Abstract
Neuropathic pain indicates pain caused by damage to the somatosensory system and is difficult to manage and treat. A new treatment strategy urgently needs to be developed. Both autophagy and apoptosis are critical adaptive mechanisms when neurons encounter stress or damage. Recent studies have shown that, after nerve damage, both autophagic and apoptotic activities in the injured nerve, dorsal root ganglia, and spinal dorsal horn change over time. Many studies have shown that upregulated autophagic activities may help myelin clearance, promote nerve regeneration, and attenuate pain behavior. On the other hand, there is no direct evidence that the inhibition of apoptotic activities in the injured neurons can attenuate pain behavior. Most studies have only shown that agents can simultaneously attenuate pain behavior and inhibit apoptotic activities in the injured dorsal root ganglia. Autophagy and apoptosis can crosstalk with each other through various proteins and proinflammatory cytokine expressions. Proinflammatory cytokines can promote both autophagic/apoptotic activities and neuropathic pain formation, whereas autophagy can inhibit proinflammatory cytokine activities and further attenuate pain behaviors. Thus, agents that can enhance autophagic activities but suppress apoptotic activities on the injured nerve and dorsal root ganglia can treat neuropathic pain. Here, we summarized the evolving changes in apoptotic and autophagic activities in the injured nerve, dorsal root ganglia, spinal cord, and brain after nerve damage. This review may help in further understanding the treatment strategy for neuropathic pain during nerve injury by modulating apoptotic/autophagic activities and proinflammatory cytokines in the nervous system.
1. Introduction
Neuropathic pain is induced by damage to the somatosensory nervous system []. Neuropathic pain is difficult to manage, and epidemiological studies have shown that the prevalence of neuropathic pain ranges from 3 to 17% [,,]. Current treatments of neuropathic pain include tricyclic antidepressants (TCAs), serotonin–noradrenaline reuptake inhibitors (SNRIs), and the α2δ subunit of voltage-dependent calcium channel ligands (pregabalin/gabapentin) [,]. However, most of these medication treatments only have moderate efficacy based on the number needed to treat (NNT), and pharmacological treatments for neuropathic pain are effective in <50% of patients, according to previous reviews [,,]. A new strategy to treat neuropathic pain is urgent and worth developing.
Autophagy and apoptosis are two crucial processes when cells encounter damage or stress [,]. Autophagy is a self-digestion process involved in protein and organelle degradation to improve the survival rate of cells in a stressful environment []. Apoptosis is a kind of programmed cell death and cell shrinkage when cells encounter stress or damage []. Apoptosis and autophagy both play an essential role in cancer, immunity, and neurodegenerative diseases [,,,,]. Furthermore, autophagy [,] and apoptosis [,,,,,,,,] are also both involved in nerve damage and the process of neuropathic pain formation. It is easy to predict that injured neurons will have apoptotic changes after nerve damage. Some animal studies have demonstrated increased apoptotic changes in the injured nerve, the dorsal root ganglia [,,,,], and spinal cord [,,,] after peripheral nerve injury. Studies have found that different interventions, including medicines and hyperbaric oxygen therapy, can simultaneously attenuate neuropathic pain and suppress apoptotic activities in the dorsal root ganglia or spinal cord [,,,,,]. However, there is no direct evidence that inhibition of apoptotic activities in the injured neurons can attenuate pain behavior. In contrast to apoptosis, many studies with different animal models have shown changed autophagic activities in the injured nerve [,,,,,,], spinal cord [,,,,,,,,,,,,], and even brain [] after nerve injury. These studies also showed that upregulated autophagic activities can directly alleviate neuropathic pain [,,,,,,]. Additionally, autophagy and apoptosis have crosstalk through various proteins [,] and are both regulated by proinflammatory cytokines [,,,,,]. Studies have shown that proinflammatory cytokines play an essential role in promoting apoptotic activities [,] and neuropathic pain formation [,], while autophagy can inhibit proinflammatory cytokine activities [,,,]. Thus, agents that can upregulate autophagic activities and suppress proinflammatory cytokines activities have the potential to treat neuropathic pain. Liu’s review summarized the complex proteins and molecular modifications of the autophagy process in neuropathic pain conditions []. However, there has been no systematic review of the role of apoptosis and the interaction between autophagy, apoptosis, and proinflammatory cytokines in neuropathic pain formation. In this review, we will discuss autophagic/apoptotic activity changes and their interactions with proinflammatory cytokines based on the different anatomical locations of the nervous system, including the injured nerve, Schwann cells, dorsal root ganglion, spinal cord, and brain. We will also summarize the alteration of pain behaviors after modulating autophagic/apoptotic activities.
