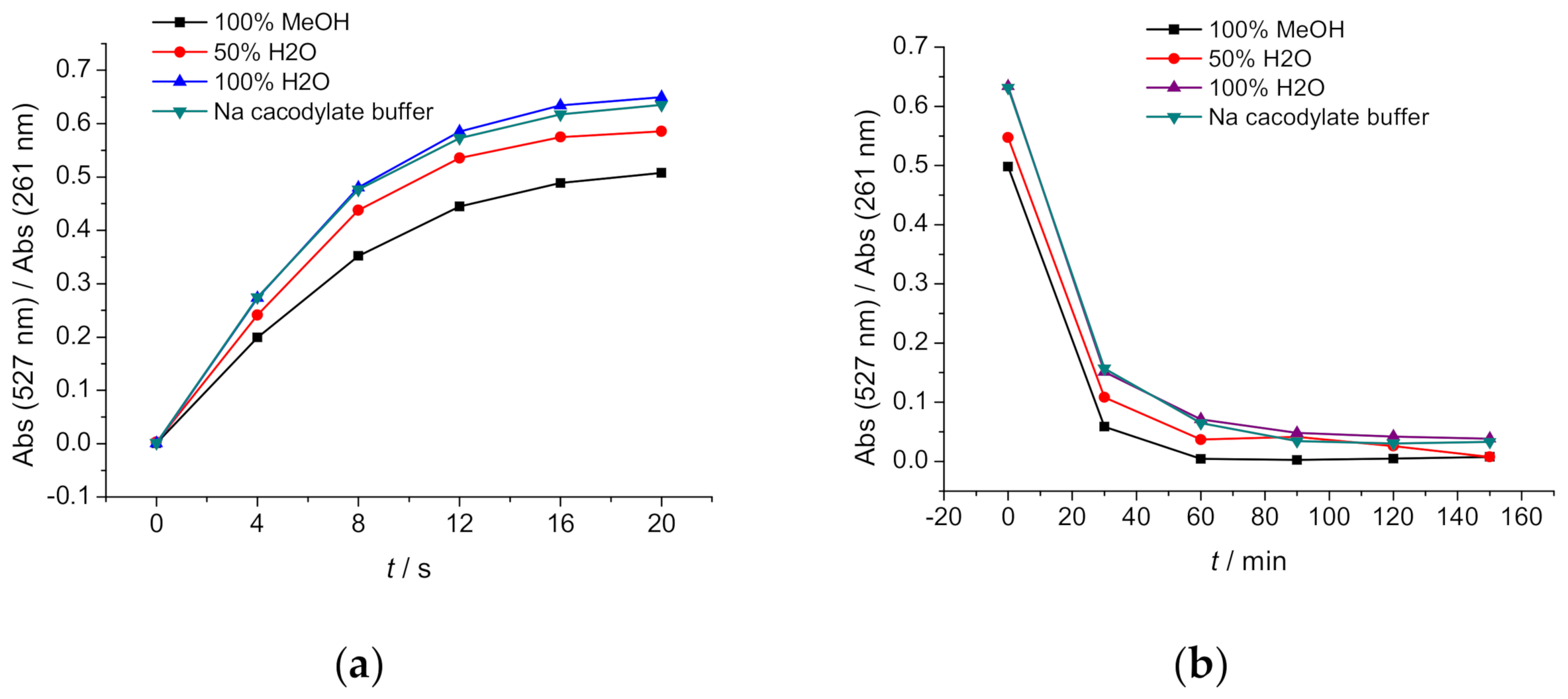
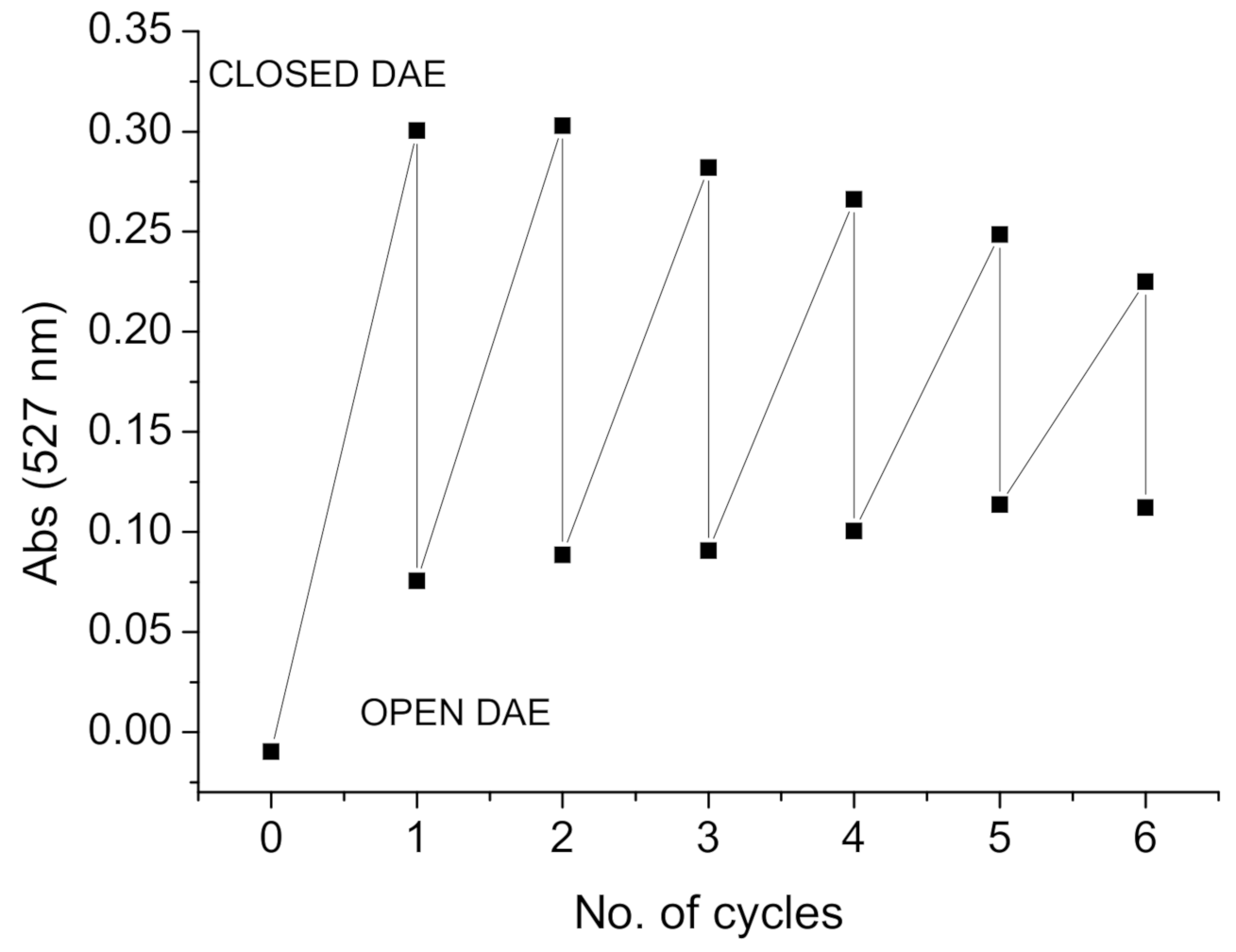
2.3. Photochemistry
2.3.1. Photo-Controlled Switching of DAE-NH-Boc, DAE-NH2 and DAE-NH-Py
2.4. Interactions with DNA/RNA
2.4.1. Thermal Denaturation of ds-DNA/RNA
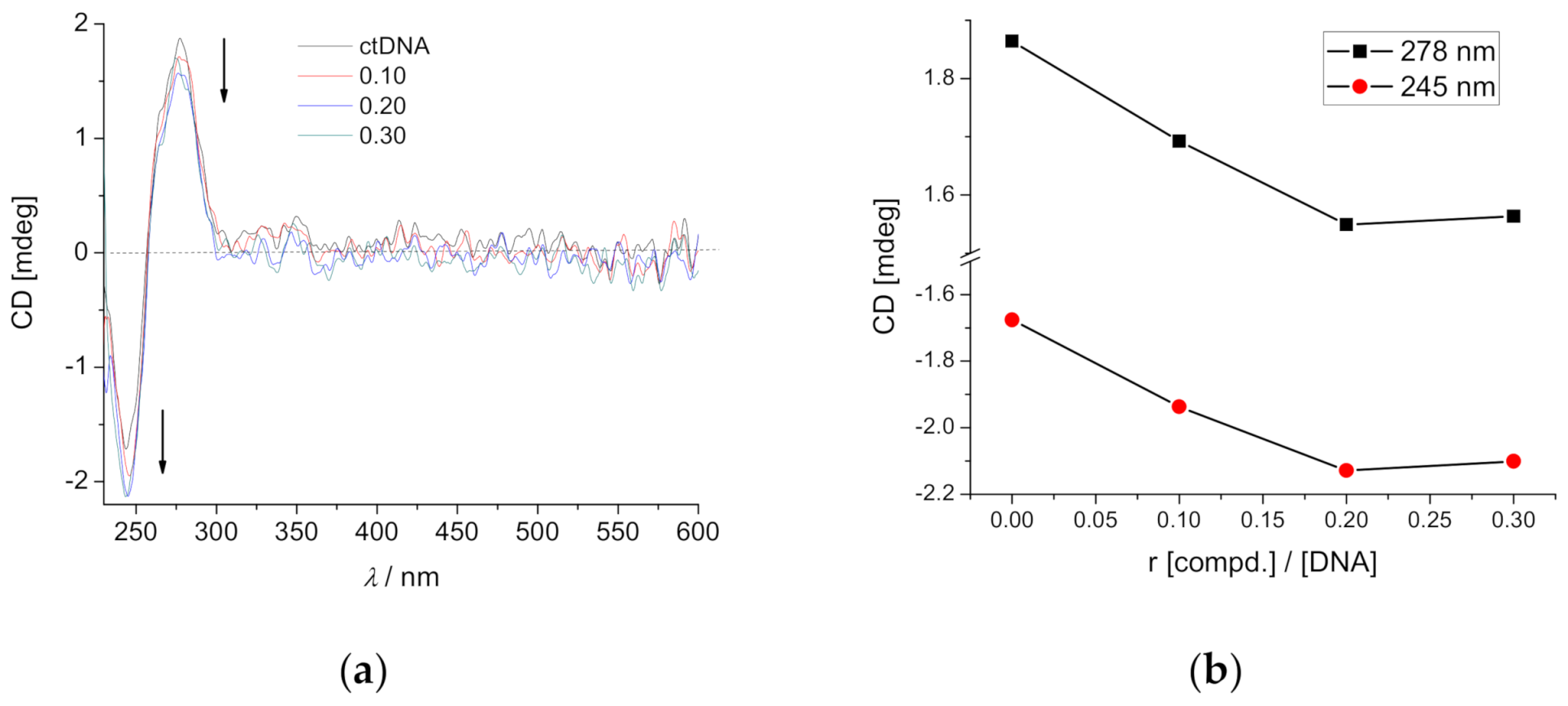
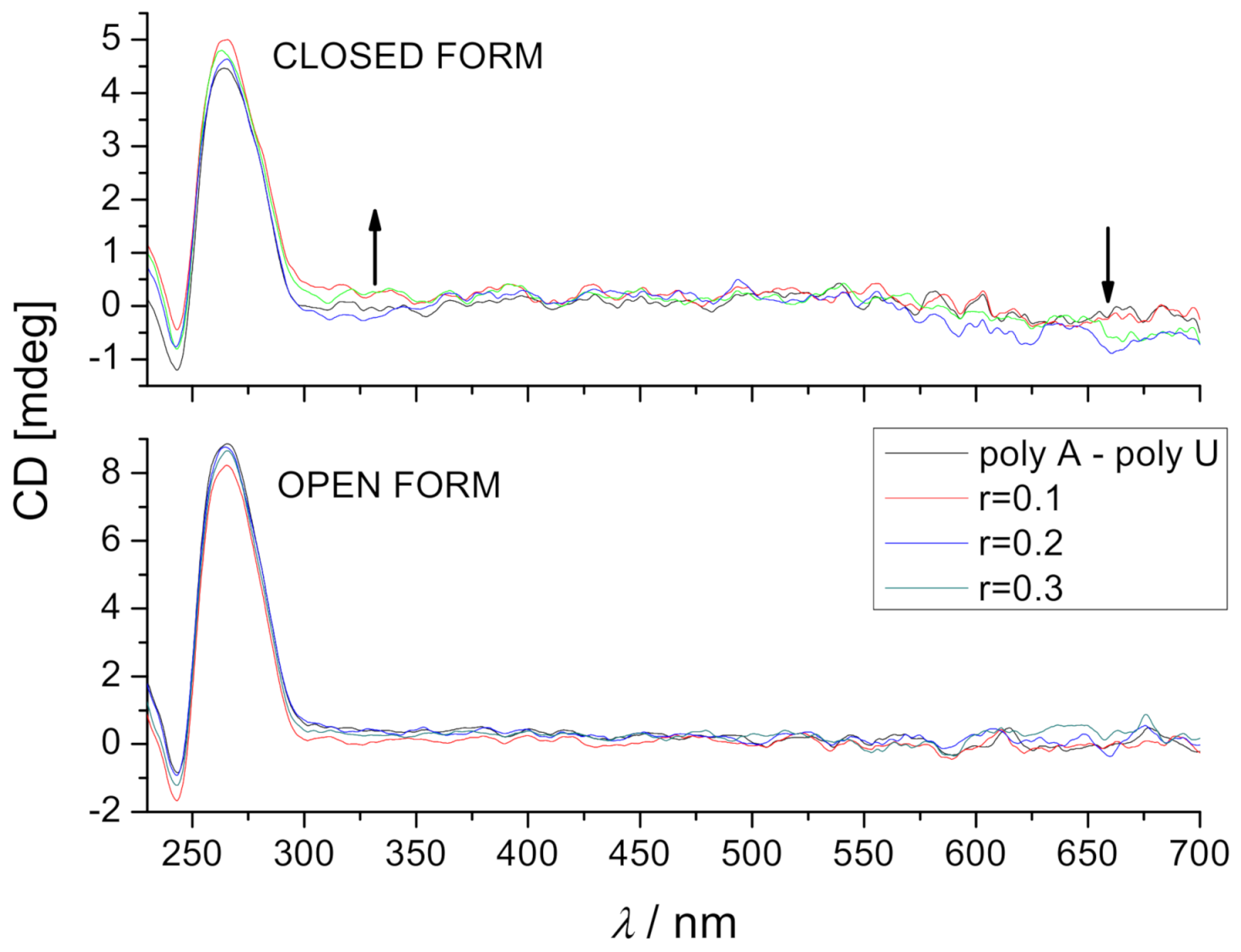
2.4.2. Circular Dichroism (CD) Experiments
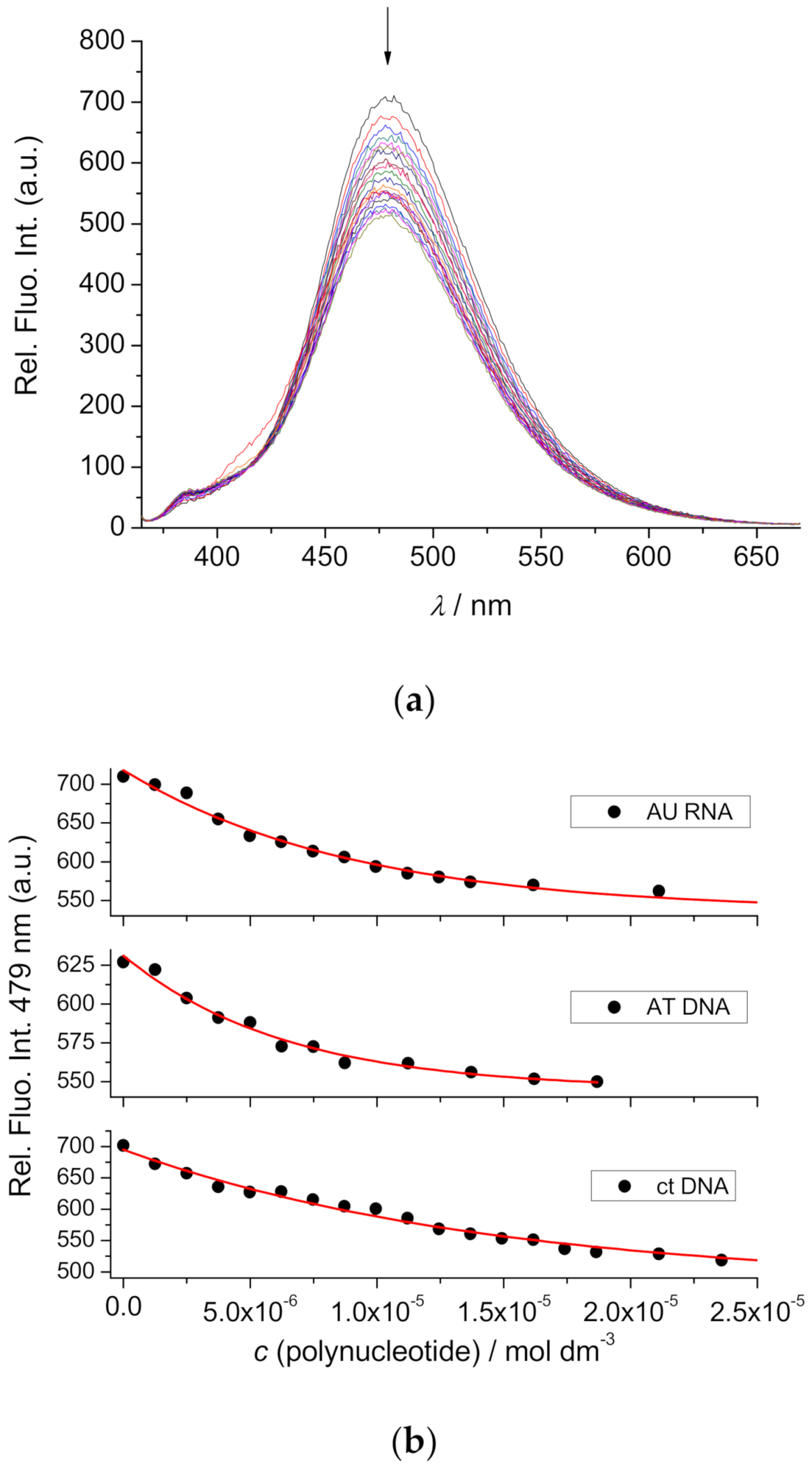
2.4.3. Fluorimetric Titrations
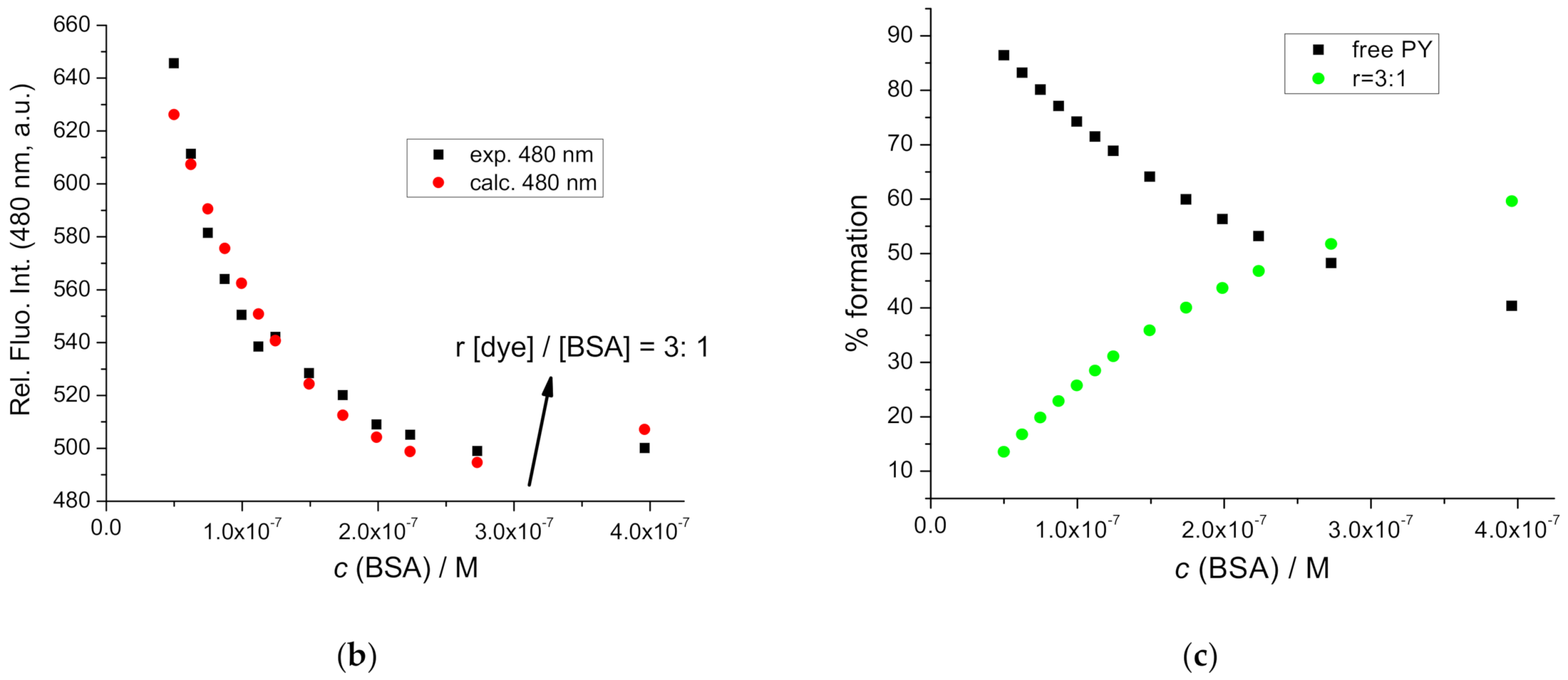
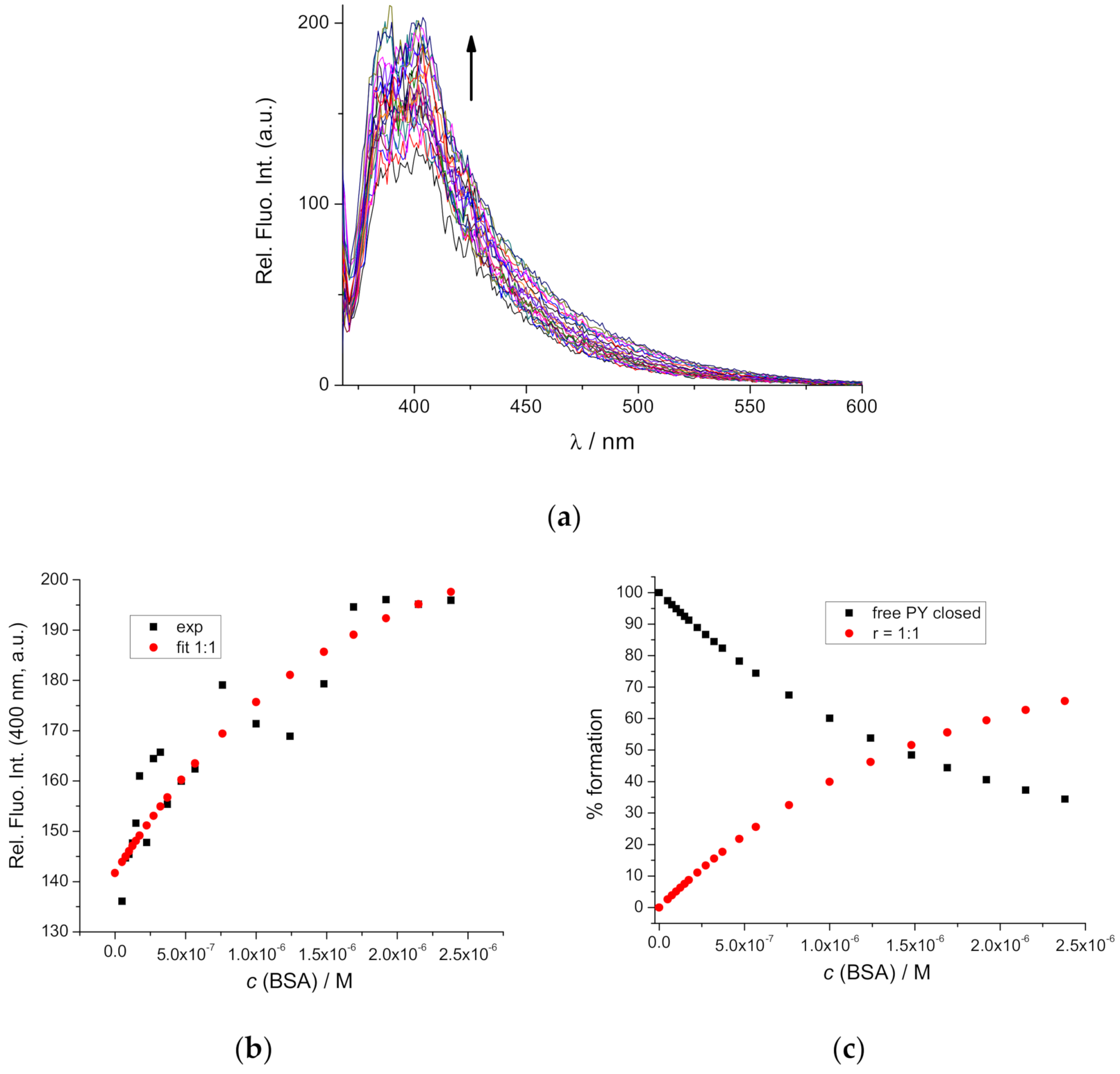
2.5. Interactions with BSA
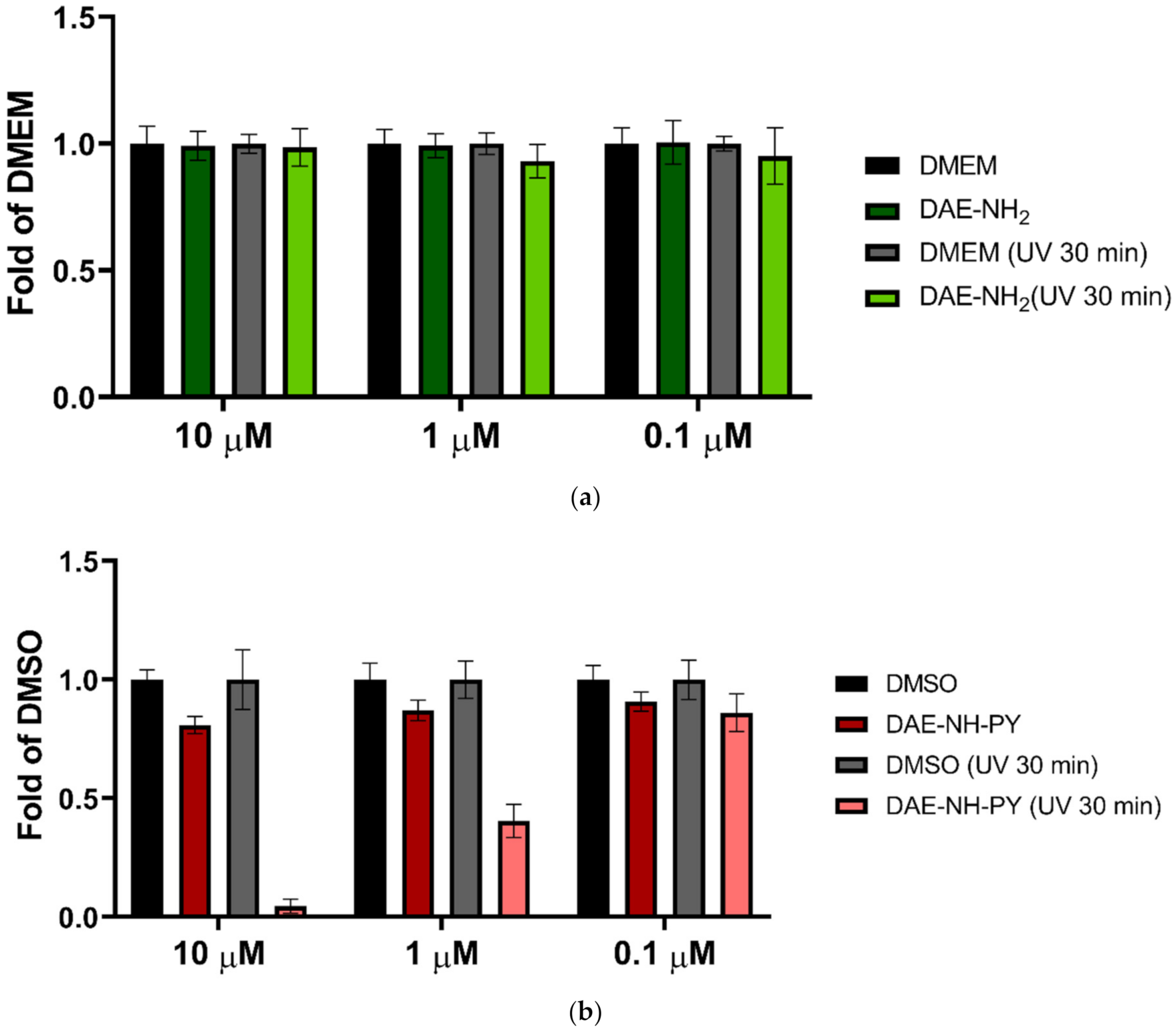
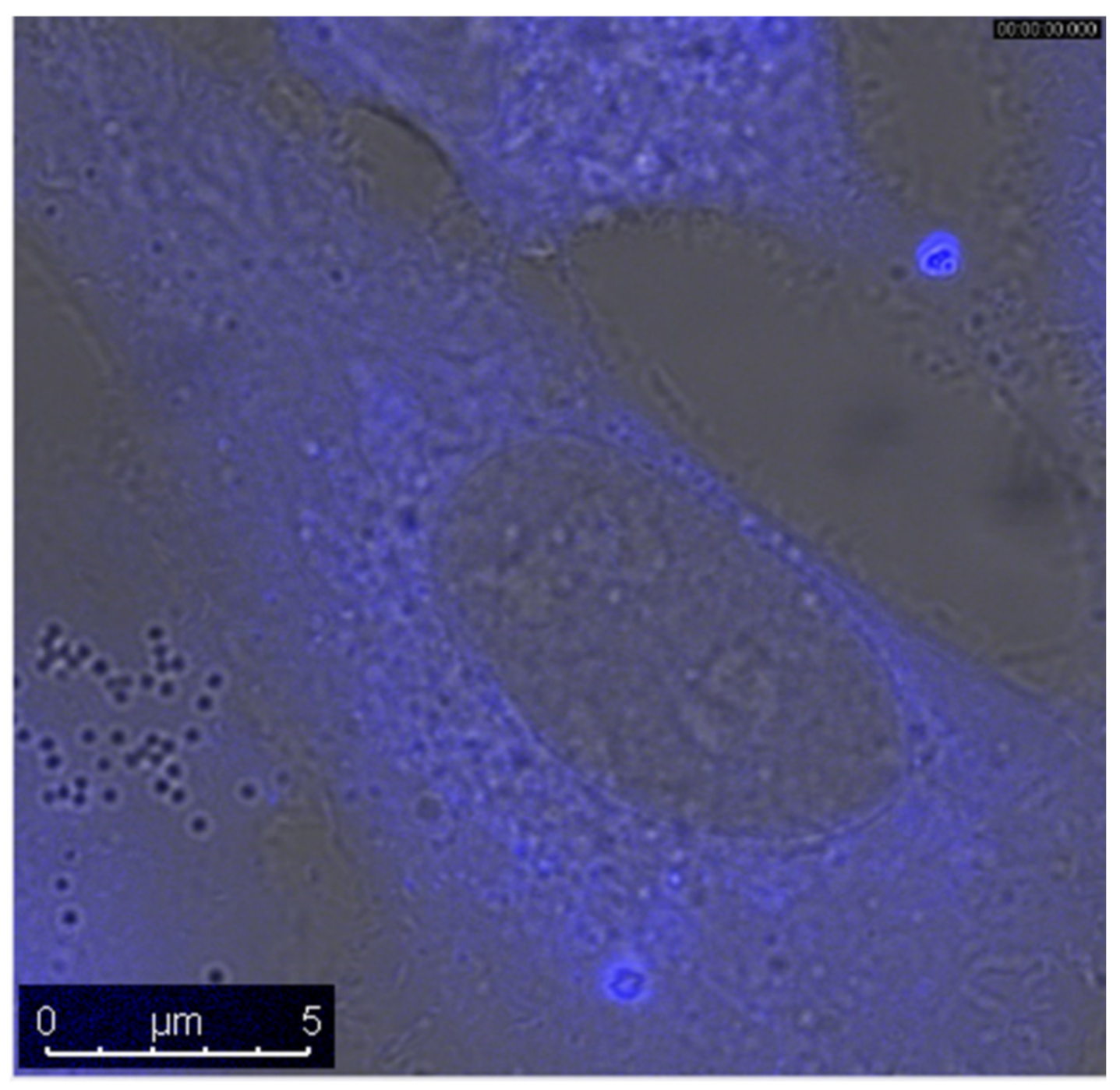
2.6. Biological Experiments
3. Conclusions
4. Materials and Methods
4.1. General
4.2. Synthesis
4.3. Study of DNA/RNA Interactions
4.4. Biology
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silverman, R.B. The Organic Chemistry of Drug Design and Drug Action; Elsevier Academic Press: New York, NY, USA, 2004. [Google Scholar]
- Lakowicz, J.R.; Masters, B.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Kluwer-Plenum: New York, NY, USA, 2008; p. 029901. [Google Scholar] [CrossRef]
- Specht, E.A.; Braselmann, E.; Palmer, A.E. A critical and comparative review of fluorescent tools for live-cell imaging. Annu. Rev. Physiol. 2017, 79, 93–117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, C.-X.; Chen, L.-G.; Yan, X.-P. Dual-stimuli responsive and reversibly activatable theranostic nanoprobe for precision tumor-targeting and fluorescence-guided photothermal therapy. Nat. Commun. 2017, 8, 14998. [Google Scholar] [CrossRef] [PubMed]
