
The Role of Autophagy and Apoptosis in Neuropathic Pain Formation

 ,
,
Abstract
:1. Introduction
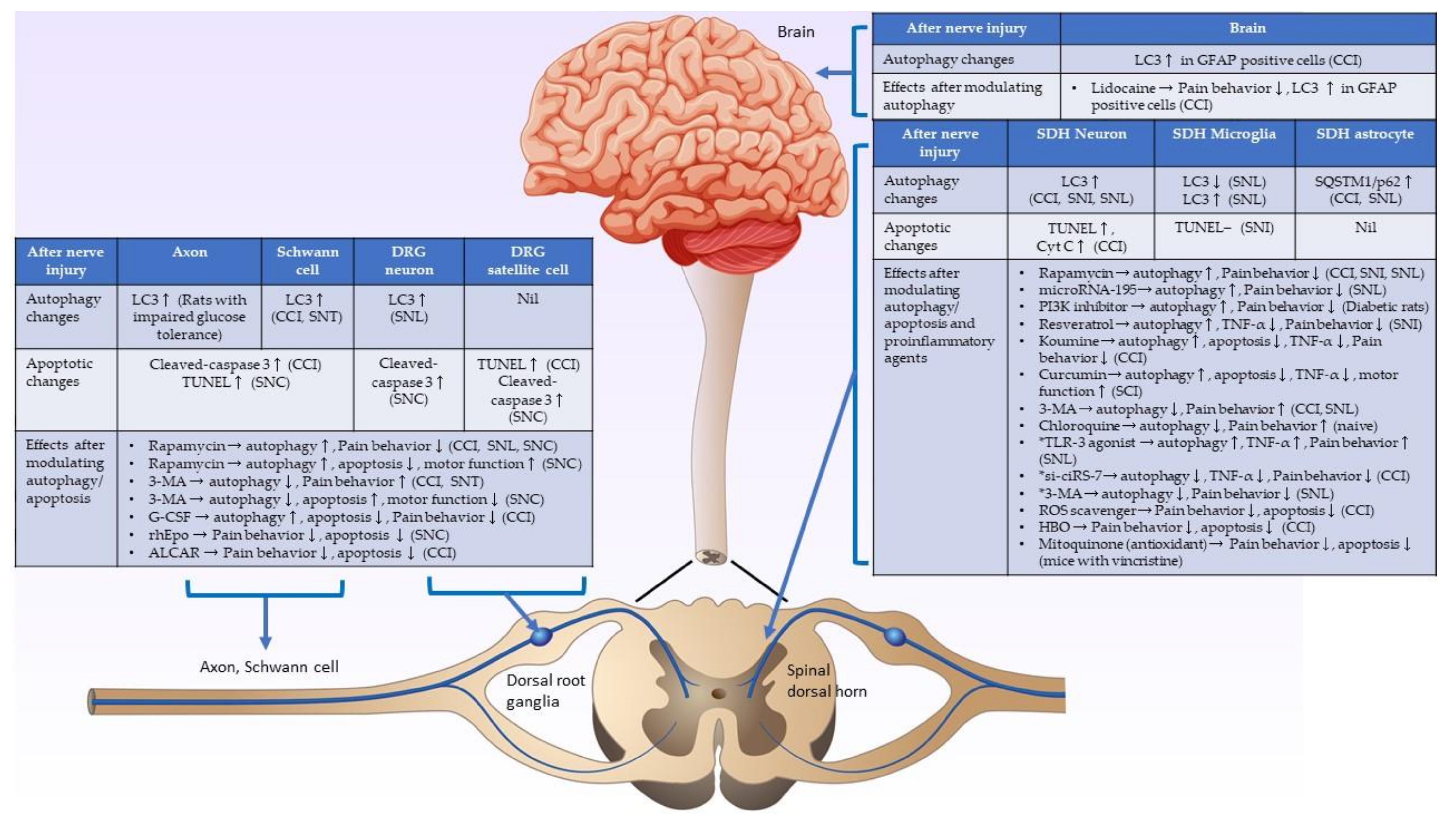
2. Autophagy in Neuropathic Pain Formation
2.1. Autophagic Activity Changes in Injured Nerves, including Schwann Cells and Dorsal Root Ganglia, after Nerve Injury
2.2. Autophagic Activity Changes in the Spinal Cord after Nerve Injury
2.2.1. Increased Autophagic Activities in the Spinal Cord Neurons, Decreased Autophagic Activities in the Spinal Cord Microglia and Astrocytes after Nerve Injury, and Autophagy Acts as a Pain Suppressor
2.2.2. Increased Autophagic Activities in the Spinal Cord Neurons and Microglia after Nerve Injury, and Autophagy Acts as a Pain Enhancer
2.3. Autophagic Activity Changes in the Brain after Nerve Injury
3. Apoptosis in Neuropathic Pain Formation
3.1. Apoptotic Activity Changes in the Injured Nerve and Dorsal Root Ganglia after Nerve Injury
3.2. Apoptotic Activity Changes in the Spinal Cord after Nerve Injury
4. Relationships between Autophagy and Apoptosis in Neuropathic Pain Formation
5. Conclusions and Perspectives on This Review
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Finnerup, N.B.; Haroutounian, S.; Kamerman, P.; Baron, R.; Bennett, D.L.; Bouhassira, D.; Cruccu, G.; Freeman, R.; Hansson, P.; Nurmikko, T.; et al. Neuropathic pain: An updated grading system for research and clinical practice. Pain 2016, 157, 1599–1606. [Google Scholar] [CrossRef] [Green Version]
- Van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 16, 17002. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.P.; Vase, L.; Hooten, W.M. Chronic pain: An update on burden, best practices, and new advances. Lancet 2021, 397, 2082–2097. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; MacLeod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular selfdigestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Sanchez, M.J.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef]
- Jung, K.T.; Lim, K.J. Autophagy: Can It be a New Experimental Research Method of Neuropathic Pain? Korean J. Pain 2015, 28, 229–230. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, M.; Ju, Y.; Li, A.; Sun, X. Autophagy dysfunction in neuropathic pain. Neuropeptides 2019, 75, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Campana, W.M.; Myers, R.R. Exogenous erythropoietin protects against dorsal root ganglion apoptosis and pain following peripheral nerve injury. Eur. J. Neurosci. 2003, 18, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, L.D.C.; Ghelardini, C.; Calvani, M.; Nicolai, R.; Mosconi, L.; Toscano, A.; Pacini, A.; Bartolini, A. Neuroprotective effects of acetyl-L-carnitine on neuropathic pain and apoptosis: A role for the nicotinic receptor. J. Neurosci. Res. 2009, 87, 200–207. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Sekiguchi, Y.; Konno, S.-I.; Kobayashi, H.; Homma, Y.; Kikuchi, S.-I. Comparison of neuropathic pain and neuronal apoptosis following nerve root or spinal nerve compression. Eur. Spine J. 2009, 18, 1978–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, L.; Patte-Mensah, C.; Eckert, A.; Mensah-Nyagan, A.G. Sciatic nerve injury induces apoptosis of dorsal root ganglion satellite glial cells and selectively modifies neurosteroidogenesis in sensory neurons. Glia 2010, 58, 169–180. [Google Scholar] [CrossRef]
- Wiberg, R.; Novikova, L.N.; Kingham, P. Evaluation of apoptotic pathways in dorsal root ganglion neurons following peripheral nerve injury. NeuroReport 2018, 29, 779–785. [Google Scholar] [CrossRef]