2. Autophagy in Neuropathic Pain Formation
There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [,]. Different animal pain models have demonstrated alterations in macroautophagic activities in the injured nerve, dorsal root ganglia [,,,,,,], spinal cord [,,,,,,,,,,,,], and brain [] after peripheral nerve damage. At the beginning of the macroautophagic process, Beclin-1 and Ambra1 (both are the upper stream proteins of the macroautophagy process) at the endoplasmic reticulum respond to the stress signaling pathway and initiate phagophore formation. In the next step, several proteins, including autophagy-related protein 5 (Atg5)–Atg12, Atg16L complex, Atg7, microtubule-associated protein 1A/1B-light chain 3 (LC3)-II, and p62, are involved in autophagosome formation [,,,]. Then, autophagosomes fuse with lysosomes, leading to autolysosome formation and further degradation [,]. Chloroquine, which blocks the fusion between autophagosomes and lysosomes, can be the autophagy inhibitor in the late stage of autophagy []. Another important autophagy regulatory pathway includes a class I phosphatidylinositol 3-kinase (PI3K) and a downstream mammalian target for rapamycin (mTOR) that inhibits the autophagic process [,]. Rapamycin, which inhibits mTOR activities, can upregulate autophagic activities, while 3-methyladenine (3-MA), which inhibits PI3K activities, can downregulate autophagic activities [,]. Microautophagy needs to be studied by electron microscopy and is still obscure in mammalian cells []. On the other hand, CMA is only observed in mammalian cells and has protein markers, including heat shock-cognate chaperone of 70 kDa (HSC70) and lysosome-associated membrane protein type-2A (LAMP2A) [,]. Both macroautophagy and CMA play essential roles in central neurological degeneration diseases []. The CMA pathway compensates when the macroautophagy process is inhibited; in contrast, CMA blockage induces compensative upregulation of macroautophagy [].
In peripheral nerve injury, only a few studies discussed the role of CMA in the damaged nerve. LAMP2A expressions increased on the sciatic nerve of rats with chronic inflammatory demyelinating polyneuropathy (CIDP) []. Many studies focused on macroautophagy changes in the neurons of animals after nerve damage with rapamycin or 3-MA treatment [,,,,,,,,,,,,,,,,,,,]. Most studies proposed that when macroautophagic activities increase, the expression levels of LC3-II, Bclin-1, and Atg5-Atg12 increase, but p62 levels (delivering ubiquitinated cargoes to autophagosomes) decrease [,,,,,,,,,,,,,,,,,,]. However, some authors stated that, lately, increased LC3-II expressions indicate that the autophagic flux is blocked and autophagic activities are decreased []. Herein, we summarized studies and discussed the alteration of autophagic activities at different anatomical locations in various animal pain models.
2.1. Autophagic Activity Changes in Injured Nerves, including Schwann Cells and Dorsal Root Ganglia, after Nerve Injury
It is easy to understand that injured nerves, including Schwann cells and dorsal root ganglia, which undergo stress and damage, show autophagic activity changes to adapt to peripheral nerve damage. Elevated autophagic activity may help myelin debris clearance, which is possibly related to chronic pain modulation. Many studies that used different animal models (including chronic constriction injury (CCI), sciatic nerve transection (SNT), spinal nerve ligation (SNL), and sciatic nerve crush injury (SNC)) have shown consistent results that the elevated autophagic activities are measured by different autophagic proteins (Beclin-1, Atg5, Atg7, LC3II) in the injured nerve axons, Schwann cells, and dorsal root ganglia from 6 h to 21 days after nerve injury [,,,,,,]. Furthermore, rapamycin can upregulate the autophagic activities in the dorsal root ganglia and Schwann cells and suppress pain behavior. On the other hand, 3-MA or transgenic methods that downregulate autophagic activities in the dorsal root ganglia and Schwann cells can enhance pain behavior [,,,,]. Table 1 summarizes the findings of those studies.

Table 1.
Summary of the alterations of autophagic activities in the injured nerve, including Schwann cells and dorsal root ganglia, after nerve injury in different models of rats and mice.
2.2. Autophagic Activity Changes in the Spinal Cord after Nerve Injury
The spinal dorsal horn involved in central sensitization is another critical area in neuropathic pain formation [,,]. Proinflammatory cytokines activated in the spinal dorsal horn (SDH) after peripheral nerve damage promote neuropathic pain formation [,]. Many studies have discussed the relations between proinflammatory cytokines and autophagic activity changes in the SDH in neuropathic pain by different animal models. Researchers studied the pain behavior and autophagic protein expressions in the spinal cord after direct administration of rapamycin and 3-MA [,,,]. Other studies utilized indirect methods; researchers monitored the effects of different proinflammatory mediators on pain behavior and autophagic protein expression in the spinal dorsal horn [,,,,,]. Thus, these findings hypothesized that autophagic activity could affect neuropathic pain indirectly.
Unlike the studies of dorsal root ganglia and injured nerves, surveys of autophagic expression changes in the spinal dorsal horn have shown inconsistent results. Some studies showed upregulated autophagic activities in the different types of cells in the spinal cord after nerve injury [,,,,,,,]; other studies demonstrated the opposite results [,,,,]. Although those studies did not have consistent results for the autophagic activity changes in the spinal cord after nerve injury, most studies showed that upregulation of autophagy could suppress pain behavior [,,,,,,,,]. These inconsistent results are probably caused by different animal models and study designs. Below, we summarized those studies according to their findings in Table 2.
Table 2.
Summary of the alterations of autophagic activities in the spinal cord after nerve injury in different models of rats and mice.
