
Environmental Factors Exacerbate Parkinsonian Phenotypes in an Asian-Specific Knock-In LRRK2 Risk Variant in Mice

 , , , ,
, , , ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
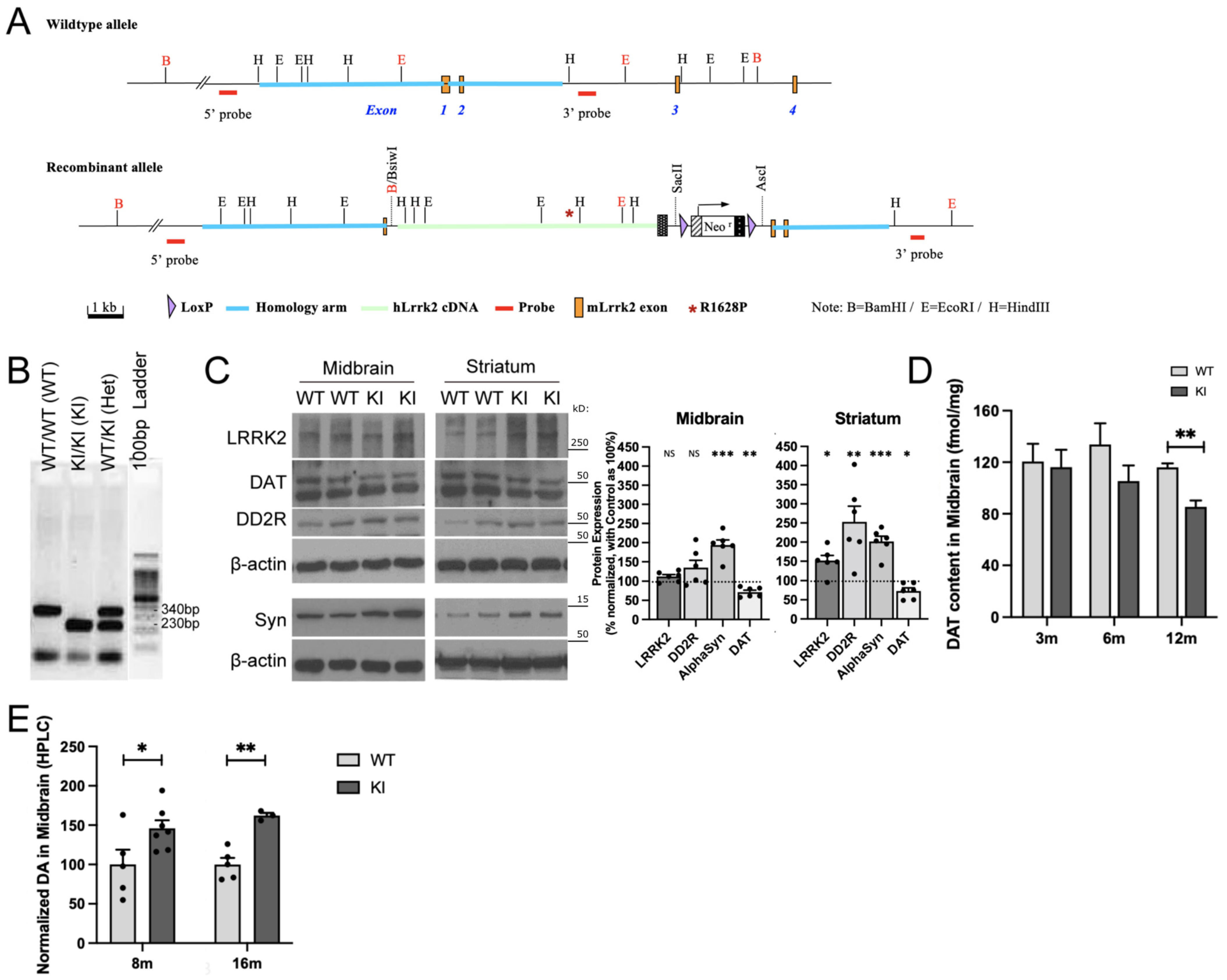
2.1. R1628P Risk Variant in Mice Differentially Regulates Key PD-Related Proteins
2.2. Combination of Paraquat/Maneb and Unpredictable Chronic Mild Stress Accelerates Behavioural Deficits in LRRK2 R1628P KI Mice
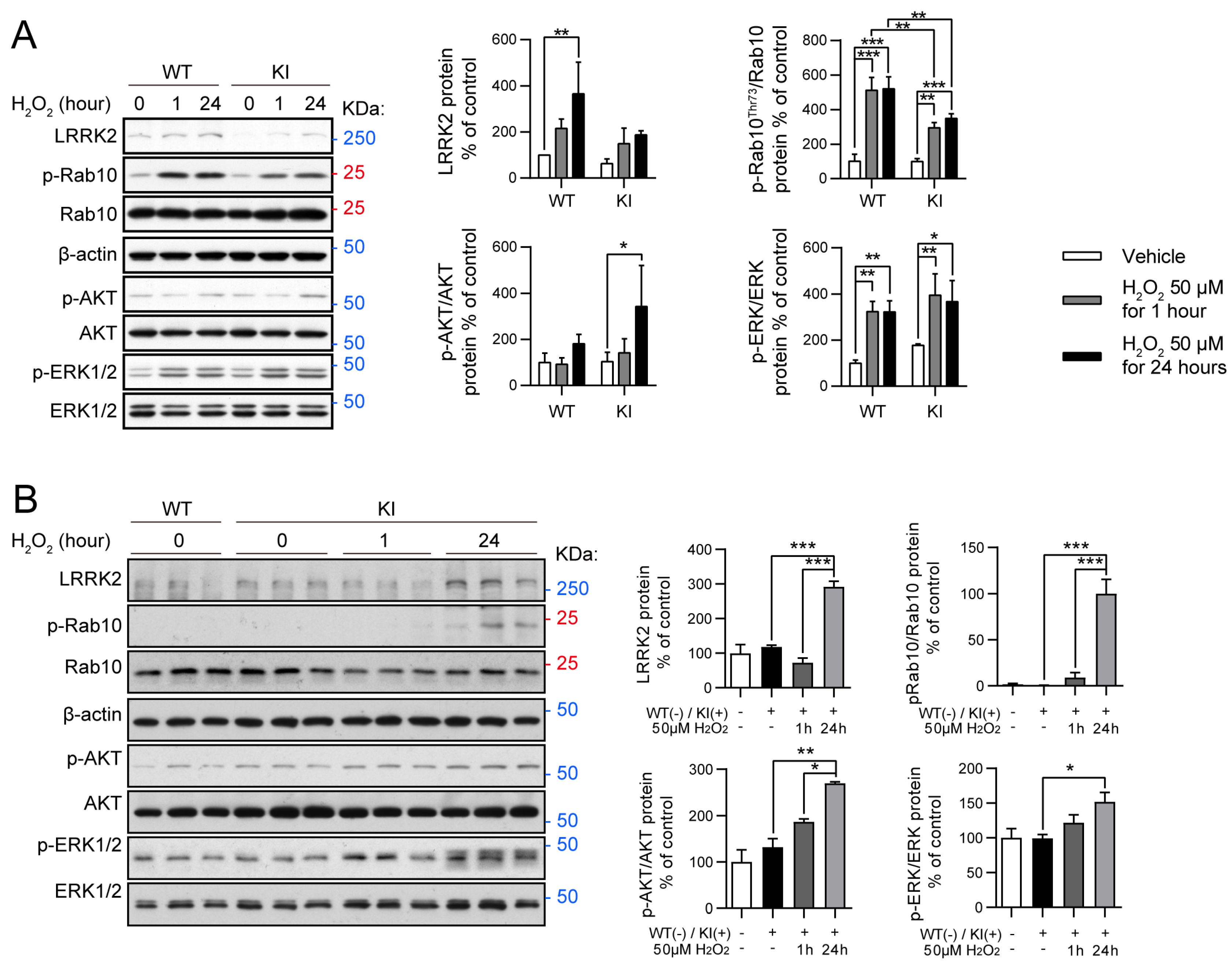
2.3. Oxidative Stress Modulates RAB10, AKT, and ERK Activity in LRRK2 R1628P WT and KI Primary Fibroblast Cells and Primary Cortical Neurons
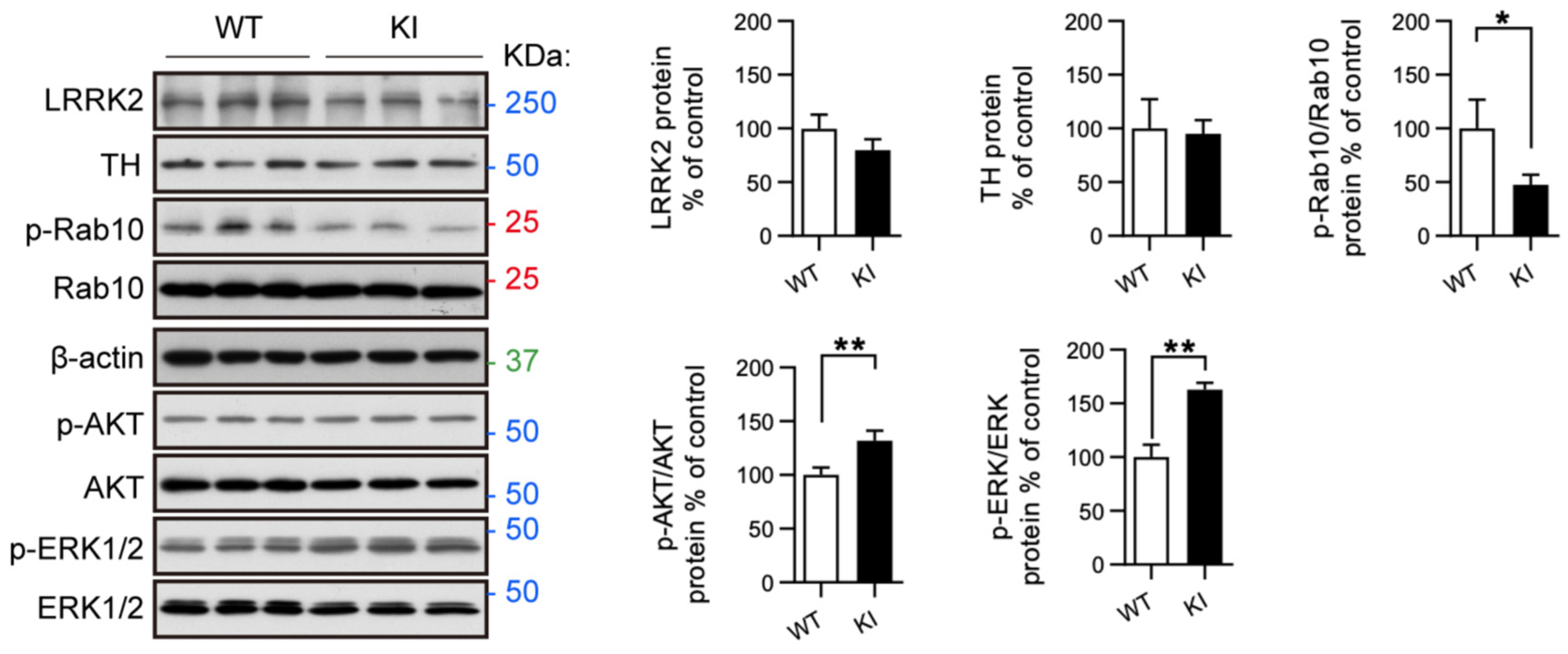
2.4. RAB10, AKT, and ERK Activation Is Altered in Primary Midbrain Neurons from LRRK2 R1628P KI Mice
3. Discussion
4. Methods and Materials
4.1. Animals
4.2. Animal Treatment
4.3. HPLC Analysis
4.4. Autoradiography Analysis
4.5. Immunohistochemistry
4.6. Cell Culture
4.7. Behavioural Testing
4.8. Protein Extraction and Western Blotting
4.9. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- DeMaagd, G.; Philip, A. Parkinson’s Disease and Its Management: Part 1: Disease Entity, Risk Factors, Pathophysiology, Clinical Presentation, and Diagnosis. Pharm. Ther. 2015, 40, 504–532. [Google Scholar]
- Rogers, G.; Davies, D.; Pink, J.; Cooper, P. Parkinson’s disease: Summary of updated NICE guidance. BMJ 2017, 358, j1951. [Google Scholar] [CrossRef]
- Rizzo, G.; Copetti, M.; Arcuti, S.; Martino, D.; Fontana, A.; Logroscino, G. Accuracy of clinical diagnosis of Parkinson disease: A systematic review and meta-analysis. Neurology 2016, 86, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; De Jager, P.L.; Feany, M.B. Parkinson’s disease: Genetics and pathogenesis. Annu. Rev. Pathol. 2011, 6, 193–222. [Google Scholar] [CrossRef]
- Reichmann, H.; Brandt, M.D.; Klingelhoefer, L. The nonmotor features of Parkinson’s disease: Pathophysiology and management advances. Curr. Opin. Neurol. 2016, 29, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef]
- Lee, F.J.; Liu, F. Genetic factors involved in the pathogenesis of Parkinson’s disease. Brain Res. Rev. 2008, 58, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Pan-Montojo, F.; Reichmann, H. Considerations on the role of environmental toxins in idiopathic Parkinson’s disease pathophysiology. Transl. Neurodegener. 2014, 3, 10. [Google Scholar] [CrossRef]