- Siniscalco, D.; Fuccio, C.; Giordano, C.; Ferraraccio, F.; Palazzo, E.; Luongo, L.; Rossi, F.; Roth, K.; Maione, S.; Denovellis, V. Role of reactive oxygen species and spinal cord apoptotic genes in the development of neuropathic pain. Pharmacol. Res. 2007, 55, 158–166. [Google Scholar] [CrossRef]
- Hu, Q.; Fang, L.; Li, F.; Thomas, S.; Yang, Z. Hyperbaric oxygenation treatment alleviates CCI-induced neuropathic pain and decreases spinal apoptosis. Eur. J. Pain 2014, 19, 920–928. [Google Scholar] [CrossRef]
- Fu, H.; Li, F.; Thomas, S.; Yang, Z. Hyperbaric oxygenation alleviates chronic constriction injury (CCI)-induced neuropathic pain and inhibits GABAergic neuron apoptosis in the spinal cord. Scand. J. Pain 2017, 17, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-J.; Wang, L.; Song, X.-Y. Mitoquinone alleviates vincristine-induced neuropathic pain through inhibiting oxidative stress and apoptosis via the improvement of mitochondrial dysfunction. Biomed. Pharmacother. 2020, 125, 110003. [Google Scholar] [CrossRef] [PubMed]
- Kosacka, J.; Nowicki, M.; Blüher, M.; Baum, P.; Stockinger, M.; Toyka, K.; Klöting, I.; Stumvoll, M.; Serke, H.; Bechmann, I. Increased autophagy in peripheral nerves may protect Wistar Ottawa Karlsburg W rats against neuropathy. Exp. Neurol. 2013, 250, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Nazio, F.; Tinari, A.; Ciarlo, L.; D’Amelio, M.; Pieroni, L.; Vacca, V.; Urbani, A.; Cecconi, F.; Malorni, W.; et al. Schwann cell autophagy counteracts the onset and chronification of neuropathic pain. Pain 2014, 155, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, J.A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M.; Griffith, M.; Hantke, J.; Macias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.-S.; Jing, P.-B.; Wang, J.-A.; Zhang, R.; Jiang, B.-C.; Gao, Y.-J.; Zhang, Z.-J. Increased autophagic activity in dorsal root ganglion attenuates neuropathic pain following peripheral nerve injury. Neurosci. Lett. 2015, 599, 158–163. [Google Scholar] [CrossRef]
- Huang, H.-C.; Chen, L.; Zhang, H.-X.; Li, S.-F.; Liu, P.; Zhao, T.-Y.; Li, C.-X. Autophagy Promotes Peripheral Nerve Regeneration and Motor Recovery Following Sciatic Nerve Crush Injury in Rats. J. Mol. Neurosci. 2016, 58, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.Y.; Shin, Y.K.; Park, S.Y.; Park, J.Y.; Lee, H.J.; Yoo, Y.H.; Kim, J.K.; Park, H.T. Autophagic myelin destruction by schwann cells during wallerian degeneration and segmental demyelination. Glia 2016, 64, 730–742. [Google Scholar] [CrossRef]
- Liao, M.-F.; Yeh, S.-R.; Lu, K.-T.; Hsu, J.-L.; Chao, P.-K.; Hsu, H.-C.; Peng, C.-H.; Lee, Y.-L.; Hung, Y.-H.; Ro, L.-S. Interactions between Autophagy, Proinflammatory Cytokines, and Apoptosis in Neuropathic Pain: Granulocyte Colony Stimulating Factor as a Multipotent Therapy in Rats with Chronic Constriction Injury. Biomedicines 2021, 9, 542. [Google Scholar] [CrossRef]
- Berliocchi, L.; Russo, R.; Maiarù, M.; Levato, A.; Bagetta, G.; Corasaniti, M.T. Autophagy Impairment in a Mouse Model of Neuropathic Pain. Mol. Pain 2011, 7, 83. [Google Scholar] [CrossRef] [Green Version]
- Berliocchi, L.; Maiarù, M.; Varano, G.P.; Russo, R.; Corasaniti, M.T.; Bagetta, G.; Tassorelli, C. Spinal Autophagy is Differently Modulated in Distinct Mouse Models of Neuropathic Pain. Mol. Pain 2015, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Hu, Y.; Xie, K.; Chen, Y.; Wang, H.; Bian, Y.; Wang, Y.; Dong, A.; Yu, Y. Effect of autophagy on allodynia, hyperalgesia and astrocyte activation in a rat model of neuropathic pain. Int. J. Mol. Med. 2018, 42, 2009–2019. [Google Scholar] [CrossRef]