2.2.1. Increased Autophagic Activities in the Spinal Cord Neurons, Decreased Autophagic Activities in the Spinal Cord Microglia and Astrocytes after Nerve Injury, and Autophagy Acts as a Pain Suppressor
Several studies from neuropathic pain models (including CCI, SNL, spared nerve injury (SNI)) and diabetic rats had similar results, showing that autophagic activities (LC3-II and Becline-1 expressions) on the spinal cord (mainly on neurons) were elevated from day 1 to day 21 after nerve injury. Furthermore, autophagy inhibition by chloroquine and the PI3K inhibitor can enhance pain behavior, while autophagy upregulation by rapamycin can suppress pain behavior [,,,]. In contrast to previous results that showed that autophagic activities on the spinal neurons were upregulated in neuropathic pain, other research showed that autophagic activities on the spinal cord decreased (mainly on microglia and astrocytes) from day 2 to day 28 after nerve injury [,,,,]. Furthermore, those studies demonstrated that autophagic upregulation could suppress pain behavior, possibly by suppressing inflammatory responses [,,,,]. However, many of those studies did not observe the direct effects of autophagy inducers and autophagy suppressors. Instead, they followed the impact of different anti-inflammatory agents (e.g., microRNA-195 inhibitor, Koumine, resveratrol) on pain behavior and autophagic activities on the spinal cord and then hypothesized the analgesic effects of autophagic upregulation on neuropathic pain [,,,,].
2.2.2. Increased Autophagic Activities in the Spinal Cord Neurons and Microglia after Nerve Injury, and Autophagy Acts as a Pain Enhancer
However, a few studies that used CCI and SNL models showed that autophagy might act as a pain enhancer [,,,]. A study showed that LC3 expression (co-stained with NeuN) elevated on the SDH of rats with spinal nerve ligation (SNL) and direct intrathecal 3-MA could decrease the pain behavior of rats with SNL []. Other studies did not observe the direct effects of autophagy inducers and autophagy suppressors; they studied the pain behavior, proinflammatory cytokine, and autophagic activity changes after administration of different proinflammatory and anti-inflammatory agents (e.g., modified citrus pectin, a TLR-3 agonist, si-ciRS-7) [,,]. They found that autophagy inducers can reverse the anti-inflammatory agent effects and hypothesized that autophagy acts as a pain inducer and has proinflammatory effects [,,].
2.3. Autophagic Activity Changes in the Brain after Nerve Injury
In addition to the dorsal root ganglia and spinal dorsal horn, different brain areas, including the anterior cingulate cortex, insula, amygdala, and nucleus accumbens, are also involved in neuropathic pain perception formation [,,]. Neural cellular circuits and molecular components change in those brain areas when neuropathic pain forms [,]. A study showed that LC3/GFAP (astrocyte marker)-positive cells increased in the brain tissue (however, the authors did not mention which brain areas) of rats with CCI []. Furthermore, lidocaine treatment attenuated mechanical allodynia and increased LC3/GFAP-positive cells in the brain of rats with CCI []. Lidocaine treatment further increased the LC3-II/LC3-I ratio but decreased proinflammatory cytokine expression (IL-1β and TNF-α) in astrocyte cultures that received a lipopolysaccharide (LPS) challenge []. 3-MA reversed the effects of lidocaine on astrocyte cultures after the LPS challenge. This study hinted that brain tissue might also exhibit increased autophagic activities under neuropathic conditions. Further upregulated autophagic activities in brain tissue may suppress proinflammatory responses and attenuate neuropathic pain [].
3. Apoptosis in Neuropathic Pain Formation
3.1. Apoptotic Activity Changes in the Injured Nerve and Dorsal Root Ganglia after Nerve Injury
The extrinsic apoptotic pathway is triggered by extracellular death ligands (such as TNF-α) and the intrinsic apoptotic pathway mediated by B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax)/Bak on the mitochondrial membrane initiates further apoptosis processes. Subsequently, cytochrome c is released from mitochondria and activates the final caspase-3 cascades of apoptosis []. It is reasonable to predict that apoptotic activities in injured nerve and dorsal root ganglia increase after nerve injury. Several studies have demonstrated that apoptotic activities in injured dorsal root ganglia increased after injury and some agents could simultaneously suppress pain behavior and apoptotic activities [,,,,]. Most studies measured the levels of antiapoptotic proteins (Bcl-2) and proapoptotic proteins (Bax, caspase-3, caspase-7, caspase-9, cytochrome-c) involved in the apoptotic pathway [] and the activities of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays, which can detect apoptotic deoxyribonucleic acid (DNA) fragmentation [] to indicate apoptotic activities in dorsal root ganglia.
Several studies that used CCI, SNC, and SNT models demonstrated consistent results that apoptotic activities (measured by cleaved caspase-3, cytochrome c, and the TUNEL assay) on dorsal root ganglia neurons and satellite cells increased from day 2 to day 28 after nerve injury [,,,,]. They also demonstrated that different agents (including ALCAR, estradiol, and recombinant human erythropoietin) could suppress pain behavior and decrease apoptotic activities in injured dorsal root ganglia at the same time [,,,,]. However, there is no direct evidence that agents showing inhibition of apoptotic activities in the injured neurons could attenuate pain behavior. Those results are summarized in Table 3.
Table 3.
Summary of the alterations of apoptotic activities in the injured nerve and dorsal root ganglia after nerve injury in different rat models.
3.2. Apoptotic Activity Changes in the Spinal Cord after Nerve Injury
In contrast to the dorsal root ganglia, different studies have shown inconsistent results for the apoptotic changes in the spinal dorsal horn in neuropathic pain. Studies that used SNC and CCI models demonstrated increased apoptotic activities in the dorsal root ganglia but not in the spinal cord after nerve injury [,]. Another study that used the SNI model demonstrated similar results; the TUNEL assay and cleaved caspase-3 activities by immunohistochemical studies were not revealed in the neurons of the dorsal spinal cord one week after nerve injury. However, they found that TUNEL activities were co-stained with Iba1 (a microglial marker) [].