- He, P.-P.; Li, X.-D.; Wang, L.; Wang, H. Bispyrene-Based Self-Assembled Nanomaterials: In Vivo Self-Assembly, Transformation, and Biomedical Effects. Acc. Chem. Res. 2019, 52, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Folgado, L.; Schmuck, C.; Tomić, S.; Piantanida, I. A novel pyrene-guanidiniocarbonyl-pyrrole cation efficiently differentiates between ds-DNA and ds-RNA by two independent, sensitive spectroscopic methods. Bioorg. Med. Chem. Lett. 2008, 18, 2977–2981. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Folgado, L.; Bareticd, D.; Piantanida, I.; Marjanovicm, M.; Kralj, M.; Rehm, T.; Schmuck, C. Guanidiniocarbonylpyrrole-Aryl Derivatives: Structure Tuning for Spectrophotometric Recognition of Specific DNA and RNA Sequences and for Antiproliferative Activity. Chem. Eur. J. 2010, 16, 3036–3056. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zou, Y.; Li, C.; Sicking, W.; Piantanida, I.; Yi, T.; Schmuck, C. A Molecular Peptide Beacon for the Ratiometric Sensing of Nucleic Acids. J. Am. Chem. Soc. 2012, 134, 1958–1961. [Google Scholar] [CrossRef]
- Starcevic, K.; Karminski-Zamola, G.; Piantanida, I.; Zinic, M.; Suman, A.L.; Kralj, M. Photoinduced Switch of a DNA/RNA Inactive Molecule into a Classical Intercalator. J. Am. Chem. Soc. 2005, 127, 1074–1075. [Google Scholar] [CrossRef]
- Berdnikova, D.V.; Gulakova, E.; Fedorova, O.; Ihmels, H. Photoinduced in situ generation of a DNA-binding benzothiazoloquinolinium derivative. Chem. Commun. 2012, 48, 4603–4605. [Google Scholar] [CrossRef]
- Szymanski, W.; Beierle, J.M.; Kistemaker, H.A.V.; Velema, W.A.; Feringa, B.L. Reversible Photocontrol of Biological Systems by the Incorporation of Molecular Photoswitches. Chem. Rev. 2013, 113, 6114–6178. [Google Scholar] [CrossRef]