- Liu, K.; Yang, Y.; Zhou, F.; Xiao, Y.; Shi, L. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy and relieves hyperalgesia in diabetic rats. NeuroReport 2020, 31, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Shi, J.; Liu, K.; Liu, N.; Wang, Y.; Fu, Z.; Ding, J.; Jia, L.; Yuan, W. Increased miR-195 aggravates neuropathic pain by inhibiting autophagy following peripheral nerve injury. Glia 2013, 61, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.-L.; Yue, R.-C.; He, S.-D.; Hong, L.-M.; Xu, Y.; Yu, C.-X. Koumine Decreases Astrocyte-Mediated Neuroinflammation and Enhances Autophagy, Contributing to Neuropathic Pain From Chronic Constriction Injury in Rats. Front. Pharmacol. 2018, 9, 989. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, Y.; Huang, Y.; Liu, W.; Cai, G.; Huang, S.; Zeng, Y.; Ren, S.; Zhan, H.; Wu, W. Resveratrol mediates mechanical allodynia through modulating inflammatory response via the TREM2-autophagy axis in SNI rat model. J. Neuroinflamm. 2020, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, X.; Cheng, J.; Kong, F.; Xia, H.; Wu, J. Mammalian target of rapamycin signaling pathway is involved in synaptic plasticity of the spinal dorsal horn and neuropathic pain in rats by regulating autophagy. NeuroReport 2021, 32, 925–935. [Google Scholar] [CrossRef]
- Li, J.; Tian, M.; Hua, T.; Wang, H.; Yang, M.; Li, W.; Zhang, X.; Yuan, H. Combination of autophagy and NFE2L2/NRF2 activation as a treatment approach for neuropathic pain. Autophagy 2021, 17, 4062–4082. [Google Scholar] [CrossRef]
- Zhang, E.; Yi, M.-H.; Ko, Y.; Kim, H.-W.; Seo, J.H.; Lee, Y.H.; Lee, W.; Kim, D.W. Expression of LC3 and Beclin 1 in the spinal dorsal horn following spinal nerve ligation-induced neuropathic pain. Brain Res. 2013, 1519, 31–39. [Google Scholar] [CrossRef]
- Ma, Z.; Han, Q.; Wang, X.; Ai, Z.-S.; Zheng, Y. Galectin-3 Inhibition Is Associated with Neuropathic Pain Attenuation after Peripheral Nerve Injury. PLoS ONE 2016, 11, e0148792. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Lu, Z. Upregulated TLR3 Promotes Neuropathic Pain by Regulating Autophagy in Rat With L5 Spinal Nerve Ligation Model. Neurochem. Res. 2016, 42, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Zhang, Y.; Su, Z. ciRS-7 targeting miR-135a-5p promotes neuropathic pain in CCI rats via inflammation and autophagy. Gene 2020, 736, 144386. [Google Scholar] [CrossRef]
- Yuan, J.; Fei, Y. Lidocaine activates autophagy of astrocytes and ameliorates chronic constriction injury-induced neuropathic pain. J. Biochem. 2021, 170, 25–31. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.D.; Kimchi, A. Life in the balance – A mechanistic view of the crosstalk between autophagy and apoptosis. J. Cell Sci. 2012, 125, 5259–5268. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.-S.; Shenderov, K.; Huang, N.-N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Huang, M.; Yao, Y.-M. Autophagy and proinflammatory cytokines: Interactions and clinical implications. Cytokine Growth Factor Rev. 2018, 43, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Fang, X.; Wang, X. Autophagy and inflammation. Clin. Transl. Med. 2017, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Netea-Maier, R.T.; Plantinga, T.; Van De Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [Green Version]
