
The Cellular Composition of Bovine Coccygeal Intervertebral Discs: A Comprehensive Single-Cell RNAseq Analysis


 ,
,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Single-Cell RNAseq Experimental Design
2.2. Bulk RNAseq Experimental Design
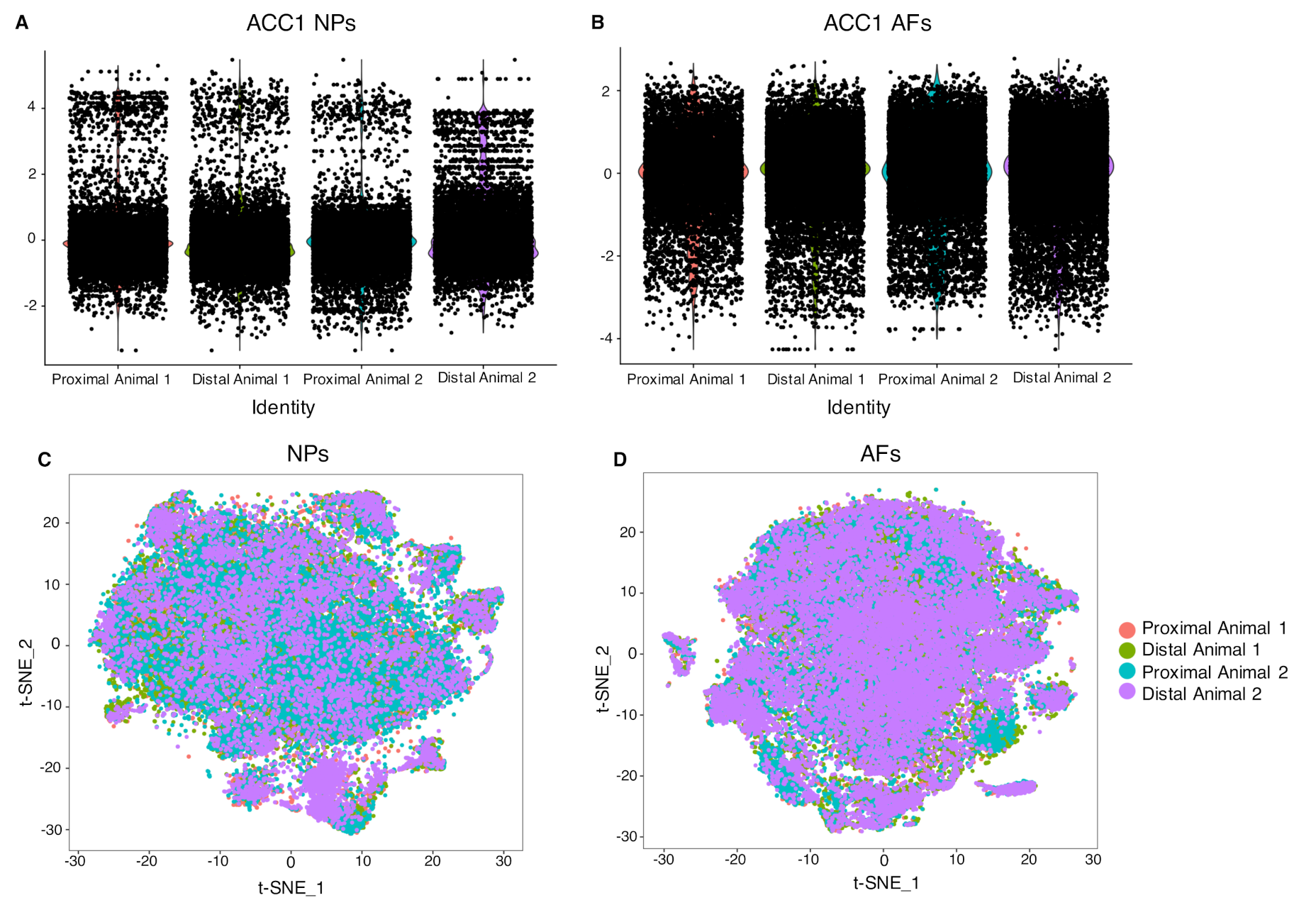
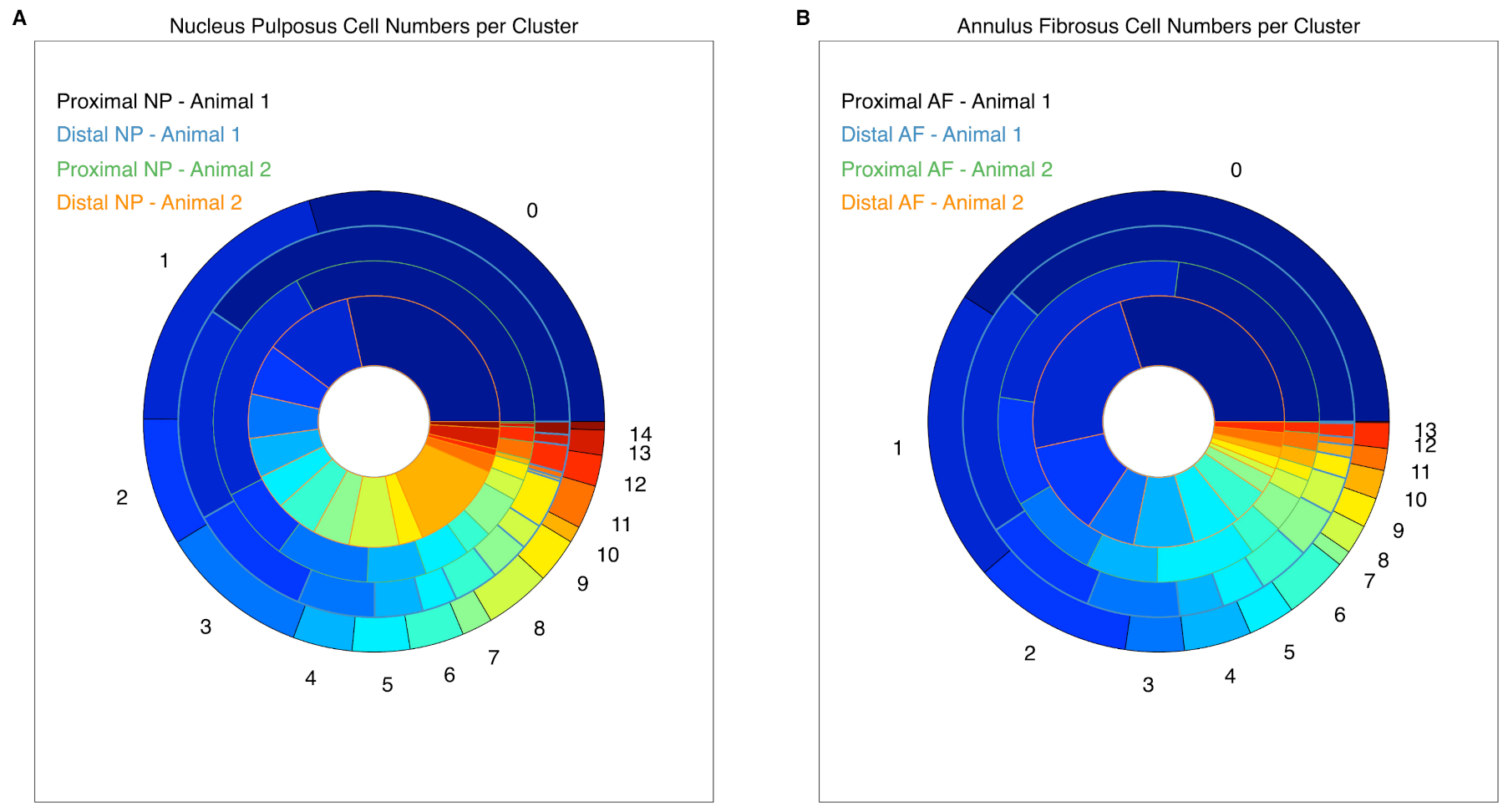
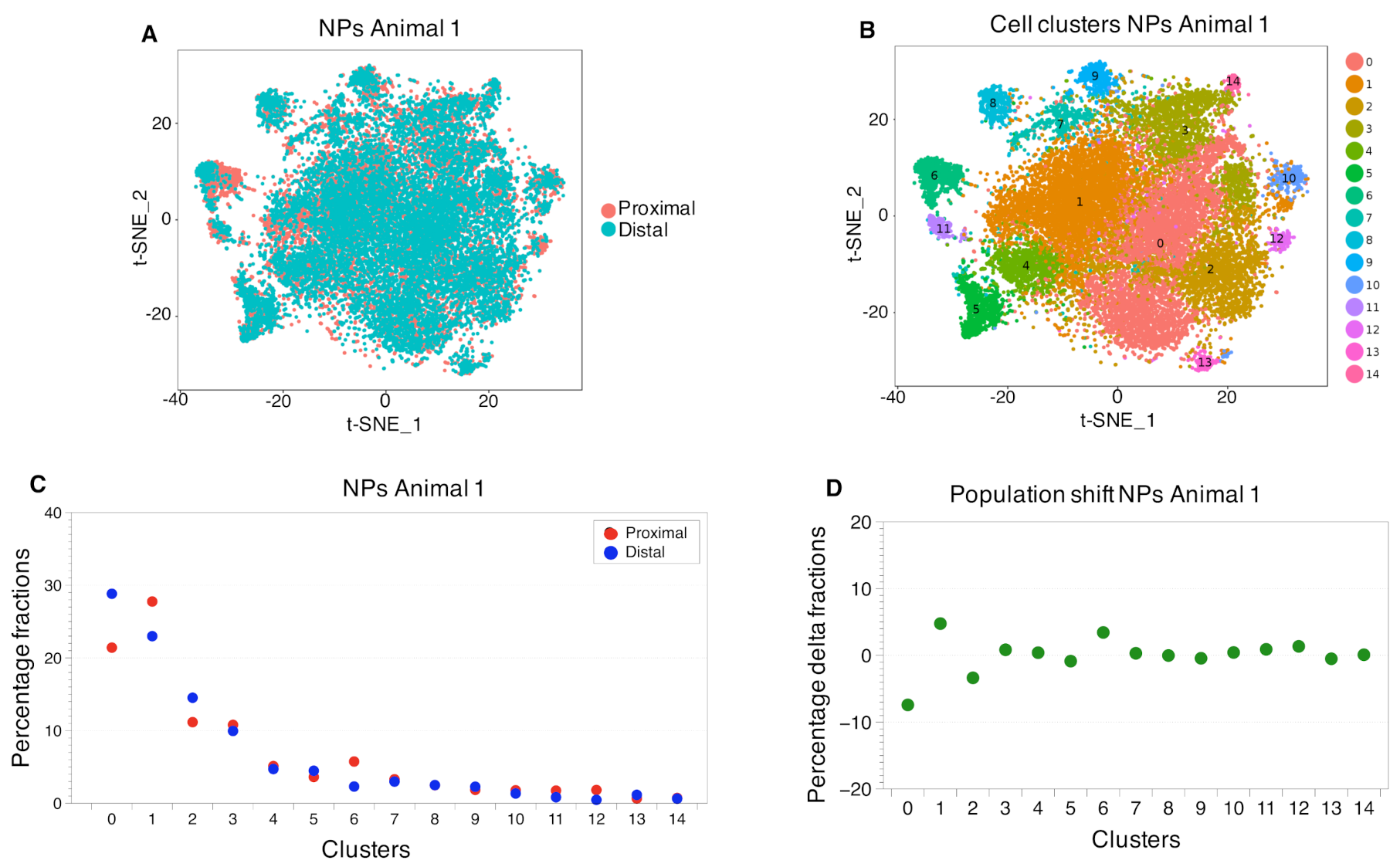
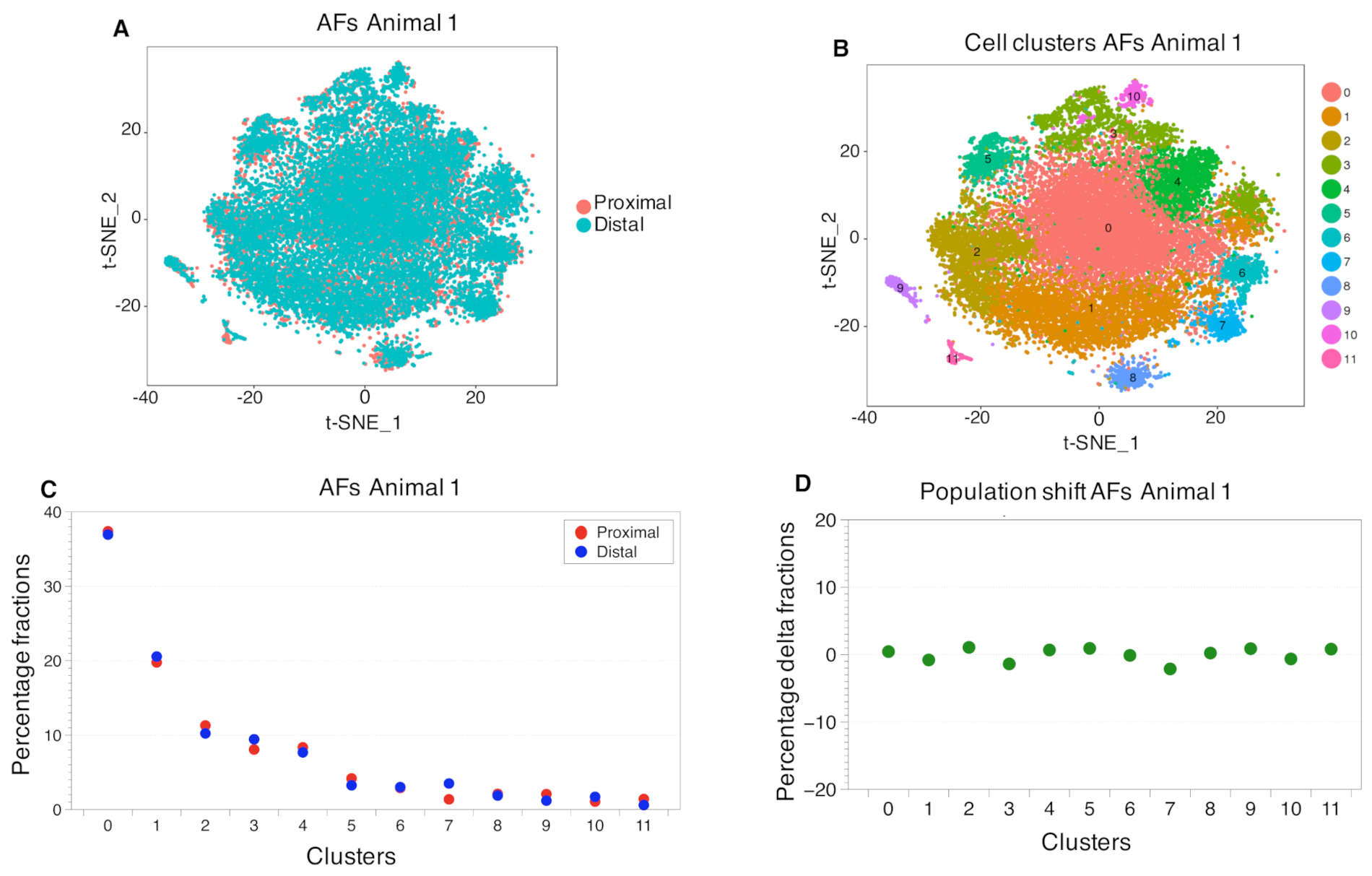
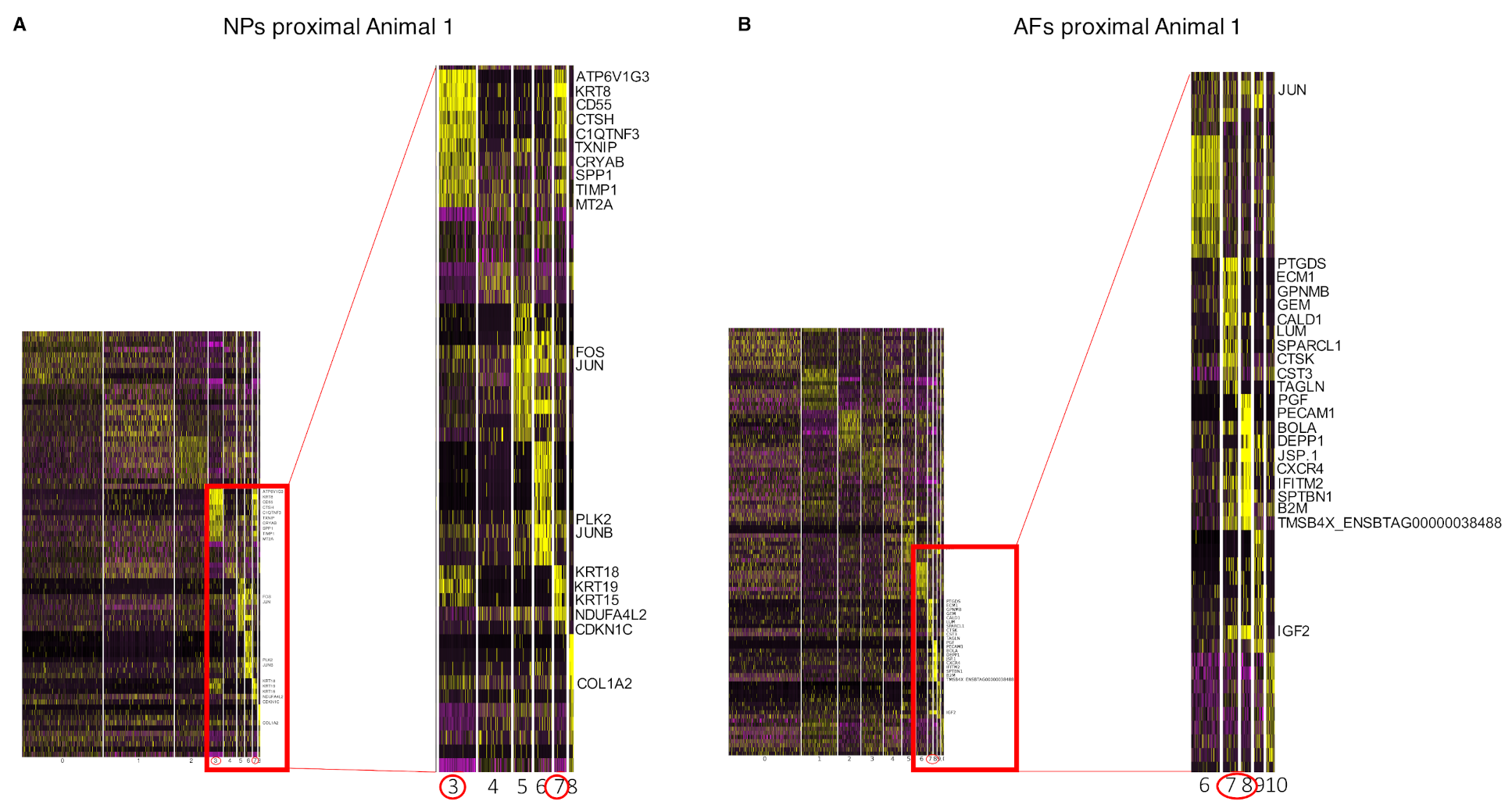
2.3. Cell Clustering Using Single-Cell RNAseq Data
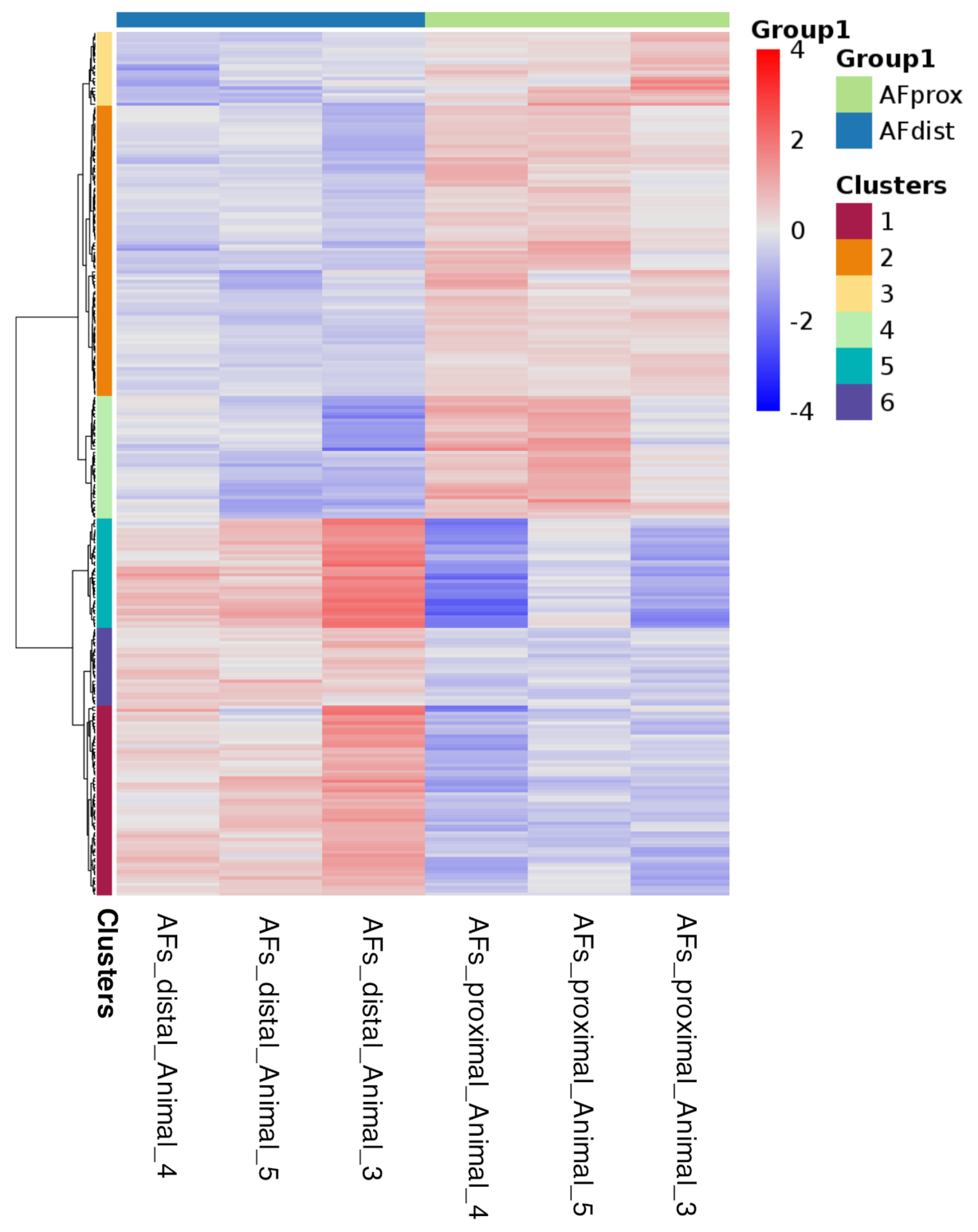
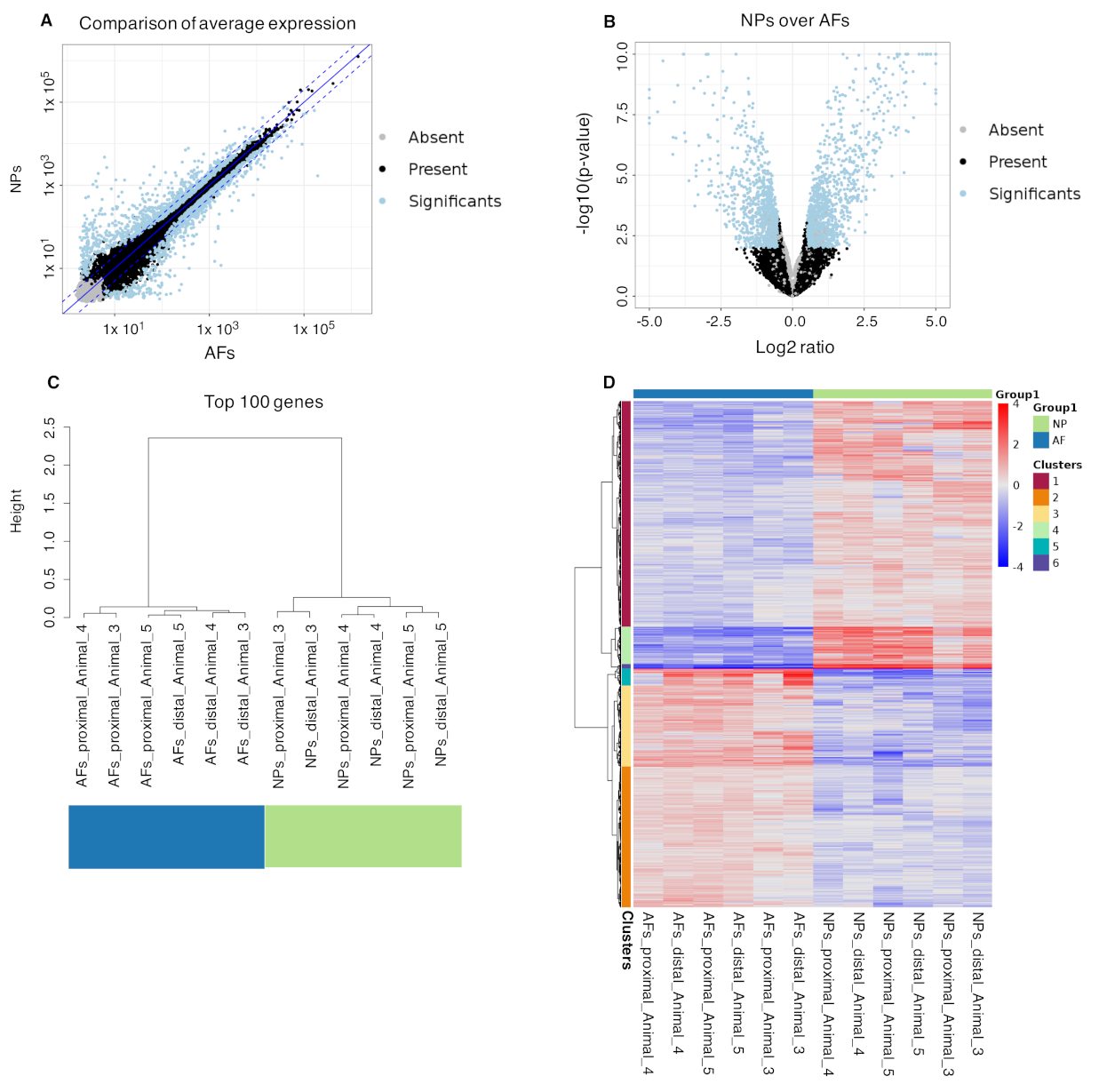
2.4. Differential Gene Expression Analysis from Bulk RNAseq
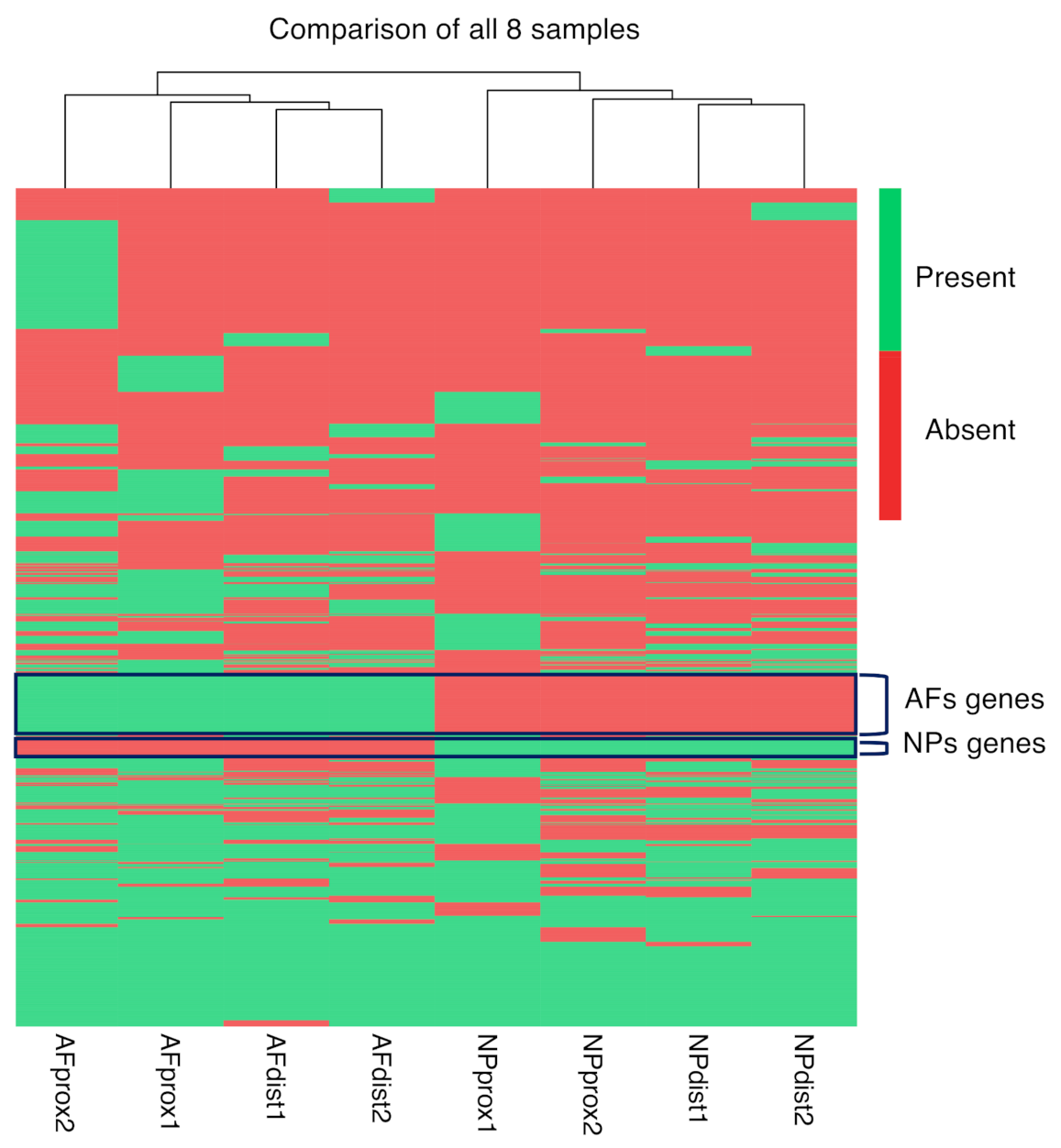
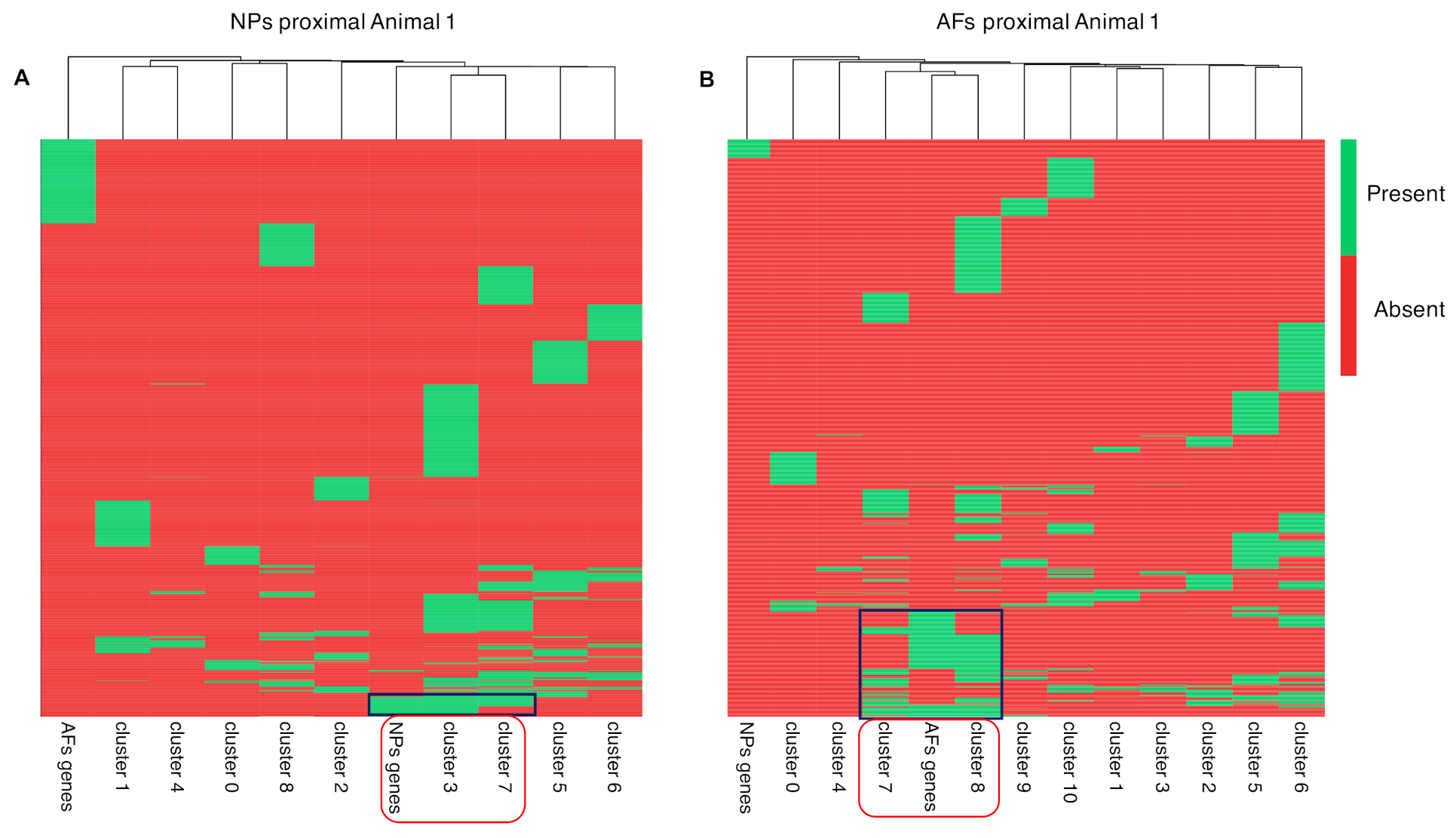
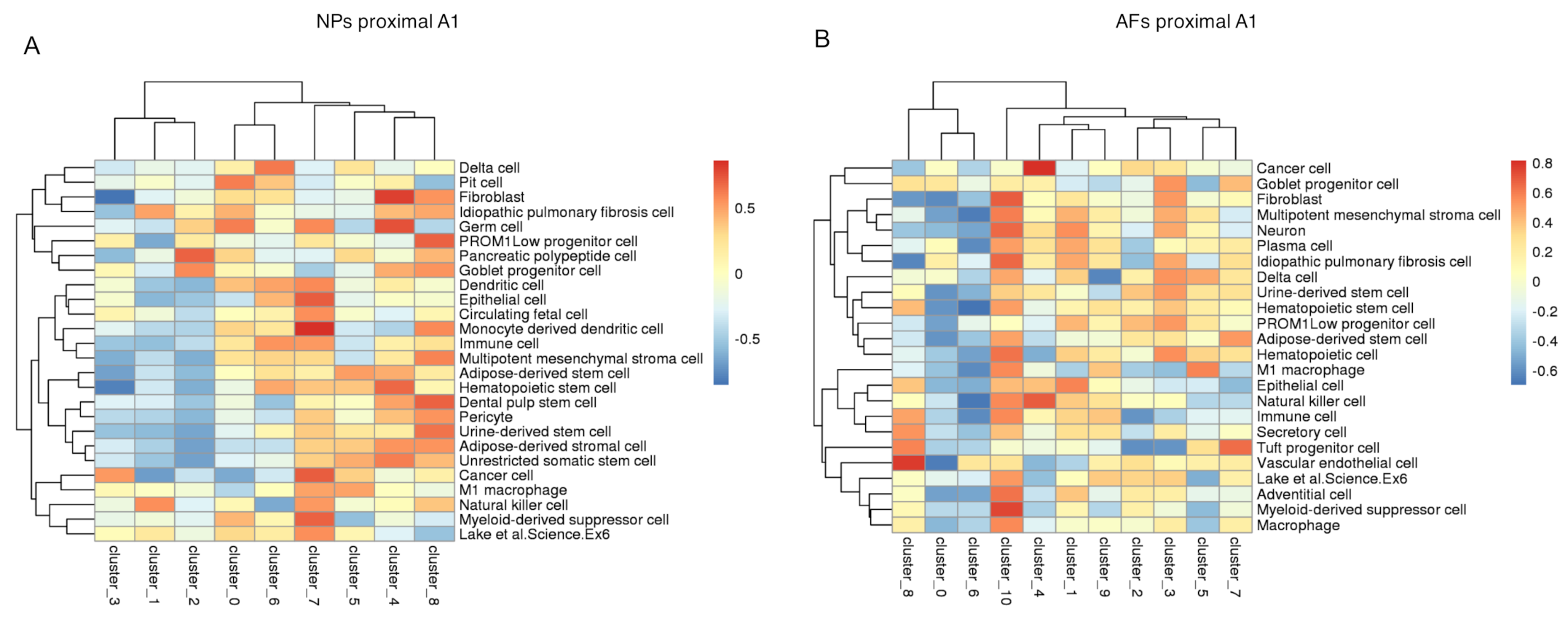
2.5. Integration of the Single-Cell RNAseq Data with the Bulk RNAseq Data
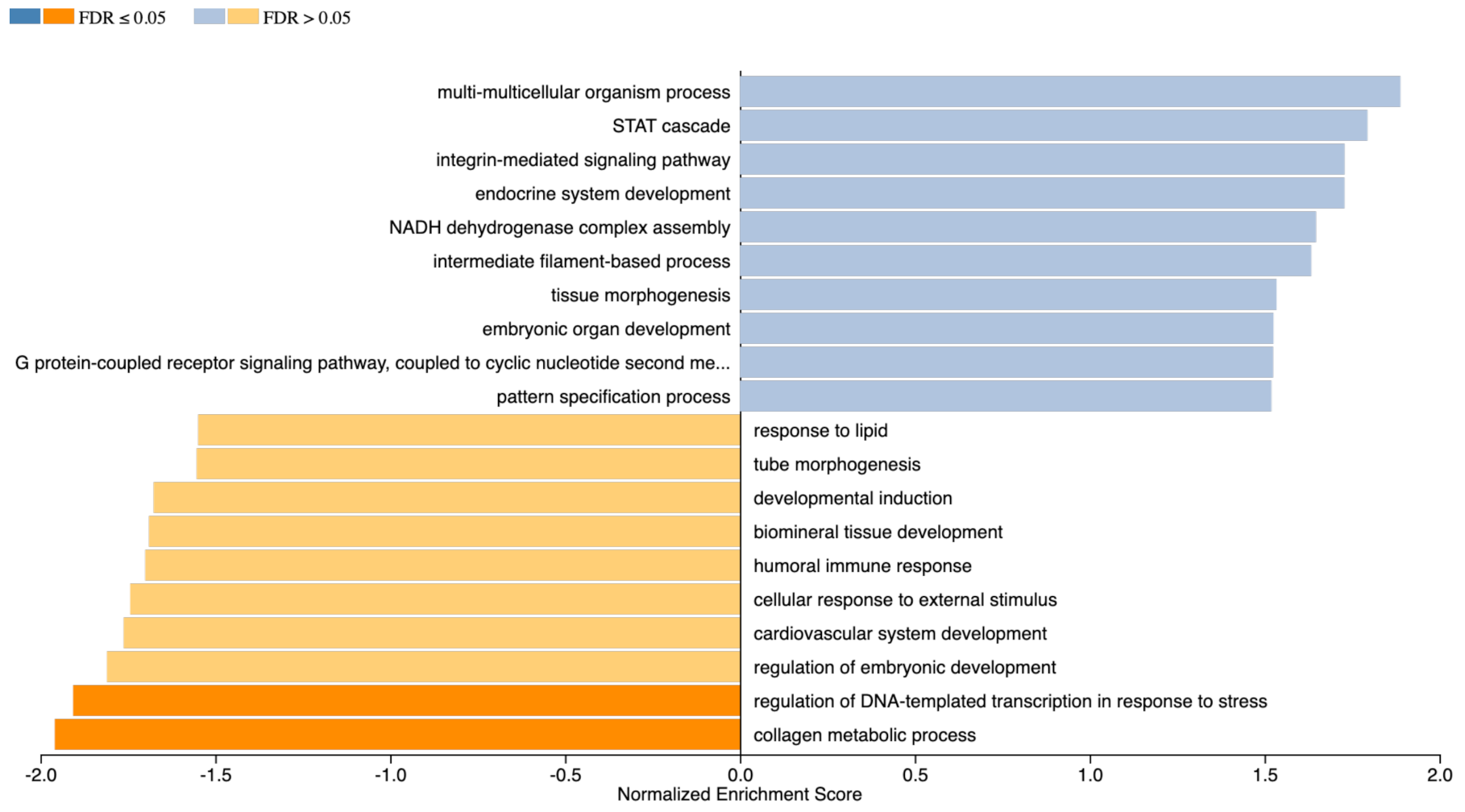
2.6. Gene Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Samples Collection and Processing
4.2. Bulk RNAseq
4.3. Bulk RNAseq Data Analysis
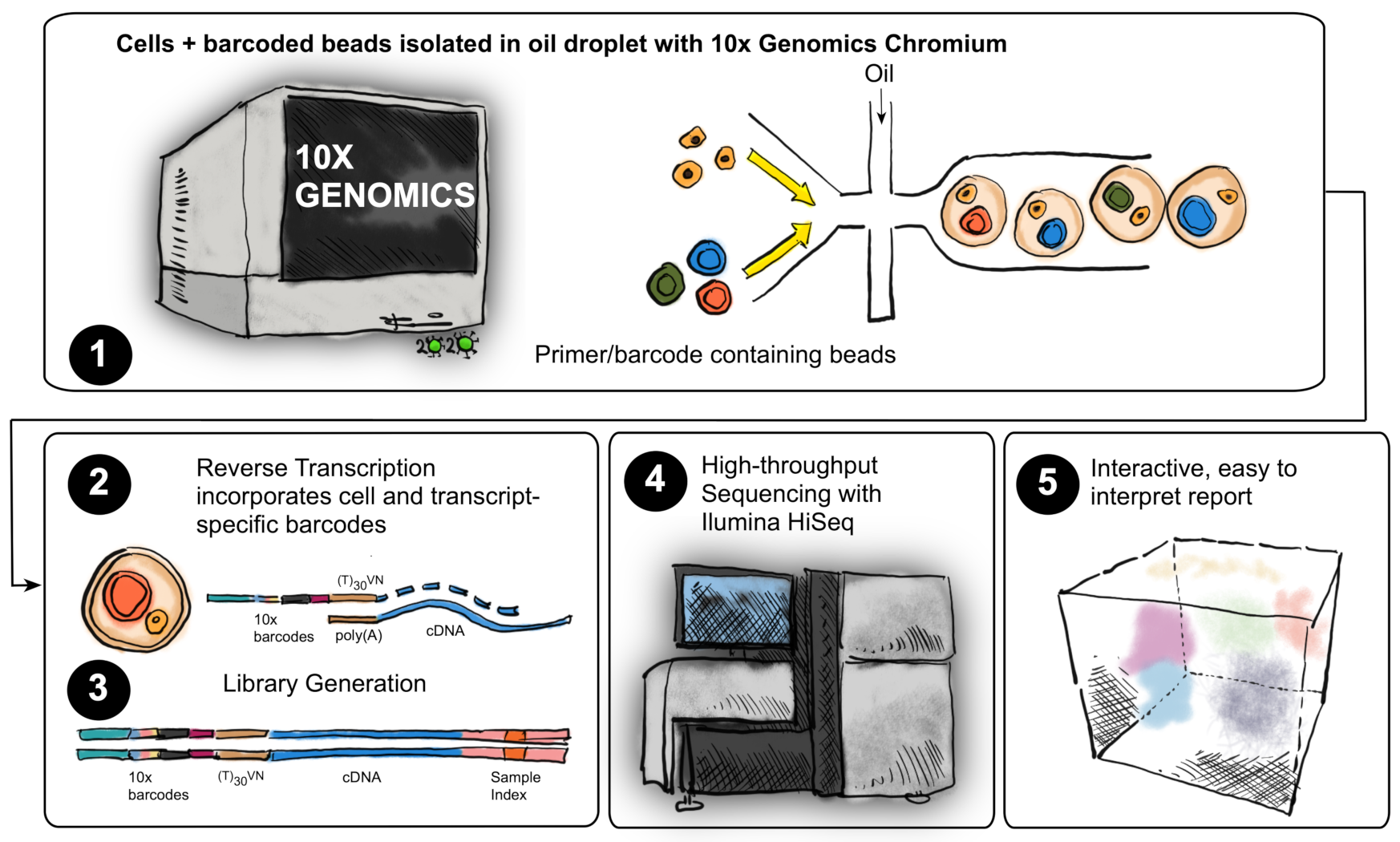
4.4. Single-Cell RNAseq
4.5. Single-Cell RNAseq Analysis
4.6. Pathways Analyses
- http://geneontology.org (accessed on 29 April 2019)
- https://www.genome.jp/kegg (accessed on 29 April 2019)
- https://www.wikipathways.org (accessed on 29 April 2019)
- http://biocc.hrbmu.edu.cn/CellMarker (accessed on 29 April 2019)
4.7. Multiple Samples Integration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AF | Annulus Fibrosus |
| AFC | Annulus Fibrosus Cell |
| CCA | Canonical Correlation Analysis |
| CEP | Cartilage Endplate |
| GSEA | Gene Set Enrichment analysis |
| GSVA | Gene Set Variant Analysis |
| IVD | Intervertebral disc |
| KNN | K-nearest neighbors |
| MNN | Mutual Nearest Neighbors |
| NP | Nucleus Pulposus |
| NPC | Nucleus Pulposus Cell |
| PCA | Principal Component Analysis |
| RISH | Hybridization on paraffin sections |
| SEA | Singular Enrichment Analysis |
| t-SNE | t-stochastic neighbor embedding |
| UMI | Unique Molecular Identifier |
References
- Risbud, M.V.; Schaer, T.P.; Shapiro, I.M. Toward an Understanding of the Role of Notochordal Cells in the Adult Intervertebral Disc: From Discord to Accord. Dev. Dyn. 2010, 239, 2141–2148. [Google Scholar] [CrossRef]
- Tessier, S.; Risbud, M.V. Understanding Embryonic Development for Cell-Based Therapies of Intervertebral Disc Degeneration: Toward an Effort to Treat Disc Degeneration Subphenotypes. Dev. Dyn. 2020, dvdy.217. [Google Scholar] [CrossRef]
- Cox, M.K.; Serra, R. Development of the Intervertebral Disc. In The Intervertebral Disc: Molecular and Structural Studies of the Disc in Health and Disease; Shapiro, I.M., Risbud, M.V., Eds.; Springer: Vienna, Austria, 2014; pp. 33–51. [Google Scholar] [CrossRef]
- Madhu, V.; Boneski, P.K.; Silagi, E.; Qiu, Y.; Kurland, I.; Guntur, A.R.; Shapiro, I.M.; Risbud, M.V. Hypoxic Regulation of Mitochondrial Metabolism and Mitophagy in Nucleus Pulposus Cells Is Dependent on HIF-1α–BNIP3 Axis. J. Bone Miner. Res. 2020, jbmr.4019. [Google Scholar] [CrossRef]
- Baldit, A. Micromechanics of the intervertebral disk. In Multiscale Biomechanics; Elsevier: Oxford, UK, 2018; pp. 455–467. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Risbud, M.V. Transcriptional Profiling of the Nucleus Pulposus: Say Yes to Notochord. Arthritis Res. Ther. 2010, 12, 117. [Google Scholar] [CrossRef][Green Version]
- D’Este, M.; Eglin, D.; Alini, M. Lessons to Be Learned and Future Directions for Intervertebral Disc Biomaterials. Acta Biomater. 2018, 78, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, J.J.; Hiltner, A.; Baer, E. Hierarchical Structure of the Intervertebral Disc. Connect. Tissue Res. 1989, 23, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.J. The Vertebral Endplate: Disc Degeneration, Disc Regeneration. Eur. Spine J. 2006, 15, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.E.; Riley, F.E.; Hoelscher, G.L.; Ingram, J.A.; Bullock, L.; Hanley, E.N. Human Annulus Progenitor Cells: Analyses of This Viable Endogenous Cell Population: Progenitor Cells in the Human annulus. J. Orthop. Res. 2016, 34, 1351–1360. [Google Scholar] [CrossRef]