- Velema, W.A.; Szymanski, W.; Feringa, B. Photopharmacology: Beyond Proof of Principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. [Google Scholar] [CrossRef]
- Mammana, A.; Carroll, G.T.; Areephong, J.; Feringa, B.L. A Chiroptical Photoswitchable DNA Complex. J. Phys. Chem. B 2011, 115, 11581–11587. [Google Scholar] [CrossRef]
- Deiana, M.; Pokladek, Z.; Olesiak-Banska, J.; Mlynarz, P.; Samoc, M.; Matczyszyn, K. Photochromic switching of the DNA helicity induced by azobenzene derivatives. Sci. Rep. 2016, 6, 28605. [Google Scholar] [CrossRef]
- Muller, S.; Paulus, J.; Mattay, J.; Ihmels, H.; Dodero, V.I.; Sewald, N. Photocontrolled DNA minor groove interactions of imidazole/pyrrole polyamides. Beilstein J. Org. Chem. 2020, 16, 60–70. [Google Scholar] [CrossRef]
- Kolsch, S.; Ihmels, H.; Mattay, J.; Sewald, N.; Patrick, B.O. Reversible photoswitching of the DNA-binding properties of styrylquinolizinium derivatives through photochromic [2 + 2] cycloaddition and cycloreversion. Beilstein J. Org. Chem. 2020, 16, 111–124. [Google Scholar] [CrossRef]
- Ihmels, H.; Mattay, J.; May, F.; Thomas, L. Photoswitchable DNA-binding properties of a photochromic spirooxazine derivative. Org. Biomol. Chem. 2013, 11, 5184. [Google Scholar] [CrossRef]
- Berdnikova, D.V. Design, synthesis and investigation of water-soluble hemi-indigo photoswitches for bioapplications. Beilstein J. Org. Chem. 2019, 15, 2822–2829. [Google Scholar] [CrossRef]