On the other hand, other studies that used CCI models and mice with vincristine showed different results [,,,]. They demonstrated increased apoptotic activities (measured by Bax, apoptotic protease-activating factor-1 (apaf-1), caspase-3, caspase-9, cytochrome c, Bax, and TUNEL activities) in the spinal dorsal horn (mainly neurons) from day 3 to day 16 after nerve injury. Furthermore, those studies showed that different methods, including antioxidants, reactive oxygen species (ROS) scavengers, mitoquinone, and hyperbaric oxygen (HBO) therapy, can suppress pain behavior and decrease apoptotic activities in the spinal dorsal horn at the same time [,,,]. However, there is no direct evidence demonstrating that agents inhibiting apoptotic activities in the spinal dorsal horn could attenuate pain behavior. Table 4 summarizes the findings of those studies.
Table 4.
Summary of the alterations of apoptotic activities in the spinal dorsal horn in different models of rats and mice.
4. Relationships between Autophagy and Apoptosis in Neuropathic Pain Formation
Autophagy and apoptosis have crosstalk through various proteins, including Atg5–Atg12, Beclin-1/Bcl-2, and caspase-3 under stress conditions [,]. Additionally, a non-classical autophagy process called LC3-associated phagocytosis (LAP) can help remove apoptotic cells by macrophages and inhibit proinflammatory processes [,]. In addition, autophagy and apoptosis are both regulated by proinflammatory cytokines [,,,,,]. Autophagy has strong interactions with proinflammatory and anti-inflammatory cytokines, mainly in immune cells, including lymphocytes and macrophages [,,,]. Proinflammatory cytokines, including IFN-γ, TNF-α, IL-1β, and IL-6, have been shown to have the ability to activate autophagic processes, while anti-inflammatory cytokines, including IL-4 and IL-10, could inhibit autophagic activities [,,]. Comparatively, autophagy could suppress the activities of proinflammatory cytokines, mainly the IL-1 family, but also IL-6, TNF-α, and IL-17 [,,,]. TNF-α itself can activate the extrinsic apoptosis pathway []. Proinflammatory cytokines could directly activate apoptosis in non-neuronal cells, including β-cells and epithelial cells [,]. The contributions of proinflammatory cytokines, including IL-1 and TNF-α, to apoptosis in neurodegenerative diseases such as Alzheimer’s disease and amyotrophic lateral sclerosis are also well established [,]. In addition to autophagy and apoptosis, proinflammatory cytokines play important roles in neuropathic pain modulation, and inhibiting proinflammatory cytokines may help to attenuate neuropathic pain [,]. Therefore, proinflammatory cytokines could be the key regulators between autophagy and apoptosis in neuropathic pain formation [,,,,,,,] (Figure 1).
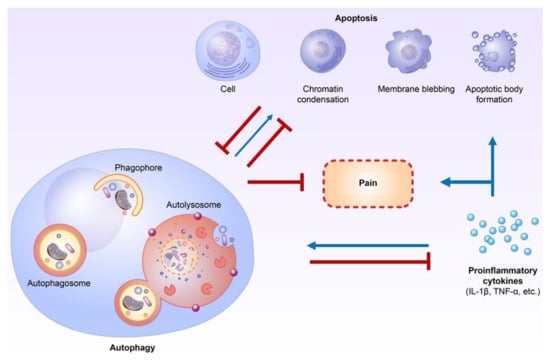
Figure 1.
Crosstalk between autophagy, apoptosis, and proinflammatory cytokines during neuropathic pain formation. Autophagy plays an important role in inflammation and proinflammatory cytokine regulation. Furthermore, proinflammatory cytokines directly influence apoptosis in non-neuronal cells and probably neurons of the central nervous system. The crosstalk or interaction of autophagy and apoptosis in neuropathic pain formation occurs mainly through the modulation of proinflammatory cytokines. (Blue lines: activation; Red lines: inhibition).