- Choi, K.S.; Cohn, M.J.; Harfe, B.D. Identification of Nucleus Pulposus Precursor Cells and Notochordal Remnants in the Mouse: Implications for Disk Degeneration and Chordoma Formation. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2008, 237, 3953–3958. [Google Scholar] [CrossRef]
- McCann, M.R.; Tamplin, O.J.; Rossant, J.; Séguin, C.A. Tracing Notochord-Derived Cells Using a Noto-Cre Mouse: Implications for Intervertebral Disc Development. Dis. Model. Mech. 2012, 5, 73–82. [Google Scholar] [CrossRef]
- Purmessur, D.; Guterl, C.C.; Cho, S.K.; Cornejo, M.C.; Lam, Y.W.; Ballif, B.A.; Laudier, J.C.I.; Iatridis, J.C. Dynamic Pressurization Induces Transition of Notochordal Cells to a Mature Phenotype While Retaining Production of Important Patterning Ligands from Development. Arthritis Res. Ther. 2013, 15, R122. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Risbud, M.V. Introduction to the Structure, Function, and Comparative Anatomy of the Vertebrae and the Intervertebral Disc. In The Intervertebral Disc: Molecular and Structural Studies of the Disc in Health and Disease; Shapiro, I.M., Risbud, M.V., Eds.; Springer: Vienna, Austria, 2014; pp. 3–15. [Google Scholar] [CrossRef]
- Risbud, M.V.; Shapiro, I.M. Notochordal Cells in the Adult Intervertebral Disc: New Perspective on an Old Question. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 29–41. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Risbud, M.V. (Eds.) The Intervertebral Disc: Molecular and Structural Studies of the Disc in Health and Disease; Springer: Wien, Austria, 2014. [Google Scholar] [CrossRef]
- Torre, O.M.; Mroz, V.; Benitez, A.R.M.; Huang, A.H.; Iatridis, J.C. Neonatal Annulus Fibrosus Regeneration Occurs via Recruitment and Proliferation of Scleraxis-Lineage Cells. NPJ Regen. Med. 2019, 4, 23. [Google Scholar] [CrossRef]
- Clouet, J.; Grimandi, G.; Pot-Vaucel, M.; Masson, M.; Fellah, H.B.; Guigand, L.; Cherel, Y.; Bord, E.; Rannou, F.; Weiss, P.; et al. Identification of Phenotypic Discriminating Markers for Intervertebral Disc Cells and Articular Chondrocytes. Rheumatology 2009, 48, 1447–1450. [Google Scholar] [CrossRef]
- Li, K.; Kapper, D.; Mondal, S.; Lufkin, T.; Kraus, P. Quantitative Single-Cell Transcript Assessment of Biomarkers Supports Cellular Heterogeneity in the Bovine IVD. Vet. Sci. 2019, 6, 42. [Google Scholar] [CrossRef]
- van den Akker, G.G.H.; Koenders, M.I.; van de Loo, F.A.J.; van Lent, P.L.E.M.; Blaney Davidson, E.; van der Kraan, P.M. Transcriptional Profiling Distinguishes Inner and Outer Annulus Fibrosus from Nucleus Pulposus in the Bovine Intervertebral Disc. Eur. Spine J. 2017, 26, 2053–2062. [Google Scholar] [CrossRef]
- Tam, V.; Chen, P.; Yee, A.; Solis, N.; Klein, T.; Kudelko, M.; Sharma, R.; Chan, W.C.; Overall, C.M.; Haglund, L.; et al. DIPPER: A Spatiotemporal Proteomics Atlas of Human Intervertebral Discs for Exploring Ageing and Degeneration Dynamics. bioRxiv 2020. [Google Scholar] [CrossRef]
- Reed, C.C.; Iozzo, R.V. The Role of Decorin in Collagen Fibrillogenesis and Skin Homeostasis. Glycoconj. J. 2002, 19, 249–255. [Google Scholar] [CrossRef]
- Chen, S.; Birk, D.E. The Regulatory Roles of Small Leucine-Rich Proteoglycans in Extracellular Matrix Assembly. FEBS J. 2013, 280, 2120–2137. [Google Scholar] [CrossRef]
- Hayes, A.J.; Isaacs, M.D.; Hughes, C.; Caterson, B.; Ralphs, J.R. Collagen Fibrillogenesis in the Development of the Annulus Fibrosus of the Intervertebral Disc. Eur. Cells Mater. 2011, 22, 226–241. [Google Scholar] [CrossRef]
- Trout, J.J.; Buckwalter, J.A.; Moore, K.C. Ultrastructure of the Human Intervertebral Disc: II. Cells of the Nucleus Pulposus. Anat. Rec. 1982, 204, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Schol, J.; Bach, F.C.; Tekari, A.; Sagawa, N.; Nakamura, Y.; Chan, S.C.W.; Nakai, T.; Creemers, L.B.; Frauchiger, D.A.; et al. Successful Fishing for Nucleus Pulposus Progenitor Cells of the Intervertebral Disc across Species. JOR Spine 2018, 1, e1018. [Google Scholar] [CrossRef]
- Brisby, H.; Papadimitriou, N.; Brantsing, C.; Bergh, P.; Lindahl, A.; Barreto Henriksson, H. The Presence of Local Mesenchymal Progenitor Cells in Human Degenerated Intervertebral Discs and Possibilities to Influence These In Vitro: A Descriptive Study in Humans. Stem Cells Dev. 2013, 22, 804–814. [Google Scholar] [CrossRef]