- Heinrich, B.; Bouazoune, K.; Wojcik, M.; Bakowsky, U.; Vazquez, O. ortho-Fluoroazobenzene derivatives as DNA intercalators for photocontrol of DNA and nucleosome binding by visible light. Org. Biomol. Chem. 2019, 17, 1827–1833. [Google Scholar] [CrossRef]
- Bergen, A.; Rudiuk, S.; Morel, M.; le Saux, T.; Ihmels, H.; Baigl, D. Photodependent Melting of Unmodified DNA Using a Photosensitive Intercalator: A New and Generic Tool for Photoreversible Assembly of DNA Nanostructures at Constant Temperature. Nano Lett. 2016, 16, 773–780. [Google Scholar] [CrossRef]
- Moratz, J.; Samanta, A.; Voskuhl, J.; Nalluri, S.K.M.; Ravoo, B.J. Light-Triggered Capture and Release of DNA and Proteins by Host–Guest Binding and Electrostatic Interaction. Chem. Eur. J. 2015, 21, 3271–3277. [Google Scholar] [CrossRef]
- Dohno, C.; Uno, S.-N.; Sakai, S.; Oku, M.; Nakatani, K. The effect of linker length on binding affinity of a photoswitchable molecular glue for DNA. Bioorg. Med. Chem. 2009, 17, 2536–2543. [Google Scholar] [CrossRef]
- Weston, C.E.; Richardson, R.D.; Haycock, P.R.; White, A.J.P.; Fuchter, M.J. Arylazopyrazoles: Azoheteroarene Photoswitches Offering Quantitative Isomerization and Long Thermal Half-Lives. J. Am. Chem. Soc. 2014, 136, 11878–11881. [Google Scholar] [CrossRef]
- Schleper, A.L.; Bossi, M.L.; Belov, V.N.; Hell, S.W. Mono- and bithiophene-substituted diarylethene photoswitches with emissive open or closed forms. Beilstein J. Org. Chem. 2019, 15, 2344–2354. [Google Scholar] [CrossRef]