- Marras, C.; Canning, C.G.; Goldman, S.M. Environment, lifestyle, and Parkinson’s disease: Implications for prevention in the next decade. Mov. Disord. 2019, 34, 801–811. [Google Scholar] [CrossRef]
- Robles, T.F.; Carroll, J.E. Restorative biological processes and health. Soc. Personal. Psychol. Compass 2011, 5, 518–537. [Google Scholar] [CrossRef]
- Djamshidian, A.; Lees, A.J. Can stress trigger Parkinson’s disease? J. Neurol. Neurosurg. Psychiatry 2014, 85, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.J.; Espinosa-Oliva, A.M.; Carrillo-Jiménez, A.; Oliva-Martín, M.J.; García-Revilla, J.; García-Quintanilla, A.; de Pablos, R.M.; Venero, J.L. Relevance of chronic stress and the two faces of microglia in Parkinson’s disease. Front. Cell Neurosci. 2015, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Hemmerle, A.M.; Dickerson, J.W.; Herman, J.P.; Seroogy, K.B. Stress exacerbates experimental Parkinson’s disease. Mol. Psychiatry 2014, 19, 638–640. [Google Scholar] [CrossRef]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaar5429. [Google Scholar] [CrossRef]
- Chia, S.J.; Tan, E.K.; Chao, Y.X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [PubMed]
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661. [Google Scholar] [CrossRef]
- Volta, M.; Beccano-Kelly, D.A.; Paschall, S.A.; Cataldi, S.; MacIsaac, S.E.; Kuhlmann, N.; Kadgien, C.A.; Tatarnikov, I.; Fox, J.; Khinda, J.; et al. Initial elevations in glutamate and dopamine neurotransmission decline with age, as does exploratory behavior, in LRRK2 G2019S knock-in mice. Elife 2017, 6, e28377. [Google Scholar] [CrossRef]
- Cookson, M.R.; Xiromerisiou, G.; Singleton, A. How genetics research in Parkinson’s disease is enhancing understanding of the common idiopathic forms of the disease. Curr. Opin. Neurol. 2005, 18, 706–711. [Google Scholar] [CrossRef]
- Taymans, J.M.; Van den Haute, C.; Baekelandt, V. Distribution of PINK1 and LRRK2 in rat and mouse brain. J. Neurochem. 2006, 98, 951–961. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Shu, Y.; Ming, J.; Zhang, P.; Wang, Q.; Jiao, F.; Tian, B. Parkinson-Related LRRK2 Mutation R1628P Enables Cdk5 Phosphorylation of LRRK2 and Upregulates Its Kinase Activity. PLoS ONE 2016, 11, e0149739. [Google Scholar] [CrossRef] [PubMed]