Several studies have focused on autophagic and apoptotic activity changes in damaged nerves simultaneously and discussed the relationship between autophagy and apoptosis after nerve damage. A study that used the SNC model showed that rats with SNC had higher autophagic activities (LC3-II/LC3-1 ratios and autophagosome numbers) and apoptotic activities (TUNEL assays) in the injured nerve than sham rats one week after nerve injury []. Furthermore, rapamycin treatment (for five continuous days after nerve injury) improved rats’ motor functions from one to two weeks after nerve injury, upregulated autophagic activities on the injured nerve, and promoted nerve regeneration (measured by Neurofilament 200 [NF200] and myelin basic protein [MBP] density) one week after nerve injury. Rapamycin treatment also decreased TUNEL assay activities in the injured nerve one week after nerve injury. Conversely, 3-MA had the opposite effect. This study demonstrated that autophagy upregulation could possibly inhibit apoptotic activities in the injured nerve. However, there were no further explanations of the detailed mechanisms in their study []. Another study that used the CCI model demonstrated that Koumine treatment could suppress the pain behavior of rats with CCI, downregulate proinflammatory cytokine (IL-1β and TNF-α) expression, and upregulate autophagic activities in the spinal cord. Autophagy inhibitors (chloroquine) can reverse these effects []. In this study, Koumine treatment also suppressed apoptotic activities (cleaved caspase 3, Bcl-2/Bax) in the spinal cord of rats with CCI at the same time (day 9 after nerve injury). The authors suggested that the antiapoptotic abilities of Koumine were probably mediated by suppressing proinflammatory cytokine expressions []. Our recent studies demonstrated similar findings []. We found that granulocyte-colony-stimulating factor (G-CSF) treatment can suppress pain behavior, upregulate autophagic activities (at the early phase after nerve injury), suppress proinflammatory cytokine expression, and downregulate apoptotic activities (at the late phase after nerve injury) in the injured nerve and dorsal root ganglia. We emphasized that upregulated autophagic activities developed first, followed by suppression of proinflammatory cytokine expression, and, furthermore, demonstrated the attenuation of apoptotic activities last, after G-CSF treatment []. Another recent study that used the SCI model showed curcumin (a traditional antibacterial agent) promoted motor recovery in rats with SCI, reduced apoptotic activities, suppressed proinflammatory cytokine expressions, and enhanced autophagic activities in the spinal cord of rats with SCI. Furthermore, chloroquine (an autophagy inhibitor) eliminated the protective effect of curcumin, including the suppression of proinflammatory cytokines in rats with SCI []. Previous studies, including ours, demonstrated the interactions of autophagy, apoptosis, and proinflammatory cytokine expression when neurons encounter stress and damage. Medications such as Koumine, G-CSF, and curcumin, which could suppress proinflammatory cytokines and apoptotic activities and enhance autophagic activities on the injured neurons, have the potential to treat neuropathic pain.
5. Conclusions and Perspectives on This Review
Most studies have shown consistent findings, suggesting that autophagic and apoptotic activities in injured nerves and dorsal root ganglia increase after nerve damage. Evidence has shown that the upregulated autophagic activities on the injured nerve and dorsal root ganglia after nerve damage could possibly help myelin clearance and promote nerve regeneration, which can attenuate pain behavior. However, there is less convincing evidence demonstrating that the pain behavior could be suppressed by agents that directly inhibit apoptosis. On the other hand, the autophagic and apoptotic changes in the spinal cord after peripheral nerve injury have shown inconsistent results in different studies. Complex central sensitization mechanisms in the spinal cord, different animal models, different study designs and times, and the use of various treatment agents can cause these inconsistent findings. The crosstalk or interaction of autophagy and apoptosis in spinal cord neuropathic pain formation mainly involves the modulation of proinflammatory cytokines. Alterations in autophagic/apoptotic and proinflammatory cytokine activities over time in the spinal dorsal horn play essential roles in neuropathic pain formation after nerve damage. The alterations of autophagic/apoptotic activities and their interactions with proinflammatory agents in the injured nerve, dorsal root ganglia, spinal cord, and brain are summarized in Figure 2. Further detailed studies of autophagy, apoptosis, proinflammatory cytokines, and their interactions in neuropathic pain formation in the spinal cord after nerve injury are warranted to develop a new strategy to treat neuropathic pain.
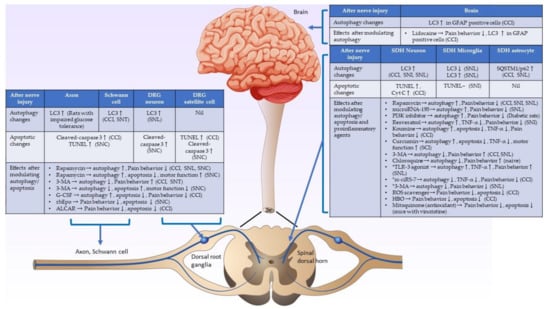
Figure 2.
Schematic diagram showing the underlying mechanisms of autophagic and apoptotic activities in the nerve injury. Summary of autophagic/apoptotic activity changes in the injured axon, Schwann cell, spinal dorsal horn, and brain after nerve injury, and the effects after modulating autophagic/apoptotic and proinflammatory cytokines activities after nerve injury. (CCI: chronic constriction injury, SCI: spinal cord injury, SNC: sciatic nerve crush injury, SNI: spared nerve injury, SNL: spinal nerve ligation, ↑: activities increase, ↓: activities decrease, -: activities do not change. *: studies showed that autophagy is a pain inducer.).