- Nevskyi, O.; Sysoiev, D.; Dreier, J.; Stein, S.C.; Oppermann, A.; Lemken, F.; Janke, T.; Enderlein, J.; Testa, I.; Huhn, T.; et al. Fluorescent Diarylethene Photoswitches-A Universal Tool for Super-Resolution Microscopy in Nanostructured Materials. Small 2018, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Kostenko, E.; Dobrikov, M.; Pyshnyi, D.; Petyuk, V.; Komarova, N.; Vlassov, V.; Zenkova, M. 5′-bis-pyrenylated oligonucleotides displaying excimer fluorescence provide sensitive probes of RNA sequence and structure. Nucleic Acids Res. 2001, 29, 3611–3620. [Google Scholar] [CrossRef]
- Østergaard, M.E.; Hrdlicka, P.J. Pyrene-functionalized oligonucleotides and locked nucleic acids (LNAs): Tools for fundamental research, diagnostics, and nanotechnology. Chem. Soc. Rev. 2011, 40, 5771–5788. [Google Scholar] [CrossRef]
- Mukherjee, S.; das Sarma, J.; Shunmugam, R. pH-Sensitive Nanoaggregates for Site-Specific Drug-Delivery as Well as Cancer Cell Imaging. ACS Omega 2016, 1, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Niko, Y.; Moritomo, H.; Sugihara, H.; Suzuki, Y.; Kawamata, J.; Konishi, G.-I. A novel pyrene-based two-photon active fluorescent dye efficiently excited and emitting in the ‘tissue optical window (650–1100 nm)’. J. Mater. Chem. B 2015, 3, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Groger, K.; Baretić, D.; Piantanida, I.; Marjanović, M.; Kralj, M.; Grabar, M.; Tomić, S.; Schmuck, C. Guanidiniocarbonyl-pyrrole-aryl conjugates as nucleic acid sensors: Switch of binding mode and spectroscopic responses by introducing additional binding sites into the linker. Org. Biomol. Chem. 2011, 9, 198–209. [Google Scholar] [CrossRef]
- Smidlehner, T.; Badovinac, M.; Piantanida, I. Pyrene–cyanine conjugas multipurpose fluorescent probes for non-covalent recognition of ds-DNA, RNA and proteins. New J. Chem. 2018, 42, 6655–6663. [Google Scholar] [CrossRef]