- Gopalai, A.A.; Lim, S.Y.; Chua, J.Y.; Tey, S.; Lim, T.T.; Mohamed Ibrahim, N.; Tan, A.H.; Eow, G.B.; Abdul Aziz, Z.; Puvanarajah, S.D.; et al. LRRK2 G2385R and R1628P mutations are associated with an increased risk of Parkinson’s disease in the Malaysian population. Biomed. Res. Int. 2014, 2014, 867321. [Google Scholar] [CrossRef]
- Simpson, C.; Vinikoor-Imler, L.; Nassan, F.L.; Shirvan, J.; Lally, C.; Dam, T.; Maserejian, N. Prevalence of ten LRRK2 variants in Parkinson’s disease: A comprehensive review. Park. Relat. Disord. 2022, 98, 103–113. [Google Scholar] [CrossRef]
- Zhang, D.; Yao, J.; Ma, J.; Gao, L.; Sun, J.; Fang, J.; He, H.; Wu, T. Connectivity of corticostriatal circuits in nonmanifesting LRRK2 G2385R and R1628P carriers. CNS Neurosci. Ther. 2022, 28, 2024–2031. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Lee, J.W.; Tapias, V.; Di Maio, R.; Greenamyre, J.T.; Cannon, J.R. Behavioral, neurochemical, and pathologic alterations in bacterial artificial chromosome transgenic G2019S leucine-rich repeated kinase 2 rats. Neurobiol. Aging 2015, 36, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.E.S.; Ho, P.W.; Liu, H.F.; Pang, S.Y.; Leung, C.T.; Malki, Y.; Choi, Z.Y.; Ramsden, D.B.; Ho, S.L. LRRK2 mutant knock-in mouse models: Therapeutic relevance in Parkinson’s disease. Transl. Neurodegener. 2022, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Seegobin, S.P.; Heaton, G.R.; Liang, D.; Choi, I.; Blanca Ramirez, M.; Tang, B.; Yue, Z. Progress in LRRK2-Associated Parkinson’s Disease Animal Models. Front. Neurosci. 2020, 14, 674. [Google Scholar] [CrossRef]
- Li, Y.; Liu, W.; Oo, T.F.; Wang, L.; Tang, Y.; Jackson-Lewis, V.; Zhou, C.; Geghman, K.; Bogdanov, M.; Przedborski, S.; et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 2009, 12, 826–828. [Google Scholar] [CrossRef]
- Weng, Y.H.; Chen, C.Y.; Lin, K.J.; Chen, Y.L.; Yeh, T.H.; Hsiao, I.T.; Chen, I.J.; Lu, C.S.; Wang, H.L. (R1441C) LRRK2 induces the degeneration of SN dopaminergic neurons and alters the expression of genes regulating neuronal survival in a transgenic mouse model. Exp. Neurol. 2016, 275, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Neifert, S.; Karuppagounder, S.S.; Liu, Q.; Stankowski, J.N.; Lee, B.D.; Ko, H.S.; Lee, Y.; Grima, J.C.; Mao, X.; et al. Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc. Natl. Acad. Sci. USA 2018, 115, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, K.M.; Yue, M.; Behrouz, B.; Dächsel, J.C.; Lincoln, S.J.; Bowles, E.E.; Beevers, J.E.; Dugger, B.; Winner, B.; Prots, I.; et al. LRRK2 knockout mice have an intact dopaminergic system but display alterations in exploratory and motor co-ordination behaviors. Mol. Neurodegener. 2012, 7, 25. [Google Scholar] [CrossRef]
- Giaime, E.; Tong, Y.; Wagner, L.K.; Yuan, Y.; Huang, G.; Shen, J. Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron 2017, 96, 796–807.e6. [Google Scholar] [CrossRef]
- Volta, M.; Cataldi, S.; Beccano-Kelly, D.; Munsie, L.; Tatarnikov, I.; Chou, P.; Bergeron, S.; Mitchell, E.; Lim, R.; Khinda, J.; et al. Chronic and acute LRRK2 silencing has no long-term behavioral effects, whereas wild-type and mutant LRRK2 overexpression induce motor and cognitive deficits and altered regulation of dopamine release. Park. Relat. Disord. 2015, 21, 1156–1163. [Google Scholar] [CrossRef]
- Desplats, P.; Patel, P.; Kosberg, K.; Mante, M.; Patrick, C.; Rockenstein, E.; Fujita, M.; Hashimoto, M.; Masliah, E. Combined exposure to Maneb and Paraquat alters transcriptional regulation of neurogenesis-related genes in mice models of Parkinson’s disease. Mol. Neurodegener. 2012, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.; Cockburn, M.; Bronstein, J.; Zhang, X.; Ritz, B. Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am. J. Epidemiol. 2009, 169, 919–926. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, X.; Zhang, Y.; Zhang, L.; Feng, L. Chronic mild stress accelerates the progression of Parkinson’s disease in A53T α-synuclein transgenic mice. Exp. Neurol. 2016, 285 Pt A, 61–71. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Palomo-Garo, C.; Gómez-Gálvez, Y.; García, C.; Fernández-Ruiz, J. Targeting the cannabinoid CB2 receptor to attenuate the progression of motor deficits in LRRK2-transgenic mice. Pharmacol. Res. 2016, 110, 181–192. [Google Scholar] [CrossRef]
- Kozina, E.; Sadasivan, S.; Jiao, Y.; Dou, Y.; Ma, Z.; Tan, H.; Kodali, K.; Shaw, T.; Peng, J.; Smeyne, R.J. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain 2018, 141, 1753–1769. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef]