Author Contributions
Writing—original draft: M.-F.L.; writing—review and editing: L.-S.R.; resources: K.-T.L., J.-L.H., M.-Y.C. and C.-H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Chang Gung Memorial Hospital under the Chang Gung Medical Research Program Grant (CMRPG3H0281), and the Ministry of Science and Technology, Taiwan, ROC, under the grant (MOST108-2320-B-182A-021-, MOST108-2314-B-182A-051-, MOST109-2314-B-182A-085-, and MOST110-2314-B-182A-075-MY2).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We wish to thank Springer Nature Author Services and Yu-Lin Hung for her help in preparing the figures.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
3-methyladenine (3-MA); Acetyl-L-Carnitine (ALCAR); apoptotic protease-activating factor-1 (apaf-1); autophagy-related protein (Atg); B-cell lymphoma 2 (Bcl-2); Bcl2-associated X protein (Bax); chronic constriction injury (CCI); circular RNAs-7 (ciRS-7); deoxyribonucleic acid (DNA); endoplasmic reticulum (ER); gamma-aminobutyric acid-releasing (GABAergic); granulocyte-colony-stimulating factor (G-CSF); glial fibrillary acidic protein (GFAP); green fluorescent protein (GFP); hyperbaric oxygen (HBO); heat shock-cognate chaperone of 70 kDa (HSC70); International Association for the Study of Pain (IASP); ionized calcium binding adaptor molecule 1 (Iba1); interleukin-1 β (IL-1β); interleukin-6 (IL-6); in situ Oligo labeling (ISOL); Janus kinase 2 (JAK2); lysosome-associated membrane protein type-2A (LAMP2A); microtubule-associated protein 1A/1B-light chain 3 (LC3); lipopolysaccharide (LPS); myelin basic protein (MBP); modified citrus pectin (MCP); messenger ribonucleic acid (mRNA); neuronal nuclear protein (NeuN); neurofilament 200 (NF200]; number needed to treat (NNT); phospho-protein kinase B (p-AKT); protein gene product 9.5 (PGP 9.5); mammalian target of rapamycin (mTOR); phospho-mammalian target of rapamycin (p-mTOR); phosphatidylinositol 3-kinase (PI3K); phospho-phosphatidylinositol-3-kinase (p-PI3K); recombinant human erythropoietin (rhEpo); reactive oxygen species (ROS); spinal cord injury (SCI); sciatic nerve crush injury (SNC); spared nerve injury (SNI); spinal nerve ligation (SNL); serotonin–noradrenaline reuptake inhibitor (SNRI); sciatic nerve transection (SNT); Streptozotocin (STZ); Tumor Necrosis Factor-α (TNF-α); triggering receptor expressed on myeloid cells 2 (TREM2); spinal nerve crush (SNC); sequestosome 1 (SQSTM1); tricyclic antidepressants (TCAs); terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL); vacuolar protein sorting 4 (Vps 4).
References
- Finnerup, N.B.; Haroutounian, S.; Kamerman, P.; Baron, R.; Bennett, D.L.; Bouhassira, D.; Cruccu, G.; Freeman, R.; Hansson, P.; Nurmikko, T.; et al. Neuropathic pain: An updated grading system for research and clinical practice. Pain 2016, 157, 1599–1606. [Google Scholar] [CrossRef] [Green Version]
- Van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 16, 17002. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.P.; Vase, L.; Hooten, W.M. Chronic pain: An update on burden, best practices, and new advances. Lancet 2021, 397, 2082–2097. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; MacLeod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular selfdigestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Sanchez, M.J.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef]
- Jung, K.T.; Lim, K.J. Autophagy: Can It be a New Experimental Research Method of Neuropathic Pain? Korean J. Pain 2015, 28, 229–230. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, M.; Ju, Y.; Li, A.; Sun, X. Autophagy dysfunction in neuropathic pain. Neuropeptides 2019, 75, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Campana, W.M.; Myers, R.R. Exogenous erythropoietin protects against dorsal root ganglion apoptosis and pain following peripheral nerve injury. Eur. J. Neurosci. 2003, 18, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, L.D.C.; Ghelardini, C.; Calvani, M.; Nicolai, R.; Mosconi, L.; Toscano, A.; Pacini, A.; Bartolini, A. Neuroprotective effects of acetyl-L-carnitine on neuropathic pain and apoptosis: A role for the nicotinic receptor. J. Neurosci. Res. 2009, 87, 200–207. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Sekiguchi, Y.; Konno, S.-I.; Kobayashi, H.; Homma, Y.; Kikuchi, S.-I. Comparison of neuropathic pain and neuronal apoptosis following nerve root or spinal nerve compression. Eur. Spine J. 2009, 18, 1978–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, L.; Patte-Mensah, C.; Eckert, A.; Mensah-Nyagan, A.G. Sciatic nerve injury induces apoptosis of dorsal root ganglion satellite glial cells and selectively modifies neurosteroidogenesis in sensory neurons. Glia 2010, 58, 169–180. [Google Scholar] [CrossRef]
- Wiberg, R.; Novikova, L.N.; Kingham, P. Evaluation of apoptotic pathways in dorsal root ganglion neurons following peripheral nerve injury. NeuroReport 2018, 29, 779–785. [Google Scholar] [CrossRef]