- Hunter, C.J.; Matyas, J.R.; Duncan, N.A. Cytomorphology of Notochordal and Chondrocytic Cells from the Nucleus Pulposus: A Species Comparison. J. Anat. 2004, 205, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Risbud, M.V.; Guttapalli, A.; Tsai, T.T.; Lee, J.Y.; Danielson, K.G.; Vaccaro, A.R.; Albert, T.J.; Gazit, Z.; Gazit, D.; Shapiro, I.M. Evidence for Skeletal Progenitor Cells in the Degenerate Human Intervertebral Disc. Spine 2007, 32, 2537–2544. [Google Scholar] [CrossRef] [PubMed]
- Leung, V.Y.L.; Aladin, D.M.K.; Lv, F.; Tam, V.; Sun, Y.; Lau, R.Y.C.; Hung, S.C.; Ngan, A.H.W.; Tang, B.; Lim, C.T.; et al. Mesenchymal Stem Cells Reduce Intervertebral Disc Fibrosis and Facilitate Repair. Stem Cells 2014, 32, 2164–2177. [Google Scholar] [CrossRef] [PubMed]
- Kraus, P.; Yerden, R.; Kocsis, V.; Lufkin, T. RNA in Situ Hybridization Characterization of Non-Enzymatic Derived Bovine Intervertebral Disc Cell Lineages Suggests Progenitor Cell Potential. Acta Histochem. 2017, 119, 150–160. [Google Scholar] [CrossRef]
- Kraus, P.; Yerden, R.; Sipes, D.; Sur, S.; Lufkin, T. A Quantitative and Qualitative RNA Expression Profiling Assay for Cell Culture with Single Cell Resolution. Cytotechnology 2018, 70, 185–192. [Google Scholar] [CrossRef]
- Kraus, P.; Li, K.; Sipes, D.; Varden, L.; Yerden, R.; Henderson, A.; Sur, S.; Lufkin, T. Single-Cell Phenotyping of Complex Heterogeneous Tissue; Springer Nature: Singapore, 2019; p. 17. [Google Scholar]
- De Luca, P.; Castagnetta, M.; de Girolamo, L.; Coco, S.; Malacarne, M.; Ragni, E.; Viganò, M.; Lugano, G.; Brayda-Bruno, M.; Coviello, D.; et al. Intervertebral Disc and Endplate Cell Characterisation Highlights Annulus Fibrosus Cells as the Most Promising for Tissue-Specific Disc Degeneration Therapy. Eur. Cells Mater. 2020, 39, 156–170. [Google Scholar] [CrossRef]
- Veras, M.A.; McCann, M.R.; Tenn, N.A.; Séguin, C.A. Transcriptional Profiling of the Murine Intervertebral Disc and Age-Associated Changes in the Nucleus Pulposus. Connect. Tissue Res. 2020, 61, 63–81. [Google Scholar] [CrossRef]
- Rodrigues-Pinto, R.; Ward, L.; Humphreys, M.; Zeef, L.A.H.; Berry, A.; Hanley, K.P.; Hanley, N.; Richardson, S.M.; Hoyland, J.A. Human Notochordal Cell Transcriptome Unveils Potential Regulators of Cell Function in the Developing Intervertebral Disc. Sci. Rep. 2018, 8, 12866. [Google Scholar] [CrossRef]
- Richardson, S.M.; Kalamegam, G.; Pushparaj, P.N.; Matta, C.; Memic, A.; Khademhosseini, A.; Mobasheri, R.; Poletti, F.L.; Hoyland, J.A.; Mobasheri, A. Mesenchymal Stem Cells in Regenerative Medicine: Focus on Articular Cartilage and Intervertebral Disc Regeneration. Methods A Companion Methods Enzymol. 2016, 99, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Weiler, C.; Nerlich, A.G.; Schaaf, R.; Bachmeier, B.E.; Wuertz, K.; Boos, N. Immunohistochemical Identification of Notochordal Markers in Cells in the Aging Human Lumbar Intervertebral Disc. Eur. Spine J. Off. Publ. Eur. Spine Soc. Eur. Spinal Deform. Soc. Eur. Sect. Cerv. Spine Res. Soc. 2010, 19, 1761–1770. [Google Scholar] [CrossRef]
- Rodrigues-Pinto, R.; Berry, A.; Piper-Hanley, K.; Hanley, N.; Richardson, S.M.; Hoyland, J.A. Spatiotemporal Analysis of Putative Notochordal Cell Markers Reveals CD24 and Keratins 8, 18, and 19 as Notochord-Specific Markers during Early Human Intervertebral Disc Development. J. Orthop. Res. 2016, 34, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zheng, Z.; Qi, J.; Wang, J.; Zhou, Q.; Hu, F.; Liang, J.; Li, C.; Zhang, W.; Zhang, X. CD24 Identifies Nucleus Pulposus Progenitors/Notochordal Cells for Disc Regeneration. J. Biol. Eng. 2018, 12, 35. [Google Scholar] [CrossRef]
- Minogue, B.M.; Richardson, S.M.; Zeef, L.A.; Freemont, A.J.; Hoyland, J.A. Transcriptional Profiling of Bovine Intervertebral Disc Cells: Implications for Identification of Normal and Degenerate Human Intervertebral Disc Cell Phenotypes. Arthritis Res. Ther. 2010, 12, 1–20. [Google Scholar] [CrossRef]