- Masaki, O.; Chuang, G. (Eds.) Albumin in Medicine. Pathological and Clinical Applications; Springer: Berlin/Heidelberg, Germany, 2016; ISBN 978-981-10-2116-9. [Google Scholar]
- Peters, T., Jr. All About Albumin. Biochemistry, Genetics, and Medical Applications; Elsevier Inc.: Amsterdam, The Netherlands, 1995; ISBN 978-0-12-552110-9. [Google Scholar]
- Varshney, A.; Sen, P.; Ahmad, E.; Rehan, M.; Subbarao, N.; Khan, R.H. Ligand binding strategies of human serum albumin: How can the cargo be utilized? Chirality 2010, 22, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Van Dijken, D.J.; Beierle, J.M.; Stuart, M.C.A.; Szymanski, W.; Browne, W.R.; Feringa, B.L. Autoamplification of molecular chirality through the induction of supramolecular chirality Angew. Chem. Int. Ed. 2014, 53, 5073–5077. [Google Scholar]
- Kaminski, Z.J. 2-Chloro-4, 6-disubstituted-1, 3, 5-triazines a novel group of condensing reagents. Tetrahedron Lett. 1985, 26, 2901. [Google Scholar] [CrossRef]
- Hartwig, S.; Nguyen, M.M.; Hecht, S. Exponential growth of functional poly(glutamic acid)dendrimers with variable stereochemistry. Polym. Chem. 2010, 1, 69–71. [Google Scholar] [CrossRef]
- Logan, S.R. Does a Photochemical Reaction Have a Reaction Order? J. Chem. Educ. 1997, 74, 1303. [Google Scholar] [CrossRef]
- Mergny, J.-L.; Lacroix, L. Analysis of Thermal Melting Curves. Oligonucleotides 2003, 13, 515–537. [Google Scholar] [CrossRef]