- Grunnet, L.G.; Aikin, R.; Tonnesen, M.F.; Paraskevas, S.; Blaabjerg, L.; Størling, J.; Rosenberg, L.; Billestrup, N.; Maysinger, D.; Mandrup-Poulsen, T. Proinflammatory Cytokines Activate the Intrinsic Apoptotic Pathway in β-Cells. Diabetes 2009, 58, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Bruewer, M.; Luegering, A.; Kucharzik, T.; Parkos, C.A.; Madara, J.L.; Hopkins, A.; Nusrat, A. Proinflammatory Cytokines Disrupt Epithelial Barrier Function by Apoptosis-Independent Mechanisms. J. Immunol. 2003, 171, 6164–6172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fimia, G.M.; Corazzari, M.; Antonioli, M.; Piacentini, M. Ambra1 at the crossroad between autophagy and cell death. Oncogene 2012, 32, 3311–3318. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 382. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Jointed by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Chen, S.; Ammar, A.-B.; Xu, J.; Wu, Q.; Pan, K.; Zhang, J.; Hong, Y. Crosstalk Between Macroautophagy and Chaperone-Mediated Autophagy: Implications for the Treatment of Neurological Diseases. Mol. Neurobiol. 2015, 52, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Brun, S.; Schall, N.; Jeltsch-David, H.; De Sèze, J.; Muller, S. Assessing Autophagy in Sciatic Nerves of a Rat Model that Develops Inflammatory Autoimmune Peripheral Neuropathies. Cells 2017, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolf, C.J.; Mannion, R.J. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999, 353, 1959–1964. [Google Scholar] [CrossRef]
- Latremoliere, A.; Woolf, C.J. Central Sensitization: A Generator of Pain Hypersensitivity by Central Neural Plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuner, R.; Kuner, T. Cellular Circuits in the Brain and Their Modulation in Acute and Chronic Pain. Physiol. Rev. 2021, 101, 213–258. [Google Scholar] [CrossRef]
- Lindsay, N.M.; Chen, C.; Gilam, G.; Mackey, S.; Scherrer, G. Brain circuits for pain and its treatment. Sci. Transl. Med. 2021, 13, 7360. [Google Scholar] [CrossRef]
- Crowley, L.; Marfell, B.J.; Waterhouse, N.J. Detection of DNA Fragmentation in Apoptotic Cells by TUNEL. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Polgár, E.; I Hughes, D.; Arham, A.Z.; Todd, A.J. Loss of Neurons from Laminas I-III of the Spinal Dorsal Horn Is Not Required for Development of Tactile Allodynia in the Spared Nerve Injury Model of Neuropathic Pain. J. Neurosci. 2005, 25, 6658–6666. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, B.L.; Boada-Romero, E.; Cunha, L.D.; Magne, J.; Green, D.R. LC3-Associated Phagocytosis and Inflammation. J. Mol. Biol. 2017, 429, 3561–3576. [Google Scholar] [CrossRef]
- Eid, N.; Ito, Y. Oxoglaucine alleviates osteoarthritis by activation of autophagy via blockade of Ca 2+ influx and TRPV5/calmodulin/CAMK-II pathway. J. Cereb. Blood Flow Metab. 2021. [Google Scholar] [CrossRef]
- Allan, S.; Rothwell, N.J. Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2001, 2, 734–744. [Google Scholar] [CrossRef]
- Li, W.; Yao, S.; Li, H.; Meng, Z.; Sun, X. Curcumin promotes functional recovery and inhibits neuronal apoptosis after spinal cord injury through the modulation of autophagy. J. Spinal Cord Med. 2021, 44, 37–45. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Reference | Animal Models | Western Blot Findings | IHC Findings | Effects of Therapeutic Agents |
|---|---|---|---|---|
| Kosacka et al., 2013 [23] | WOKW rats (hyperinsulinemia and impaired glucose tolerance). | Atg 5, Atg7, LC3-II/LC3-I ratio in the sciatic nerve of WOKW rats increased. | Autophagic proteins (LC3) were co-stained with S100 (Schwann cell), PGP9.5 (nerve fiber), and Iba1 (macrophage). | Nil. |