- Rutges, J.; Creemers, L.; Dhert, W.; Milz, S.; Sakai, D.; Mochida, J.; Alini, M.; Grad, S. Variations in Gene and Protein Expression in Human Nucleus Pulposus in Comparison with Annulus Fibrosus and Cartilage Cells: Potential Associations with Aging and Degeneration. Osteoarthr. Cartil. 2010, 18, 416–423. [Google Scholar] [CrossRef]
- Peck, S.H.; McKee, K.K.; Tobias, J.W.; Malhotra, N.R.; Harfe, B.D.; Smith, L.J. Whole Transcriptome Analysis of Notochord-Derived Cells during Embryonic Formation of the Nucleus Pulposus. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Sakai, D.; Nakamura, Y.; Nakai, T.; Mishima, T.; Kato, S.; Grad, S.; Alini, M.; Risbud, M.V.; Chan, D.; Cheah, K.S.E.; et al. Exhaustion of Nucleus Pulposus Progenitor Cells with Ageing and Degeneration of the Intervertebral Disc. Nat. Commun. 2012, 3, 1211–1264. [Google Scholar] [CrossRef]
- Richardson, S.M.; Ludwinski, F.E.; Gnanalingham, K.K.; Atkinson, R.A.; Freemont, A.J.; Hoyland, J.A. Notochordal and Nucleus Pulposus Marker Expression Is Maintained by Sub-Populations of Adult Human Nucleus Pulposus Cells through Aging and Degeneration. Sci. Rep. 2017, 7, 1501. [Google Scholar] [CrossRef]
- Schol, J.; Sakai, D. Cell Therapy for Intervertebral Disc Herniation and Degenerative Disc Disease: Clinical Trials. Int. Orthop. 2019, 43, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Liu, H.; Yang, M.; Zhang, Y.; Huang, B.; Zhou, Y. Disc Cell Senescence in Intervertebral Disc Degeneration: Causes and Molecular Pathways. Cell Cycle 2016, 15, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Lakstins, K.; Arnold, L.; Gunsch, G.; Khan, S.; Moore, S.; Purmessur, D. Characterization of Bovine and Canine Animal Model Cartilage Endplates and Comparison to Human Cartilage Endplate Structure, Matrix Composition, and Cell Phenotype. JOR Spine 2020, 3. [Google Scholar] [CrossRef]
- Dal Molin, A.; Baruzzo, G.; Di Camillo, B. Single-Cell RNA-Sequencing: Assessment of Differential Expression Analysis Methods. Front. Genet. 2017, 8. [Google Scholar] [CrossRef]
- Soneson, C.; Robinson, M.D. Bias, Robustness and Scalability in Single-Cell Differential Expression Analysis. Nat. Methods 2018, 15, 255–261. [Google Scholar] [CrossRef]
- Chen, J.; Yan, W.; Setton, L.A. Molecular Phenotypes of Notochordal Cells Purified from Immature Nucleus Pulposus. Eur. Spine J. 2006, 15, 303–311. [Google Scholar] [CrossRef]
- Kim, J.H.; Deasy, B.M.; Seo, H.Y.; Studer, R.K.; Vo, N.V.; Georgescu, H.I.; Sowa, G.A.; Kang, J.D. Differentiation of Intervertebral Notochordal Cells Through Live Automated Cell Imaging System In Vitro. Spine 2009, 34, 2486–2493. [Google Scholar] [CrossRef]
- Oguz, E.; Tsai, T.T.; Di Martino, A.; Guttapalli, A.; Albert, T.J.; Shapiro, I.M.; Risbud, M.V. Galectin-3 Expression in the Intervertebral Disc: A Useful Marker of the Notochord Phenotype? Spine 2007, 32, 9–16. [Google Scholar] [CrossRef]
- Poiraudeau, S.; Monteiro, I.; Anract, P.; Blanchard, O.; Revel, M.; Corvol, M.T. Phenotypic Characteristics of Rabbit Intervertebral Disc Cells. Comparison with Cartilage Cells from the Same Animals. Spine 1999, 24, 837–844. [Google Scholar] [CrossRef]
- Rastogi, A.; Thakore, P.; Leung, A.; Benavides, M.; Machado, M.; Morschauser, M.A.; Hsieh, A.H. Environmental Regulation of Notochordal Gene Expression in Nucleus Pulposus Cells. J. Cell. Physiol. 2009, 220, 698–705. [Google Scholar] [CrossRef]
- Tang, X.; Jing, L.; Chen, J. Changes in the Molecular Phenotype of Nucleus Pulposus Cells with Intervertebral Disc Aging. PLoS ONE 2012, 7, e52020. [Google Scholar] [CrossRef]
- He, S.; Wang, L.H.; Liu, Y.; Li, Y.Q.; Chen, H.T.; Xu, J.H.; Peng, W.; Lin, G.W.; Wei, P.P.; Li, B.; et al. Single-Cell Transcriptome Profiling of an Adult Human Cell Atlas of 15 Major Organs. Genome Biol. 2020, 21. [Google Scholar] [CrossRef]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A Revised Airway Epithelial Hierarchy Includes CFTR-Expressing Ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A Single-Cell Atlas of the Airway Epithelium Reveals the CFTR-Rich Pulmonary Ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science 2017, 356. [Google Scholar] [CrossRef]
- Pal, B.; Chen, Y.; Vaillant, F.; Jamieson, P.; Gordon, L.; Rios, A.C.; Wilcox, S.; Fu, N.; Liu, K.H.; Jackling, F.C.; et al. Construction of Developmental Lineage Relationships in the Mouse Mammary Gland by Single-Cell RNA Profiling. Nat. Commun. 2017, 8, 1627. [Google Scholar] [CrossRef]