- Smidlehner, T.; Piantanida, I.; Pescitelli, G. Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids–tutorial. Beilstein J. Org. Chem. 2018, 14, 84–105. [Google Scholar] [CrossRef]
- Egli, M.; Saenger, W. Principles of Nucleic Acid Structure; Springer: New York, NY, USA, 1983. [Google Scholar]
- McGhee, J.D.; Hippel, P.H.V. Theoretical Aspects of DNA-Protein Interactions—Cooperative and Non-Cooperative Binding of Large Ligands to a One-Dimensional Homogeneous Lattice. J. Mol. Biol. 1974, 86, 469–489. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Zunszain, P.; Ghuman, J.; Curry, S. Human serum albumin complexed with 4Z,15E-bilirubin-IX-alpha. Hum. Serum Albumin Complexed 4Z,15E-Bilirubin-IX-Alpha 2008, 381, 394–406. [Google Scholar] [CrossRef]
- Chaires, J.B.; Dattagupta, N.; Crothers, D.M. Studies on interaction of anthracycline antibiotics and deoxyribonucleic acid: Equilibrium binding studies on the interaction of daunomycin with deoxyribonucleic acid. Biochemistry 1982, 21, 3933–3940. [Google Scholar] [CrossRef]
- Tumir, L.M.; Piantanida, I.; Cindric, I.J.; Hrenar, T.; Meic, Z.; Zinic, M. New permanently charged phenanthridinium-nucleobase conjugates. Interactions with nucleotides and polynucleotides and recognition of ds-polyAH(+). J. Phys. Org. Chem. 2003, 16, 891–899. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λ (MeOH) /nm | ε (MeOH) /mmol−1 cm2 | λ (Buffer) /nm | ε (Buffer) /mmol−1 cm2 |
|---|---|---|---|---|
| DAE-NH-Boc | 261 | 95,211 | 266 | 22,423 |
| DAE-NH2 | - | - | 264 | 17,588 |
| DAE-NH-Py (open) | 275 | 43,206 | 280 | 29,577 |
| 341 | 27,724 | 348 | 17,637 | |
| a DAE-NH-Py (closed) | 275 | 46,910 | 280 | 36,254 |
| 347 | 26,792 | |||