- Siniscalco, D.; Fuccio, C.; Giordano, C.; Ferraraccio, F.; Palazzo, E.; Luongo, L.; Rossi, F.; Roth, K.; Maione, S.; Denovellis, V. Role of reactive oxygen species and spinal cord apoptotic genes in the development of neuropathic pain. Pharmacol. Res. 2007, 55, 158–166. [Google Scholar] [CrossRef]
- Hu, Q.; Fang, L.; Li, F.; Thomas, S.; Yang, Z. Hyperbaric oxygenation treatment alleviates CCI-induced neuropathic pain and decreases spinal apoptosis. Eur. J. Pain 2014, 19, 920–928. [Google Scholar] [CrossRef]
- Fu, H.; Li, F.; Thomas, S.; Yang, Z. Hyperbaric oxygenation alleviates chronic constriction injury (CCI)-induced neuropathic pain and inhibits GABAergic neuron apoptosis in the spinal cord. Scand. J. Pain 2017, 17, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-J.; Wang, L.; Song, X.-Y. Mitoquinone alleviates vincristine-induced neuropathic pain through inhibiting oxidative stress and apoptosis via the improvement of mitochondrial dysfunction. Biomed. Pharmacother. 2020, 125, 110003. [Google Scholar] [CrossRef] [PubMed]
- Kosacka, J.; Nowicki, M.; Blüher, M.; Baum, P.; Stockinger, M.; Toyka, K.; Klöting, I.; Stumvoll, M.; Serke, H.; Bechmann, I. Increased autophagy in peripheral nerves may protect Wistar Ottawa Karlsburg W rats against neuropathy. Exp. Neurol. 2013, 250, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Nazio, F.; Tinari, A.; Ciarlo, L.; D’Amelio, M.; Pieroni, L.; Vacca, V.; Urbani, A.; Cecconi, F.; Malorni, W.; et al. Schwann cell autophagy counteracts the onset and chronification of neuropathic pain. Pain 2014, 155, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, J.A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M.; Griffith, M.; Hantke, J.; Macias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.-S.; Jing, P.-B.; Wang, J.-A.; Zhang, R.; Jiang, B.-C.; Gao, Y.-J.; Zhang, Z.-J. Increased autophagic activity in dorsal root ganglion attenuates neuropathic pain following peripheral nerve injury. Neurosci. Lett. 2015, 599, 158–163. [Google Scholar] [CrossRef]
- Huang, H.-C.; Chen, L.; Zhang, H.-X.; Li, S.-F.; Liu, P.; Zhao, T.-Y.; Li, C.-X. Autophagy Promotes Peripheral Nerve Regeneration and Motor Recovery Following Sciatic Nerve Crush Injury in Rats. J. Mol. Neurosci. 2016, 58, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.Y.; Shin, Y.K.; Park, S.Y.; Park, J.Y.; Lee, H.J.; Yoo, Y.H.; Kim, J.K.; Park, H.T. Autophagic myelin destruction by schwann cells during wallerian degeneration and segmental demyelination. Glia 2016, 64, 730–742. [Google Scholar] [CrossRef]
- Liao, M.-F.; Yeh, S.-R.; Lu, K.-T.; Hsu, J.-L.; Chao, P.-K.; Hsu, H.-C.; Peng, C.-H.; Lee, Y.-L.; Hung, Y.-H.; Ro, L.-S. Interactions between Autophagy, Proinflammatory Cytokines, and Apoptosis in Neuropathic Pain: Granulocyte Colony Stimulating Factor as a Multipotent Therapy in Rats with Chronic Constriction Injury. Biomedicines 2021, 9, 542. [Google Scholar] [CrossRef]
- Berliocchi, L.; Russo, R.; Maiarù, M.; Levato, A.; Bagetta, G.; Corasaniti, M.T. Autophagy Impairment in a Mouse Model of Neuropathic Pain. Mol. Pain 2011, 7, 83. [Google Scholar] [CrossRef] [Green Version]
- Berliocchi, L.; Maiarù, M.; Varano, G.P.; Russo, R.; Corasaniti, M.T.; Bagetta, G.; Tassorelli, C. Spinal Autophagy is Differently Modulated in Distinct Mouse Models of Neuropathic Pain. Mol. Pain 2015, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Hu, Y.; Xie, K.; Chen, Y.; Wang, H.; Bian, Y.; Wang, Y.; Dong, A.; Yu, Y. Effect of autophagy on allodynia, hyperalgesia and astrocyte activation in a rat model of neuropathic pain. Int. J. Mol. Med. 2018, 42, 2009–2019. [Google Scholar] [CrossRef]
- Liu, K.; Yang, Y.; Zhou, F.; Xiao, Y.; Shi, L. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy and relieves hyperalgesia in diabetic rats. NeuroReport 2020, 31, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Shi, J.; Liu, K.; Liu, N.; Wang, Y.; Fu, Z.; Ding, J.; Jia, L.; Yuan, W. Increased miR-195 aggravates neuropathic pain by inhibiting autophagy following peripheral nerve injury. Glia 2013, 61, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.-L.; Yue, R.-C.; He, S.-D.; Hong, L.-M.; Xu, Y.; Yu, C.-X. Koumine Decreases Astrocyte-Mediated Neuroinflammation and Enhances Autophagy, Contributing to Neuropathic Pain From Chronic Constriction Injury in Rats. Front. Pharmacol. 2018, 9, 989. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, Y.; Huang, Y.; Liu, W.; Cai, G.; Huang, S.; Zeng, Y.; Ren, S.; Zhan, H.; Wu, W. Resveratrol mediates mechanical allodynia through modulating inflammatory response via the TREM2-autophagy axis in SNI rat model. J. Neuroinflamm. 2020, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, X.; Cheng, J.; Kong, F.; Xia, H.; Wu, J. Mammalian target of rapamycin signaling pathway is involved in synaptic plasticity of the spinal dorsal horn and neuropathic pain in rats by regulating autophagy. NeuroReport 2021, 32, 925–935. [Google Scholar] [CrossRef]
- Li, J.; Tian, M.; Hua, T.; Wang, H.; Yang, M.; Li, W.; Zhang, X.; Yuan, H. Combination of autophagy and NFE2L2/NRF2 activation as a treatment approach for neuropathic pain. Autophagy 2021, 17, 4062–4082. [Google Scholar] [CrossRef]