- Kiselev, V.Y.; Andrews, T.S.; Hemberg, M. Challenges in Unsupervised Clustering of Single-Cell RNA-Seq Data. Nat. Rev. Genet. 2019, 20, 273–282. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven Grand Challenges in Single-Cell Data Science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.; Coulombe, P.A. Of Mice and Men: Genetic Skin Diseases of Keratin. Cell 1992, 69, 899–902. [Google Scholar] [CrossRef]
- Bedore, J.; Sha, W.; McCann, M.R.; Liu, S.; Leask, A.; Séguin, C.A. Impaired Intervertebral Disc Development and Premature Disc Degeneration in Mice with Notochord-Specific Deletion of CCN2. Arthritis Rheum. 2013, 65, 2634–2644. [Google Scholar] [CrossRef]
- Russo, J.W.; Castellot, J.J. CCN5: Biology and Pathophysiology. J. Cell Commun. Signal. 2010, 4, 119–130. [Google Scholar] [CrossRef]
- Lake, A.C.; Bialik, A.; Walsh, K.; Castellot, J.J. CCN5 Is a Growth Arrest-Specific Gene That Regulates Smooth Muscle Cell Proliferation and Motility. Am. J. Pathol. 2003, 162, 219–231. [Google Scholar] [CrossRef]
- Ukita, K.; Hirahara, S.; Oshima, N.; Imuta, Y.; Yoshimoto, A.; Jang, C.W.; Oginuma, M.; Saga, Y.; Behringer, R.R.; Kondoh, H.; et al. Wnt Signaling Maintains the Notochord Fate for Progenitor Cells and Supports the Posterior Extension of the Notochord. Mech. Dev. 2009, 126, 791–803. [Google Scholar] [CrossRef]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-Tissue Growth Factor (CTGF) Modulates Cell Signalling by BMP and TGF-Beta. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef]
- Si, W.; Kang, Q.; Luu, H.H.; Park, J.K.; Luo, Q.; Song, W.X.; Jiang, W.; Luo, X.; Li, X.; Yin, H.; et al. CCN1/Cyr61 Is Regulated by the Canonical Wnt Signal and Plays an Important Role in Wnt3A-Induced Osteoblast Differentiation of Mesenchymal Stem Cells. Mol. Cell. Biol. 2006, 26, 2955–2964. [Google Scholar] [CrossRef]
- Kondo, N.; Yuasa, T.; Shimono, K.; Tung, W.; Okabe, T.; Yasuhara, R.; Pacifici, M.; Zhang, Y.; Iwamoto, M.; Enomoto-Iwamoto, M. Intervertebral Disc Development Is Regulated by Wnt/β-Catenin Signaling. Spine 2011, 36, E513–E518. [Google Scholar] [CrossRef]
- Hiyama, A.; Sakai, D.; Risbud, M.V.; Tanaka, M.; Arai, F.; Abe, K.; Mochida, J. Enhancement of Intervertebral Disc Cell Senescence by WNT/β-Catenin Signaling-Induced Matrix Metalloproteinase Expression. Arthritis Rheum. 2010, 62, 3036–3047. [Google Scholar] [CrossRef]
- Xia, Z.; Wu, S.; Wei, X.; Liao, Y.; Yi, P.; Liu, Y.; Liu, J.; Liu, J. Hypoxic ER Stress Suppresses β-Catenin Expression and Promotes Cooperation between the Transcription Factors XBP1 and HIF1α for Cell Survival. J. Biol. Chem. 2019, 294, 13811–13821. [Google Scholar] [CrossRef]
- Arkesteijn, I.T.M.; Potier, E.; Ito, K. The Regenerative Potential of Notochordal Cells in a Nucleus Pulposus Explant. Glob. Spine J. 2017, 7, 14–20. [Google Scholar] [CrossRef]
- Erwin, W.M.; Islam, D.; Inman, R.D.; Fehlings, M.G.; Tsui, F.W.L. Notochordal Cells Protect Nucleus Pulposus Cells from Degradation and Apoptosis: Implications for the Mechanisms of Intervertebral Disc Degeneration. Arthritis Res. Ther. 2011, 13, R215. [Google Scholar] [CrossRef] [PubMed]
- Matta, A.; Karim, M.Z.; Isenman, D.E.; Erwin, W.M. Molecular Therapy for Degenerative Disc Disease: Clues from Secretome Analysis of the Notochordal Cell-Rich Nucleus Pulposus. Sci. Rep. 2017, 7, 45623. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Mori, D.; Makii, Y.; Nakamoto, H.; Murahashi, Y.; Yano, F.; Chang, S.H.; Taniguchi, Y.; Kobayashi, H.; Semba, H.; et al. Hypoxia-Inducible Factor-1 Alpha Maintains Mouse Articular Cartilage through Suppression of NF-κB Signaling. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Jiang, G.; Liu, H.; Li, Z.; Pei, Y.; Wang, H.; Pan, H.; Cui, H.; Long, J.; Wang, J.; et al. Melatonin Alleviates Intervertebral Disc Degeneration by Disrupting the IL-1beta/NF-KB-NLRP3 Inflammasome Positive Feedback Loop. Bone Res. 2020, 8, 1–13. [Google Scholar] [CrossRef]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in Metabolic Regulation: Physiological Role and Therapeutic Outlook. Curr. Drug Targets 2017, 18, 1095–1103. [Google Scholar] [CrossRef]