| Marinelli et al., 2014 [24] | Ambra1 and GFP-LC3 transgenic mice with chronic constriction injury (CCI). | Rapamycin increased the LC3-II/LC3-1 ratio in the injured sciatic nerve; 3-MA decreased LC3-II/LC3-1 ratio on the injured nerve. | Autophagic proteins (LC3) were co-stained with GFAP (Schwann cell). | Rapamycin intraplantar (i.pl.) injected 3 days after CCI suppressed pain behavior; 3-MA enhanced pain behavior. |
| Liao et al., 2021 [29] | Rats with chronic constriction injury (CCI). | LC3-II in the injured sciatic nerve and dorsal root ganglia increased from day 3 to day 7 after nerve injury. | Nil. | Granulocyte-colony-stimulating factor (G-CSF) suppressed pain behavior and upregulated autophagic activities. |
| Huang et al., 2016 [27] | Rats with a sciatic nerve crush injury (SNC). | LC3-II/LC3-1 ratio and the number of autophagosomes in the injured sciatic nerve increased 1 week after nerve injury. | Nil. | Intraperitoneal injections of rapamycin improved motor function and upregulated autophagic activities. |
| Jang et al., 2016 [28] | Atg 7 knockout mice with a sciatic nerve crush injury (SNC). | LC3-II at the distal stump of the injured sciatic nerve increased from 6 h to 48 h following nerve injury. | Nil. | Pharmacological intervention of lysosomal function or inhibition of autophagy via Schwann cell-specific knockout of the Atg 7 gene caused a significant delay in myelin clearance. |
| Guo et al., 2015 [26] | Mice with L5 spinal nerve ligation (SNL). | LC3-II in the injured dorsal root ganglia of mice with SNL increased from day 3 to day 21 after nerve injury. | Increased LC3 expressions were found on the DRG neurons. | Injection of rapamycin into DRG upregulated autophagic activities and suppressed pain behavior. |
| Gomez-Sanchez et al., 2015 [25] | Atg 7fl/fl and GFP-LC3 transgenic mice with sciatic nerve transection (SNT). | LC3-II, Beclin-1, Atg5-Atg12, Atg7, Atg 16L1 in the injured nerve increased from day 2 to day 14 after nerve injury. LC3-II had the highest expressions on day 2 after nerve injury. | Autophagic proteins (LC3) were co-stained with MPZ (myelin). | Pharmacological (3-MA) and genetic inhibition (Atg7fl/fl mice) of autophagy impaired myelin clearance. |
| Reference | Animal Models | Western Blot Findings | IHC Findings | Effects of Therapeutic Agents |
|---|---|---|---|---|
| Increased autophagic activities in the spinal cord neurons, decreased autophagic activities in the spinal cord microglia and astrocytes after nerve injury, and autophagy acts as a pain suppressor. | ||||
| Berliocchi et al., 2011 [30], 2015 [31] | Mice with chronic constriction injury (CCI), spared nerve injury (SNI), and spinal nerve ligation (SNL). | LC3-II and p62 increased in the mice with spinal nerve ligation (SNL) on day 7 after nerve injury. LC3-II increased in the mice with spared nerve injury (SNI) on day 14 after nerve injury. Beclin-1 increased in the mice with chronic constriction injury (CCI) on day 14 after nerve injury. | Autophagic proteins (p62) were co-stained with NeuN in the spinal cord. | Intrathecal chloroquine (can inhibit autophagic flux and block the late stage of autophagy) enhanced pain behavior. |
| Chen et al., 2018 [32] | Rats with chronic constriction injury (CCI). | LC3-II and Beclin-1 increased from day 1 to day 7 (peak at day 3) after nerve injury. p62 decreased from day 1 to day 7 after nerve injury. | Nil. | Intraperitoneal rapamycin before chronic constriction injury suppressed pain behavior, upregulated LC3-II/Beclin-1 expressions, and suppressed astrocyte activation in the spinal cord. 3-MA showed the opposite effect. |