- Azkona, G.; López de Maturana, R.; Del Rio, P.; Sousa, A.; Vazquez, N.; Zubiarrain, A.; Jimenez-Blasco, D.; Bolaños, J.P.; Morales, B.; Auburger, G.; et al. LRRK2 Expression Is Deregulated in Fibroblasts and Neurons from Parkinson Patients with Mutations in PINK1. Mol. Neurobiol. 2018, 55, 506–516. [Google Scholar] [CrossRef]
- Wang, S.; Unnithan, S.; Bryant, N.; Chang, A.; Rosenthal, L.S.; Pantelyat, A.; Dawson, T.M.; Al-Khalidi, H.R.; West, A.B. Elevated Urinary Rab10 Phosphorylation in Idiopathic Parkinson Disease. Mov. Disord. 2022, 37, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Chuang, C.L.; Lu, Y.N.; Wang, H.C.; Chang, H.Y. Genetic dissection reveals that Akt is the critical kinase downstream of LRRK2 to phosphorylate and inhibit FOXO1, and promotes neuron survival. Hum. Mol. Genet. 2014, 23, 5649–5658. [Google Scholar] [CrossRef]
- Gao, Y.; Cui, M.; Zhong, S.; Feng, C.; Nwobodo, A.K.; Chen, B.; Song, Y.; Wang, Y. Dihydroartemisinin ameliorates LPS-induced neuroinflammation by inhibiting the PI3K/AKT pathway. Metab. Brain Dis. 2020, 35, 661–672. [Google Scholar] [CrossRef]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes. Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.J.; Chiu, G.N. Role of oxidative stress, endoplasmic reticulum stress and ERK activation in triptolide-induced apoptosis. Int. J. Oncol. 2013, 42, 1605–1612. [Google Scholar] [CrossRef]
- Verma, M.; Steer, E.K.; Chu, C.T. ERKed by LRRK2: A cell biological perspective on hereditary and sporadic Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 1273–1281. [Google Scholar] [CrossRef]
- Bohush, A.; Niewiadomska, G.; Filipek, A. Role of Mitogen Activated Protein Kinase Signaling in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 2973. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; DeFranco, D.B. Opposing roles for ERK1/2 in neuronal oxidative toxicity: Distinct mechanisms of ERK1/2 action at early versus late phases of oxidative stress. J. Biol. Chem. 2006, 281, 16436–16442. [Google Scholar] [CrossRef]
- Omura, T.; Kaneko, M.; Okuma, Y.; Matsubara, K.; Nomura, Y. Endoplasmic reticulum stress and Parkinson’s disease: The role of HRD1 in averting apoptosis in neurodegenerative disease. Oxid. Med. Cell Longev. 2013, 2013, 239854. [Google Scholar] [CrossRef]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Lindner, P.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal. 2020, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C. Role of eIF2α Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb. Perspect. Biol. 2018, 10, a032870. [Google Scholar] [CrossRef]