- Kamitori, K.; Yamaguchi, F.; Dong, Y.; Hossain, A.; Katagi, A.; Noguchi, C.; Hirata, Y.; Tsukamoto, I.; Hatano, N.; Tokuda, M. Both Ser361 Phosphorylation and the C-Arrestin Domain of Thioredoxin Interacting Protein Are Important for Cell Cycle Blockade at the G1/S Checkpoint. FEBS Open Bio 2018, 8, 1804–1819. [Google Scholar] [CrossRef]
- Young, R.A. Control of Embryonic Stem Cell State. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef]
- Stegen, S.; Laperre, K.; Eelen, G.; Rinaldi, G.; Fraisl, P.; Torrekens, S.; Van Looveren, R.; Loopmans, S.; Bultynck, G.; Vinckier, S.; et al. HIF-1α Metabolically Controls Collagen Synthesis and Modification in Chondrocytes. Nature 2019, 565, 511–515. [Google Scholar] [CrossRef]
- Yamauchi, M.; Sricholpech, M. Lysine Post-Translational Modifications of Collagen. Essays Biochem. 2012, 52, 113–133. [Google Scholar] [CrossRef]
- Myllyharju, J.; Kivirikko, K.I. Collagens, Modifying Enzymes and Their Mutations in Humans, Flies and Worms. Trends Genet. TIG 2004, 20, 33–43. [Google Scholar] [CrossRef]
- Fernandes, L.M.; Khan, N.M.; Trochez, C.M.; Duan, M.; Diaz-Hernandez, M.E.; Presciutti, S.M.; Gibson, G.; Drissi, H. Single-Cell RNA-Seq Identifies Unique Transcriptional Landscapes of Human Nucleus Pulposus and Annulus Fibrosus Cells. Sci. Rep. 2020, 10, 15263. [Google Scholar] [CrossRef]
- Risbud, M.V.; Schoepflin, Z.R.; Mwale, F.; Kandel, R.A.; Grad, S.; Iatridis, J.C.; Sakai, D.; Hoyland, J.A. Defining the Phenotype of Young Healthy Nucleus Pulposus Cells: Recommendations of the Spine Research Interest Group at the 2014 Annual ORS Meeting. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2015, 33, 283–293. [Google Scholar] [CrossRef]
- Anderson, R.M.; Lawrence, A.R.; Stottmann, R.W.; Bachiller, D.; Klingensmith, J. Chordin and Noggin Promote Organizing Centers of Forebrain Development in the Mouse. Development 2002, 129, 4975–4987. [Google Scholar] [CrossRef]
- Johnson, J.L.F.; Hall, T.E.; Dyson, J.M.; Sonntag, C.; Ayers, K.; Berger, S.; Gautier, P.; Mitchell, C.; Hollway, G.E.; Currie, P.D. Scube Activity Is Necessary for Hedgehog Signal Transduction in Vivo. Dev. Biol. 2012, 368, 193–202. [Google Scholar] [CrossRef]
- Nakamichi, R.; Kataoka, K.; Asahara, H. Essential Role of Mohawk for Tenogenic Tissue Homeostasis Including Spinal Disc and Periodontal Ligament. Mod. Rheumatol. 2018, 28, 933–940. [Google Scholar] [CrossRef]
- Harris, B.S.; Zhang, Y.; Card, L.; Rivera, L.B.; Brekken, R.A.; Bradshaw, A.D. SPARC Regulates Collagen Interaction with Cardiac Fibroblast Cell Surfaces. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H841–H847. [Google Scholar] [CrossRef]
- McCurdy, S.; Baicu, C.F.; Heymans, S.; Bradshaw, A.D. Cardiac Extracellular Matrix Remodeling: Fibrillar Collagens and Secreted Protein Acidic and Rich in Cysteine (SPARC). J. Mol. Cell. Cardiol. 2010, 48, 544–549. [Google Scholar] [CrossRef]
- Bradshaw, A.D.; Sage, E.H. SPARC, a Matricellular Protein That Functions in Cellular Differentiation and Tissue Response to Injury. J. Clin. Investig. 2001, 107, 1049–1054. [Google Scholar] [CrossRef]
- Brekken, R.A.; Sage, E.H. SPARC, a Matricellular Protein: At the Crossroads of Cell-Matrix Communication. Matrix Biol. J. Int. Soc. Matrix Biol. 2001, 19, 816–827. [Google Scholar] [CrossRef]
- Giudici, C.; Raynal, N.; Wiedemann, H.; Cabral, W.A.; Marini, J.C.; Timpl, R.; Bächinger, H.P.; Farndale, R.W.; Sasaki, T.; Tenni, R. Mapping of SPARC/BM-40/Osteonectin-Binding Sites on Fibrillar Collagens. J. Biol. Chem. 2008, 283, 19551–19560. [Google Scholar] [CrossRef]
- Sasaki, T.; Göhring, W.; Mann, K.; Maurer, P.; Hohenester, E.; Knäuper, V.; Murphy, G.; Timpl, R. Limited Cleavage of Extracellular Matrix Protein BM-40 by Matrix Metalloproteinases Increases Its Affinity for Collagens. J. Biol. Chem. 1997, 272, 9237–9243. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.E.; Ingram, J.A.; Leslie, K.; Hanley, E.N.J. Cellular, but Not Matrix, Immunolocalization of SPARC in the Human Intervertebral Disc: Decreasing Localization with Aging and Disc Degeneration. Spine 2004, 29, 2223–2228. [Google Scholar] [CrossRef] [PubMed]
