Abstract
The selenoprotein family includes 25 members, many of which are antioxidant or redox regulating enzymes. A unique member of this family is Selenoprotein I (SELENOI), which does not catalyze redox reactions, but instead is an ethanolamine phosphotransferase (Ept). In fact, the characteristic selenocysteine residue that defines selenoproteins lies far outside of the catalytic domain of SELENOI. Furthermore, data using recombinant SELENOI lacking the selenocysteine residue have suggested that the selenocysteine amino acid is not directly involved in the Ept reaction. SELENOI is involved in two different pathways for the synthesis of phosphatidylethanolamine (PE) and plasmenyl PE, which are constituents of cellular membranes. Ethanolamine phospholipid synthesis has emerged as an important process for metabolic reprogramming that occurs in pluripotent stem cells and proliferating tumor cells, and this review discusses roles for upregulation of SELENOI during T cell activation, proliferation, and differentiation. SELENOI deficiency lowers but does not completely diminish de novo synthesis of PE and plasmenyl PE during T cell activation. Interestingly, metabolic reprogramming in activated SELENOI deficient T cells is impaired and this reduces proliferative capacity while favoring tolerogenic to pathogenic phenotypes that arise from differentiation. The implications of these findings are discussed related to vaccine responses, autoimmunity, and cell-based therapeutic approaches.
1. Introduction
Selenium (Se) is an essential dietary trace mineral that is important for various aspects of human health, including optimal immunity [1]. The biological effects of Se are mainly exerted through its incorporation into selenoproteins as the 21st amino acid, selenocysteine (Sec) [2]. The 25 members of the selenoprotein family exhibit a wide variety of functions including the control of reactive oxygen species and cellular redox tone, regulating thyroid hormone metabolism, facilitating sperm maturation and protection, and promoting optimal immunity [3]. Under conditions of low Se status, the translation of selenoproteins stalls at the Sec-encoding UGA codon and both the mRNA and truncated protein become degraded through nonsense-mediated decay and destruction via C-end degrons, respectively [4,5]. In a Se deficient individual, the brain, muscle, and testes receive ‘priority’ for bioavailable Se at a cost to other tissues, such as those comprising the immune system [6]. Insufficient Se intake or other factors (e.g., defects in selenoprotein gene expression or some chronic infections that deplete Se) can impair adaptive immunity, especially T cell responses that are critical for producing effective vaccine responses and fighting infections [7,8]. Interestingly, not all types of immune responses are equivalently affected by Se deficiency or by Se supplementation [9,10]. Although the reasons for this are unclear due to an inadequate understanding of the mechanisms by which Se affects the immune system, data have recently emerged regarding roles for individual selenoproteins in immunity. This is particularly the case for T cell immunity, and a better understanding of how selenoproteins regulate T cell immunity may provide new targets for therapeutic intervention for immune based disease.
2. T Cell Immunity and Selenoprotein I Expression
CD4+ T cells constitute the topmost regulatory layer of the adaptive immune system, providing cytokine ‘help’ to CD8+ T cells (effector cells of cell mediated immunity) and B cells (effector cells of humoral immunity), thus coordinating acquired immune responses [11]. CD4+ T cells are activated through the T cell receptor (TCR), proliferate and differentiate into helper subsets, which forms the foundation for their ability to shape immune response and mediate host protection [12]. CD8+ T cells are also activated through their TCR to produce effector and memory cells required for optimal immunity [13]. Levels of Se and selenoproteins regulate T cell functions that drive both cell-mediated and humoral immunity [14,15,16,17]. However, it remains unclear which selenoproteins are involved in the different steps of T cell activation, proliferation, and differentiation. To gain insight into the roles that different selenoproteins play in T cell activation, we used real-time PCR to evaluate the selenoprotein transcriptome in naïve vs. activated T cells purified from C57BL/6 mice [18]. Results showed that a subset of selenoprotein mRNAs increased during T cell activation. These included the thioredoxin reductases (TXNRD1-3) enzymatically regenerate reduced thioredoxin and promote reducing capacity within cells, and we previously published that mRNA levels for these enzymes were increased as a mechanism for regulating redox tone in during T cell activation [7]. An interesting result found during T cell activation was an increase in the mRNA encoding selenoprotein I (SELENOI), which was upregulated nearly 3-fold [18]. Protein levels and enzyme activity for SELENOI were similarly increased in activated T cells. This raised the question: Why do SELENOI levels increase during T cell activation and what is its role in regulating T cell immunity?