| Liu et al., 2020 [33] | Rats received streptozotocin (STZ) injection (diabetic rats). | p-PI3K, p-AKT, and p-mTOR decreased, but LC3-II and Beclin1 increased 3 weeks after STZ injection. | Nil. | Intravenous PI3K inhibitor (LY294002) suppressed pain behavior and increased LC3-II expressions in the spinal cord of diabetic rats. |
| Jin et al., 2018 [35] | Rats with chronic constriction injury (CCI). | LC3-II/I ratio and p62 increased on day 9 after nerve injury. (The authors suggested that increased LC3-II indicated that autophagic flux was blocked in the late stage of autophagy and concluded that autophagic activities in the spinal cord decreased after nerve injury.) | Autophagic proteins (LC3) were co-stained with GFAP in the spinal cord. | Subcutaneously (s.c.) Koumine treatment suppressed pain behavior, upregulated autophagic activities, and downregulated proinflammatory cytokine expressions in the spinal cord of rats with CCI. Intrathecal chloroquine (autophagy inhibitor) blocked the effects of Koumine. |
| Wang et al., 2020 [36] | Rats with spared nerve injury (SNI). | LC3-II decreased but triggering receptor expressed on myeloid cells 2 (TREM2), p62, and proinflammatory cytokine increased on day 7 after nerve injury. | Nil. | Intrathecal resveratrol treatment (suppressed TREM2 expressions) suppressed pain behavior, suppressed proinflammatory cytokine, and increased LC3-II expressions in the spinal cord. 3-MA (autophagy suppressor) reduced the analgesic effects of resveratrol. |
| Hu et al., 2021 [37] | Rats with spared nerve injury (SNI). | LC3-II decreased, but Beclin-1 and p62 increased from day 7 to day 14 after nerve injury. | Nil. | Intravenous rapamycin treatment decreased pain behavior, increased LC3-II expressions and autophagosome number in the spinal cord, and suppressed C- and A-fiber-evoked field potentials from day 7 to day 14 after nerve injury. |
| Shi et al., 2013 [34] | Rats with spinal nerve ligation (SNL). | LC3-II/LC3-1 ratio of the primary microglia culture isolated from rats with SNL decreased from day 2 to day 14 after nerve injury. P62 of the primary microglia culture isolated from rats with SNL increased from day 5 to day 14 after nerve injury. | Autophagic proteins (LC3-II) were co-stained with Iba1 in primary microglia culture. | Intrathecal administration of microRNA-195 inhibitor reduced the pain behavior of rats with SNL and increased the LC3-II/LC3-I ratio in the spinal dorsal horn. |
| Li et al., 2021 [38] | Mice with spinal nerve ligation (SNL). | LC3-II/LC3-I ratio and Atg5 expressions decreased, but SQSTM1/p62 (autophagy receptor) increased from day 7 to day 28 after nerve injury. | SQSTM1 was mainly co-stained with GFAP rather than NeuN. Electron microscopic studies showed the number of autophagosomes decreased, mainly in the astrocyte of mice with SNL. | Intrathecal rapamycin treatment on day 7 to day 9 after nerve injury suppressed pain behavior on day 10 and day 14 after nerve injury and increased LC3-II/LC3-I ratio in the spinal cord. 3-MA showed the opposite effects. |
| Increased autophagic activities in the spinal cord neurons and microglia after nerve injury, and autophagy acts as a pain enhancer. | ||||