- Tajerian, M.; Alvarado, S.; Millecamps, M.; Dashwood, T.; Anderson, K.M.; Haglund, L.; Ouellet, J.; Szyf, M.; Stone, L.S. DNA Methylation of SPARC and Chronic Low Back Pain. Mol. Pain 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Schilling, T.F. Thrombospondin-4 Controls Matrix Assembly during Development and Repair of Myotendinous Junctions. eLife 2014, 3, e02372. [Google Scholar] [CrossRef] [PubMed]
- Kitaori, T.; Ito, H.; Schwarz, E.M.; Tsutsumi, R.; Yoshitomi, H.; Oishi, S.; Nakano, M.; Fujii, N.; Nagasawa, T.; Nakamura, T. Stromal Cell-Derived Factor 1/CXCR4 Signaling Is Critical for the Recruitment of Mesenchymal Stem Cells to the Fracture Site during Skeletal Repair in a Mouse Model. Arthritis Rheum. 2009, 60, 813–823. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Q.; Gong, J.; Du, J.; Zhang, Y.; Zhao, X. Yak IGF2 Promotes Fibroblast Proliferation Via Suppression of IGF1R and PI3KCG Expression. Genes 2018, 9, 169. [Google Scholar] [CrossRef]
- Patan, S. TIE1 and TIE2 Receptor Tyrosine Kinases Inversely Regulate Embryonic Angiogenesis by the Mechanism of Intussusceptive Microvascular Growth. Microvasc. Res. 1998, 56, 1–21. [Google Scholar] [CrossRef]
- Leppänen, V.M.; Saharinen, P.; Alitalo, K. Structural Basis of Tie2 Activation and Tie2/Tie1 Heterodimerization. Proc. Natl. Acad. Sci. USA 2017, 114, 4376–4381. [Google Scholar] [CrossRef]
- Singh, H.; Milner, C.S.; Aguilar Hernandez, M.M.; Patel, N.; Brindle, N.P.J. Vascular Endothelial Growth Factor Activates the Tie Family of Receptor Tyrosine Kinases. Cell. Signal. 2009, 21, 1346–1350. [Google Scholar] [CrossRef]
- Leavitt, T.; Hu, M.S.; Borrelli, M.R.; Januszyk, M.; Garcia, J.T.; Ransom, R.C.; Mascharak, S.; desJardins-Park, H.E.; Litzenburger, U.M.; Walmsley, G.G.; et al. Prrx1 Fibroblasts Represent a Pro-Fibrotic Lineage in the Mouse Ventral Dermis. Cell Rep. 2020, 33, 108356. [Google Scholar] [CrossRef]
- Solé-Boldo, L.; Raddatz, G.; Schütz, S.; Mallm, J.P.; Rippe, K.; Lonsdorf, A.S.; Rodríguez-Paredes, M.; Lyko, F. Single-Cell Transcriptomes of the Human Skin Reveal Age-Related Loss of Fibroblast Priming. Commun. Biol. 2020, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Juarez, C.F.; Dedhia, P.H.; Jin, S.; Ruiz-Vega, R.; Ma, D.; Liu, Y.; Yamaga, K.; Shestova, O.; Gay, D.L.; Yang, Z.; et al. Single-Cell Analysis Reveals Fibroblast Heterogeneity and Myeloid-Derived Adipocyte Progenitors in Murine Skin Wounds. Nat. Commun. 2019, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Parreno, J.; Raju, S.; Wu, P.H.; Kandel, R.A. MRTF-A Signaling Regulates the Acquisition of the Contractile Phenotype in Dedifferentiated Chondrocytes. Matrix Biol. 2017, 62, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Fortelny, N.; Overall, C.M.; Pavlidis, P.; Freue, G.V.C. Can We Predict Protein from mRNA Levels? Nature 2017, 547, E19–E20. [Google Scholar] [CrossRef]
- Beckstein, J.C.; Sen, S.; Schaer, T.P.; Vresilovic, E.J.; Elliott, D.M. Comparison of Animal Discs Used in Disc Research to Human Lumbar Disc. Spine 2008, 8. [Google Scholar] [CrossRef]
- O’Connell, G.D.; Vresilovic, E.J.; Elliott, D.M. Comparison of Animals Used in Disc Research to Human Lumbar Disc Geometry. Spine 2007, 32, 328–333. [Google Scholar] [CrossRef]
- Showalter, B.L.; Beckstein, J.C.; Martin, J.T.; Beattie, E.E.; Orías, A.A.E.; Schaer, T.P.; Vresilovic, E.J.; Elliott, D.M. Comparison of Animal Discs Used in Disc Research to Human Lumbar Disc: Torsion Mechanics and Collagen Content. Spine 2012, 37, E900–E907. [Google Scholar] [CrossRef]
- Chen, J.W.; Li, B.; Yang, Y.H.; Jiang, S.D.; Jiang, L.S. Significance of Hypoxia in the Physiological Function of Intervertebral Disc Cells. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 193–204. [Google Scholar] [CrossRef]
- Risbud, M.V.; Shapiro, I.M. Microenvironmental Control of Disc Cell Function: Influence of Hypoxia and Osmotic Pressure. In The Intervertebral Disc: Molecular and Structural Studies of the Disc in Health and Disease; Shapiro, I.M., Risbud, M.V., Eds.; Springer: Vienna, Austria, 2014; pp. 93–108. [Google Scholar] [CrossRef]