| 530 | 4252 |
| Compound | b r | ct-DNA | poly A—poly U |
|---|---|---|---|
| DAE-NH2 (open form) | 0.3 | −1.0 | −0.7 |
| DAE-NH-Py (open form) | 0.1 | 0.0 | |
| 0.2 | 0.0/26.1 | ||
| 0.3 | −0.7 | 0/23.4 | |
| DAE-NH-Py (closed form) | 0.3 | −1.9 | 0.3 |
| ct-DNA | AT-DNA | GC-DNA | AU-RNA |
|---|---|---|---|
| 5.6/26 | 6.3/11 | c | 6.1/22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orehovec, I.; Matković, M.; Pehar, I.; Majhen, D.; Piantanida, I. Bis-Pyrene Photo-Switch Open- and Closed-Form Differently Bind to ds-DNA, ds-RNA and Serum Albumin and Reveal Light-Induced Bioactivity. Int. J. Mol. Sci. 2021, 22, 4916. https://doi.org/10.3390/ijms22094916
Orehovec I, Matković M, Pehar I, Majhen D, Piantanida I. Bis-Pyrene Photo-Switch Open- and Closed-Form Differently Bind to ds-DNA, ds-RNA and Serum Albumin and Reveal Light-Induced Bioactivity. International Journal of Molecular Sciences. 2021; 22(9):4916. https://doi.org/10.3390/ijms22094916
Chicago/Turabian StyleOrehovec, Iva, Marija Matković, Isabela Pehar, Dragomira Majhen, and Ivo Piantanida. 2021. "Bis-Pyrene Photo-Switch Open- and Closed-Form Differently Bind to ds-DNA, ds-RNA and Serum Albumin and Reveal Light-Induced Bioactivity" International Journal of Molecular Sciences 22, no. 9: 4916. https://doi.org/10.3390/ijms22094916
APA StyleOrehovec, I., Matković, M., Pehar, I., Majhen, D., & Piantanida, I. (2021). Bis-Pyrene Photo-Switch Open- and Closed-Form Differently Bind to ds-DNA, ds-RNA and Serum Albumin and Reveal Light-Induced Bioactivity. International Journal of Molecular Sciences, 22(9), 4916. https://doi.org/10.3390/ijms22094916