| Cai et al., 2020 [42] | Rats with chronic constriction injury (CCI). | LC3-II, circular RNAs-7 (ciRS-7), and proinflammatory cytokines (IL-6, IL-12, TNF-α) levels increased from day 7 to day 20 after nerve injury. | Nil. | Intrathecal si-ciRS-7 treatment suppressed pain behaviors and decreased autophagic proteins (LC3-II) and proinflammatory cytokines (IL-12, TNF-α) expressions. |
| Zhang et al., 2013 [39] | Rats with spinal nerve ligation (SNL). | Nil. | Autophagic proteins (LC3 and Beclin-1) were co-stained with NeuN and Calretinin (a marker of GABAergic interneurons) in the spinal dorsal horn and increased on day 14 after nerve injury. | Intrathecal 3-MA 3 days after SNL decreased pain behavior from day 7 to day 10 after nerve injury. |
| Ma et al., 2016 [40] | Rats with spinal nerve ligation (SNL). | LC3-II of the primary microglia culture isolated from the rats that received SNL increased on day 10 after nerve injury. | Nil. | Intrathecal administration of modified citrus pectin (a kind of anti-inflammatory protein) suppressed pain behavior of rats with SNL, and rapamycin reversed those effects. |
| Weijia Chen et al., 2017 [41] | Rats with spinal nerve ligation (SNL). | LC3-II of the primary microglia culture isolated from the rats with SNL increased but p62 levels decreased on day 10 after nerve injury. | Autophagic proteins (LC3) were co-stained with Iba-1 (microglia marker) in the spinal cord. | Intrathecal TLR-3 agonist enhanced pain behavior of rats with SNL and increased autophagy and proinflammatory cytokines (IL-1β and TNF-α) expressions in the dorsal horn of rats with SNL. Intrathecal 3-MA administration reversed previous findings. |
| Reference | Animal Models | Apoptotic Activities | IHC Findings | Effects of Therapeutic Agents |
|---|---|---|---|---|
| Mannelli et al., 2009 [15] | Rats with chronic constriction injury (CCI). | Cleaved caspase 3, cytochrome c, DNA fragmentation levels in the injured nerve increased on day 15 after nerve injury. | Nil. | Intraperitoneal ALCAR twice per day for 15 days suppressed pan behavior and decreased cleaved-caspase 3, cytochrome c, and DNA fragmentation levels in the injured nerve. |
| Schaeffer et al., 2010 [17] | Rats with chronic constriction injury (CCI). | TUNEL assay activities in the dorsal root ganglia increased on day 30 (not day 5 and day 15) after nerve injury. | TUNEL activities were co-stained with anti-glutamine synthetase (satellite cells marker). Aromatase (estradiol-synthesizing enzyme) was co-stained with NeuN but not anti-glutamine synthetase (satellite cells marker) | Letrozole, which blocked aromatase activities, increased apoptotic activities in the dorsal root ganglia of rats with CCI. |
| Campana et al., 2003 [14] | Rats with spinal nerve crush (SNC) injury. | TUNEL assay activities in the dorsal root ganglia increased on day 2 after nerve injury. | Nil | Subcutaneous administration of recombinant human erythropoietin (rhEpo) one day before nerve injury suppressed pain behavior and apoptotic activities in the dorsal root ganglia from day 2 to day 14 after nerve injury. |
| Sekiguchi et al., 2009 [16] | Rats with spinal nerve crush (SNC) injury. | In situ Oligo labeling (ISOL) in the dorsal root ganglia increased from day 2 to day 28 after nerve injury. | Cleaved-caspase 3 on dorsal root ganglia were co-stained with NeuN and GFAP (satellite cells marker). | Nil. |
| Wiberg et al., 2018 [18] | Rats with sciatic nerve transection (SNT). | Caspase-3, caspase-8, caspase-12, caspase-7, and calpain expressions in the dorsal root anglia increased on day 7, day 14, and day 28 after nerve injury. | Nil. | Nil. |