- Choy, M.S.; Yusoff, P.; Lee, I.C.; Newton, J.C.; Goh, C.W.; Page, R.; Shenolikar, S.; Peti, W. Structural and Functional Analysis of the GADD34:PP1 eIF2α Phosphatase. Cell Rep. 2015, 11, 1885–1891. [Google Scholar] [CrossRef]
- Song, J.; Zhang, Q.; Wang, S.; Yang, F.; Chen, Z.; Dong, Q.; Ji, Q.; Yuan, X.; Ren, D. Cleavage of caspase-12 at Asp94, mediated by endoplasmic reticulum stress (ERS), contributes to stretch-induced apoptosis of myoblasts. J. Cell Physiol. 2018, 233, 9473–9487. [Google Scholar] [CrossRef]
- Pan, Z.; Xu, S. Population genomics of East Asian ethnic groups. Hereditas 2020, 157, 49. [Google Scholar] [CrossRef]
- Vanan, S.; Zeng, X.; Chia, S.Y.; Varnäs, K.; Jiang, M.; Zhang, K.; Saw, W.T.; Padmanabhan, P.; Yu, W.P.; Zhou, Z.D.; et al. Altered striatal dopamine levels in Parkinson’s disease VPS35 D620N mutant transgenic aged mice. Mol. Brain 2020, 13, 164. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109 Pt B, 249–257. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, E.; Zhao, H.T.; Cole, T.; West, A.B. LRRK2 and Rab10 coordinate macropinocytosis to mediate immunological responses in phagocytes. EMBO J. 2020, 39, e104862. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Howden, A.J.M.; Sarhan, A.R.; Lis, P.; Ito, G.; Martinez, T.N.; Brockmann, K.; Gasser, T.; Alessi, D.R.; Sammler, E.M. Interrogating Parkinson’s disease LRRK2 kinase pathway activity by assessing Rab10 phosphorylation in human neutrophils. Biochem. J. 2018, 475, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Kang, S.S.; Wang, Z.H.; Liu, X.; Day, J.X.; Wu, Z.; Peng, J.; Xiang, D.; Springer, W.; Ye, K. Akt Phosphorylates NQO1 and Triggers its Degradation, Abolishing Its Antioxidative Activities in Parkinson’s Disease. J. Neurosci. 2019, 39, 7291–7305. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Agrawal, A.; Verma, A.; Dubey, N. The PI3K-AKT pathway: A plausible therapeutic target in Parkinson’s disease. Exp. Mol. Pathol. 2023, 129, 104846. [Google Scholar] [CrossRef]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Falkenburger, B.H.; Saridaki, T.; Dinter, E. Cellular models for Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 121–130. [Google Scholar] [CrossRef]