- Li, H.; Tao, Y.; Liang, C.; Han, B.; Li, F.; Chen, G.; Chen, Q. Influence of Hypoxia in the Intervertebral Disc on the Biological Behaviors of Rat Adipose- and Nucleus Pulposus-Derived Mesenchymal Stem Cells. Cells Tissues Organs 2013, 198, 266–277. [Google Scholar] [CrossRef]
- Yang, S.H.; Hu, M.H.; Sun, Y.H.; Lin, F.H. Differential Phenotypic Behaviors of Human Degenerative Nucleus Pulposus Cells under Normoxic and Hypoxic Conditions: Influence of Oxygen Concentration during Isolation, Expansion, and Cultivation. Spine J. Off. J. N. Am. Spine Soc. 2013, 13, 1590–1596. [Google Scholar] [CrossRef]
- Wiseman, M.A.; Birch, H.L.; Akmal, M.; Goodship, A.E. Segmental Variation in the in Vitro Cell Metabolism of Nucleus Pulposus Cells Isolated from a Series of Bovine Caudal Intervertebral Discs. Spine 2005, 30, 505–511. [Google Scholar] [CrossRef]
- Simon, A. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. 2021. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 April 2019).
- Steven, W. Babraham Bioinformatics—FastQ Screen. 2021. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/ (accessed on 29 April 2019).
- The UniVec Database. Available online: https://www.ncbi.nlm.nih.gov/tools/vecscreen/univec/ (accessed on 29 April 2019).
- The UniVec Database—Refseq. Available online: https://www.ncbi.nlm.nih.gov/refseq/ (accessed on 29 April 2019).
- Silva. Available online: https://www.arb-silva.de/ (accessed on 29 April 2019).
- Hansen, K.D.; Brenner, S.E.; Dudoit, S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010, 38. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Lun, A.T.; McCarthy, D.J.; Marioni, J.C. A Step-by-Step Workflow for Low-Level Analysis of Single-Cell RNA-Seq Data with Bioconductor. F1000Research 2016, 5, 2122. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.R.; Neff, N.F.; Kalisky, T.; Dalerba, P.; Treutlein, B.; Rothenberg, M.E.; Mburu, F.M.; Mantalas, G.L.; Sim, S.; Clarke, M.F.; et al. Quantitative Assessment of Single-Cell RNA-Sequencing Methods. Nat. Methods 2014, 11, 41–46. [Google Scholar] [CrossRef]
- Jew, B.; Alvarez, M.; Rahmani, E.; Miao, Z.; Ko, A.; Garske, K.M.; Sul, J.H.; Pietiläinen, K.H.; Pajukanta, P.; Halperin, E. Accurate Estimation of Cell Composition in Bulk Expression through Robust Integration of Single-Cell Information. Nat. Commun. 2020, 11, 1971. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample N | Description | Source |
|---|---|---|
| 1 | 3 NPs proximal | Animal 1 |
| 2 | 3 AFs proximal | Animal 1 |
| 3 | 3 NPs distal | Animal 1 |
| 4 | 3 AFs distal | Animal 1 |
| 5 | 3 NPs proximal | Animal 2 |
| 6 | 3 AFs proximal | Animal 2 |
| 7 | 3 NPs distal | Animal 2 |
| 8 | 3 AFs distal | Animal 2 |
| Sample N | Description | Source |
|---|---|---|
| 1 | 2 NPs proximal | Animal 3 |
| 2 | 2 AFs proximal | Animal 3 |
| 3 | 2 NPs distal | Animal 3 |
| 4 | 2 AFs distal | Animal 3 |
| 5 | 2 NPs proximal | Animal 4 |
| 6 | 2 AFs proximal | Animal 4 |
| 7 | 2 NPs distal | Animal 4 |
| 8 | 2 AFs distal | Animal 4 |
| 9 | 2 NPs proximal | Animal 5 |
| 10 | 2 AFs proximal | Animal 5 |
| 11 | 2 NPs distal | Animal 5 |
| 12 | 2 AFs distal | Animal 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calió, M.; Gantenbein, B.; Egli, M.; Poveda, L.; Ille, F. The Cellular Composition of Bovine Coccygeal Intervertebral Discs: A Comprehensive Single-Cell RNAseq Analysis. Int. J. Mol. Sci. 2021, 22, 4917. https://doi.org/10.3390/ijms22094917
Calió M, Gantenbein B, Egli M, Poveda L, Ille F. The Cellular Composition of Bovine Coccygeal Intervertebral Discs: A Comprehensive Single-Cell RNAseq Analysis. International Journal of Molecular Sciences. 2021; 22(9):4917. https://doi.org/10.3390/ijms22094917
Chicago/Turabian StyleCalió, Martina, Benjamin Gantenbein, Marcel Egli, Lucy Poveda, and Fabian Ille. 2021. "The Cellular Composition of Bovine Coccygeal Intervertebral Discs: A Comprehensive Single-Cell RNAseq Analysis" International Journal of Molecular Sciences 22, no. 9: 4917. https://doi.org/10.3390/ijms22094917
APA StyleCalió, M., Gantenbein, B., Egli, M., Poveda, L., & Ille, F. (2021). The Cellular Composition of Bovine Coccygeal Intervertebral Discs: A Comprehensive Single-Cell RNAseq Analysis. International Journal of Molecular Sciences, 22(9), 4917. https://doi.org/10.3390/ijms22094917