| Reference | Animal Models | Apoptotic Activities | IHC Findings | Effects of Therapeutic Agents (Inhibit Apoptosis) |
|---|---|---|---|---|
| Schaeffer et al., 2010 [17] | Rats with chronic constriction injury (CCI). | TUNEL assay activities in the spinal cord did not increase on days 5, 15, and 30 after nerve injury. | Nil. | Nil. |
| Campana et al., 2003 [14] | Rats with spinal nerve crush (SNC) injury. | TUNEL assay activities in the spinal cord did not increase on day 2 after nerve injury. | Nil. | Subcutaneous administration of recombinant human erythropoietin (rhEpo) 1 day before nerve injury reduced pain behavior from day 2 to day 14 after nerve injury. |
| Siniscalco et al., 2007 [19] | Mice with chronic constriction injury (CCI). | Bax, apoptotic protease-activating factor-1 (apaf-1), caspase-9 mRNA expressions, and TUNEL and caspase-3 activities increased on day 3 after nerve injury. | TUNEL activities were co-stained with NeuN on day 3 after nerve injury. | Intraperitoneal phenyl-N-tert-butylnitrone (ROS scavenger) suppressed pain behavior from day 1 to day 3 after nerve injury and decreased apoptotic activities in the spinal cord on day 3 after nerve injury. |
| Hu et al., 2015 [20] | Rats with chronic constriction injury (CCI). | TNF-α and caspase-3 mRNA expressions and TUINEL activities increased on day 3 and day 7 after nerve injury, respectively. | Nil. | HBO suppressed pain behavior and apoptotic activities in the spinal cord of rats with CCI on day 3 and day 7 after nerve injury. |
| Fu et al., 2017 [21] | Rats with chronic constriction injury (CCI). | The number of cleaved caspase-3 and cytochrome C positive neurons increased on day 8 and day 14 after nerve injury. | Cytochrome C was co-stained with NeuN (neuron marker). | Daily HBO therapy suppressed pain behavior and apoptotic activities in the spinal dorsal horn of rats with CCI. |
| Polga’r et al., 2005 [66] | Mice with spared nerve injury (SNI). | TUNEL assay and cleaved caspase-3 activities did not demonstrate in the apoptotic neurons in the dorsal spinal cord 1 week after nerve injury. | TUNEL activities were co-stained with Iba1 (microglia marker). | Nil. |
| Chen et al., 2020 [22] | Mice with neuropathic pain induced by vincristine. | Cleaved caspase 3, Bax, and cytochrome c (Cyt-c) increased on day 9 after five consecutive vincristine administrations. | Nil. | Mitoquinone (antioxidant) treatment after vincristine injury suppressed pain behavior of mice, ROS production, and apoptotic activities in the spinal cord of mice. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, M.-F.; Lu, K.-T.; Hsu, J.-L.; Lee, C.-H.; Cheng, M.-Y.; Ro, L.-S. The Role of Autophagy and Apoptosis in Neuropathic Pain Formation. Int. J. Mol. Sci. 2022, 23, 2685. https://doi.org/10.3390/ijms23052685
Liao M-F, Lu K-T, Hsu J-L, Lee C-H, Cheng M-Y, Ro L-S. The Role of Autophagy and Apoptosis in Neuropathic Pain Formation. International Journal of Molecular Sciences. 2022; 23(5):2685. https://doi.org/10.3390/ijms23052685
Chicago/Turabian StyleLiao, Ming-Feng, Kwok-Tung Lu, Jung-Lung Hsu, Chih-Hong Lee, Mei-Yun Cheng, and Long-Sun Ro. 2022. "The Role of Autophagy and Apoptosis in Neuropathic Pain Formation" International Journal of Molecular Sciences 23, no. 5: 2685. https://doi.org/10.3390/ijms23052685
APA StyleLiao, M.-F., Lu, K.-T., Hsu, J.-L., Lee, C.-H., Cheng, M.-Y., & Ro, L.-S. (2022). The Role of Autophagy and Apoptosis in Neuropathic Pain Formation. International Journal of Molecular Sciences, 23(5), 2685. https://doi.org/10.3390/ijms23052685