- Abdulwahid Arif, I.; Ahmad Khan, H. Environmental toxins and Parkinson’s disease: Putative roles of impaired electron transport chain and oxidative stress. Toxicol. Ind. Health 2010, 26, 121–128. [Google Scholar] [CrossRef]
- De Miranda, B.R.; Goldman, S.M.; Miller, G.W.; Greenamyre, J.T.; Dorsey, E.R. Preventing Parkinson’s Disease: An Environmental Agenda. J. Park. Dis. 2022, 12, 45–68. [Google Scholar] [CrossRef]
- Wills, J.; Credle, J.; Oaks, A.W.; Duka, V.; Lee, J.H.; Jones, J.; Sidhu, A. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS ONE 2012, 7, e30745. [Google Scholar] [CrossRef]
- Vellingiri, B.; Chandrasekhar, M.; Sri Sabari, S.; Gopalakrishnan, A.V.; Narayanasamy, A.; Venkatesan, D.; Iyer, M.; Kesari, K.; Dey, A. Neurotoxicity of pesticides—A link to neurodegeneration. Ecotoxicol. Environ. Saf. 2022, 243, 113972. [Google Scholar] [CrossRef] [PubMed]
- Ducottet, C.; Griebel, G.; Belzung, C. Effects of the selective nonpeptide corticotropin-releasing factor receptor 1 antagonist antalarmin in the chronic mild stress model of depression in mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 625–631. [Google Scholar] [CrossRef]
- Isingrini, E.; Surget, A.; Belzung, C.; Freslon, J.L.; Frisbee, J.; O’Donnell, J.; Camus, V.; d’Audiffret, A. Altered aortic vascular reactivity in the unpredictable chronic mild stress model of depression in mice: UCMS causes relaxation impairment to ACh. Physiol. Behav. 2011, 103, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Mineur, Y.S.; Belzung, C.; Crusio, W.E. Effects of unpredictable chronic mild stress on anxiety and depression-like behavior in mice. Behav. Brain Res. 2006, 175, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Gil, V.; Bichler, Z.; Lee, J.K.; Seira, O.; Llorens, F.; Bribian, A.; Morales, R.; Claverol-Tinture, E.; Soriano, E.; Sumoy, L.; et al. Developmental expression of the oligodendrocyte myelin glycoprotein in the mouse telencephalon. Cereb. Cortex 2010, 20, 1769–1779. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Tu, H.; Jiang, M.; Vanan, S.; Chia, S.Y.; Jang, S.E.; Saw, W.T.; Ong, Z.W.; Ma, D.R.; Zhou, Z.D.; et al. The APP intracellular domain promotes LRRK2 expression to enable feed-forward neurodegenerative mechanisms in Parkinson’s disease. Sci. Signal. 2022, 15, eabk3411. [Google Scholar] [CrossRef]