- Zhang, E.; Yi, M.-H.; Ko, Y.; Kim, H.-W.; Seo, J.H.; Lee, Y.H.; Lee, W.; Kim, D.W. Expression of LC3 and Beclin 1 in the spinal dorsal horn following spinal nerve ligation-induced neuropathic pain. Brain Res. 2013, 1519, 31–39. [Google Scholar] [CrossRef]
- Ma, Z.; Han, Q.; Wang, X.; Ai, Z.-S.; Zheng, Y. Galectin-3 Inhibition Is Associated with Neuropathic Pain Attenuation after Peripheral Nerve Injury. PLoS ONE 2016, 11, e0148792. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Lu, Z. Upregulated TLR3 Promotes Neuropathic Pain by Regulating Autophagy in Rat With L5 Spinal Nerve Ligation Model. Neurochem. Res. 2016, 42, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Zhang, Y.; Su, Z. ciRS-7 targeting miR-135a-5p promotes neuropathic pain in CCI rats via inflammation and autophagy. Gene 2020, 736, 144386. [Google Scholar] [CrossRef]
- Yuan, J.; Fei, Y. Lidocaine activates autophagy of astrocytes and ameliorates chronic constriction injury-induced neuropathic pain. J. Biochem. 2021, 170, 25–31. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.D.; Kimchi, A. Life in the balance – A mechanistic view of the crosstalk between autophagy and apoptosis. J. Cell Sci. 2012, 125, 5259–5268. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.-S.; Shenderov, K.; Huang, N.-N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Huang, M.; Yao, Y.-M. Autophagy and proinflammatory cytokines: Interactions and clinical implications. Cytokine Growth Factor Rev. 2018, 43, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Fang, X.; Wang, X. Autophagy and inflammation. Clin. Transl. Med. 2017, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Netea-Maier, R.T.; Plantinga, T.; Van De Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [Green Version]
- Grunnet, L.G.; Aikin, R.; Tonnesen, M.F.; Paraskevas, S.; Blaabjerg, L.; Størling, J.; Rosenberg, L.; Billestrup, N.; Maysinger, D.; Mandrup-Poulsen, T. Proinflammatory Cytokines Activate the Intrinsic Apoptotic Pathway in β-Cells. Diabetes 2009, 58, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Bruewer, M.; Luegering, A.; Kucharzik, T.; Parkos, C.A.; Madara, J.L.; Hopkins, A.; Nusrat, A. Proinflammatory Cytokines Disrupt Epithelial Barrier Function by Apoptosis-Independent Mechanisms. J. Immunol. 2003, 171, 6164–6172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fimia, G.M.; Corazzari, M.; Antonioli, M.; Piacentini, M. Ambra1 at the crossroad between autophagy and cell death. Oncogene 2012, 32, 3311–3318. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 382. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Jointed by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Chen, S.; Ammar, A.-B.; Xu, J.; Wu, Q.; Pan, K.; Zhang, J.; Hong, Y. Crosstalk Between Macroautophagy and Chaperone-Mediated Autophagy: Implications for the Treatment of Neurological Diseases. Mol. Neurobiol. 2015, 52, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Brun, S.; Schall, N.; Jeltsch-David, H.; De Sèze, J.; Muller, S. Assessing Autophagy in Sciatic Nerves of a Rat Model that Develops Inflammatory Autoimmune Peripheral Neuropathies. Cells 2017, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolf, C.J.; Mannion, R.J. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999, 353, 1959–1964. [Google Scholar] [CrossRef]
- Latremoliere, A.; Woolf, C.J. Central Sensitization: A Generator of Pain Hypersensitivity by Central Neural Plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuner, R.; Kuner, T. Cellular Circuits in the Brain and Their Modulation in Acute and Chronic Pain. Physiol. Rev. 2021, 101, 213–258. [Google Scholar] [CrossRef]
- Lindsay, N.M.; Chen, C.; Gilam, G.; Mackey, S.; Scherrer, G. Brain circuits for pain and its treatment. Sci. Transl. Med. 2021, 13, 7360. [Google Scholar] [CrossRef]
- Crowley, L.; Marfell, B.J.; Waterhouse, N.J. Detection of DNA Fragmentation in Apoptotic Cells by TUNEL. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Polgár, E.; I Hughes, D.; Arham, A.Z.; Todd, A.J. Loss of Neurons from Laminas I-III of the Spinal Dorsal Horn Is Not Required for Development of Tactile Allodynia in the Spared Nerve Injury Model of Neuropathic Pain. J. Neurosci. 2005, 25, 6658–6666. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, B.L.; Boada-Romero, E.; Cunha, L.D.; Magne, J.; Green, D.R. LC3-Associated Phagocytosis and Inflammation. J. Mol. Biol. 2017, 429, 3561–3576. [Google Scholar] [CrossRef]
- Eid, N.; Ito, Y. Oxoglaucine alleviates osteoarthritis by activation of autophagy via blockade of Ca 2+ influx and TRPV5/calmodulin/CAMK-II pathway. J. Cereb. Blood Flow Metab. 2021. [Google Scholar] [CrossRef]
- Allan, S.; Rothwell, N.J. Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2001, 2, 734–744. [Google Scholar] [CrossRef]
- Li, W.; Yao, S.; Li, H.; Meng, Z.; Sun, X. Curcumin promotes functional recovery and inhibits neuronal apoptosis after spinal cord injury through the modulation of autophagy. J. Spinal Cord Med. 2021, 44, 37–45. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).