- Lü, Q.; Xu, X.L.; He, Z.; Huang, X.J.; Guo, L.J.; Wang, H.X. Guattegaumerine protects primary cultured cortical neurons against oxidative stress injury induced by hydrogen peroxide concomitant with serum deprivation. Cell Mol. Neurobiol. 2009, 29, 355–364. [Google Scholar] [CrossRef]
- Guo, L.; Du, J.; Yuan, D.F.; Zhang, Y.; Zhang, S.; Zhang, H.C.; Mi, J.W.; Ning, Y.L.; Chen, M.J.; Wen, D.L.; et al. Optimal H2O2 preconditioning to improve bone marrow mesenchymal stem cells’ engraftment in wound healing. Stem Cell Res. Ther. 2020, 11, 434. [Google Scholar] [CrossRef]
- Bichler, Z.; Lim, H.C.; Zeng, L.; Tan, E.K. Non-motor and motor features in LRRK2 transgenic mice. PLoS ONE 2013, 8, e70249. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bichler, Z.; Vanan, S.; Zhang, Z.; Dong, Q.; Lee, J.W.L.; Zhang, C.; Hang, L.; Jiang, M.; Padmanabhan, P.; Saw, W.T.; et al. Environmental Factors Exacerbate Parkinsonian Phenotypes in an Asian-Specific Knock-In LRRK2 Risk Variant in Mice. Int. J. Mol. Sci. 2025, 26, 3556. https://doi.org/10.3390/ijms26083556
Bichler Z, Vanan S, Zhang Z, Dong Q, Lee JWL, Zhang C, Hang L, Jiang M, Padmanabhan P, Saw WT, et al. Environmental Factors Exacerbate Parkinsonian Phenotypes in an Asian-Specific Knock-In LRRK2 Risk Variant in Mice. International Journal of Molecular Sciences. 2025; 26(8):3556. https://doi.org/10.3390/ijms26083556
Chicago/Turabian StyleBichler, Zoë, Sarivin Vanan, Zhiwei Zhang, Qianying (Sally) Dong, Jolene Wei Ling Lee, Chengwu Zhang, Liting Hang, Mei Jiang, Parasuraman Padmanabhan, Wuan Ting Saw, and et al. 2025. "Environmental Factors Exacerbate Parkinsonian Phenotypes in an Asian-Specific Knock-In LRRK2 Risk Variant in Mice" International Journal of Molecular Sciences 26, no. 8: 3556. https://doi.org/10.3390/ijms26083556
APA StyleBichler, Z., Vanan, S., Zhang, Z., Dong, Q., Lee, J. W. L., Zhang, C., Hang, L., Jiang, M., Padmanabhan, P., Saw, W. T., Zhou, Z., Gulyás, B., Lim, K. L., Zeng, L., & Tan, E. K. (2025). Environmental Factors Exacerbate Parkinsonian Phenotypes in an Asian-Specific Knock-In LRRK2 Risk Variant in Mice. International Journal of Molecular Sciences, 26(8), 3556. https://doi.org/10.3390/ijms26083556