To understand how SELENOI is involved in T cell activation, a bit more background on this selenoprotein is necessary. After the initial identification of the SELENOI gene in 2003 [19], the sequence was subsequently characterized through a homology search that found the cytidine diphosphate (CDP) alcohol phosphatidyltransferase signature, a common motif conserved in phospholipid synthases [20]. This study went on to demonstrate that SELENOI exhibits ethanolamine phosphotransferase (Ept) activity in vitro, providing the first data showing that SELENOI (which this group called EPT1) is an enzyme that transfers phosphoethanolamine from CDP-ethanolamine to 1,2-DAG acceptors to produce phosphatidylethanolamine (PE). SELENOI is only found in vertebrates, and in humans is expressed in a wide variety of cells [19,20]. A more recent study showed that fibroblasts from a patient with a mutation (exon skipping) leading to nonfunctional SELENOI had impaired Ept activity and reduced levels of several PE species, especially plasmenyl PE [21]. This study focused on the neurodevelopment defects exhibited by the patient, and another earlier clinical report substantiated the effects of a Arg112Pro mutation in the gene encoding SELENOI on the central nervous system (CNS) [22]. Understandably, these patients with severe CNS impairments were not evaluated in terms of immune cell function. Furthermore, there has been a paucity of data published regarding dietary Se regulating less overt changes in SELENOI. This is significant because the central nervous system retains Se under conditions Se deficiency, at a cost to the immune system [6]. Thus, individuals deficient in Se may maintain higher SELENOI levels in the brain compared to the immune system, and Se deficient T cells expressing lower levels of SELENOI may impact immunity. SELENOI protein or enzyme activity have not been measured in individuals with different Se status, but these data would be useful in determining how Se intake is related to function for this selenoprotein.
3. Structure of SELENOI Related to the Synthesis of PE and Plasmenyl PE
Plasma membrane phospholipids are distributed asymmetrically between the outer and inner leaflets of viable mammalian cells. The outer leaflet of the plasma membrane is composed primarily of sphingomyelin (SM) and phosphatidylcholine (PC), whereas the inner leaflet contains phosphatidylserine (PS) and phosphatidylethanolamine (PE) and phosphatidylinositol (PI), with cholesterol distributed equally [23,24]. In the plasma membrane and organelle membranes of mammalian cells, PE comprises 15–25% of total phospholipids [25]. PE is comprised of 1,2-diacylglycerol (DAG) and ethanolamine phosphate (Figure 1A). A structurally related ethanolamine phospholipid, plasmenyl PE, is an ether-linked lipid comprised of 1-alkyl-2-acylglycerol (AAG) and ethanolamine phosphate (Figure 1B), and is found at a lower abundance in cellular membranes compared to PE. SELENOI is involved in de novo synthesis of both PE and plasmenyl PE, carrying out the transfer of phosphoethanolamine from cytidine diphosphate (CDP)-ethanolamine to DAG to generate PE or to AAG to generate plasmenyl PE. In particular, SELENOI catalyzes the final step of the Kennedy pathway in the interface of the cytosol and endoplasmic reticulum (ER) that is responsible for synthesizing PE [26]. Although an alternative pathway operates in the mitochondria for synthesizing PE from PS, the importance of SELENOI in the synthesis of PE for cellular membranes through the Kennedy pathway has been clearly demonstrated in primary human fibroblasts and HeLa cells [21]. Plasmenyl PE is synthesized through a separate pathway beginning in peroxisomes (Reactions 1–3) and finishing in the ER membrane (Reactions 4–7). SELENOI catalyzes the sixth reaction to generate plasmenyl PE [27]. The vinyl ether bond at the sn-1 position of glycerol backbone contributes to a difference in biophysical properties compared to PE, and plasmenyl PE species are enriched in lipid rafts [28,29,30]. Thus, synthesis of PE and plasmenyl PE species are largely dependent on SELENOI and serve structural functions in cellular membranes, but possible roles in signaling pathways and in metabolic regulation are beginning to emerge [31,32]. Related to SELENOI (aka ethanolamine phosphotransferase 1 or EPT1) is the enzyme choline/ethanolamine phosphotransferase (CEPT1). CEPT1 is not a selenoprotein and can use both CDP-ethanolamine and CDP-choline as substrates, while SELENOI only uses CDP-ethanolamine [6]. This seems to suggest that SELENOI uses the selenocysteine residue to select cytidine diphosphate (CDP)-ethanolamine instead of CDP-choline as a substrate, although there are no data to include or exclude this possibility. Experimental evidence strongly suggests that CEPT1 can partially compensate for a lack of SELENOI expression [33], and PE may be generated by converting other phospholipids like phosphatidylserine to PE [34]. This other pathway occurs within the mitochondrial inner membrane, involving the conversion of the serine base to ethanolamine by PS decarboxylase (PSD) [26]. The PS decarboxylation pathway generates different PE species from CDP-ethanolamine pathway (i.e., the Kennedy pathway) [35]. This implies that phospholipid synthesis may occur by different routes and highlights the priority that cells place on maintaining balanced membrane composition.
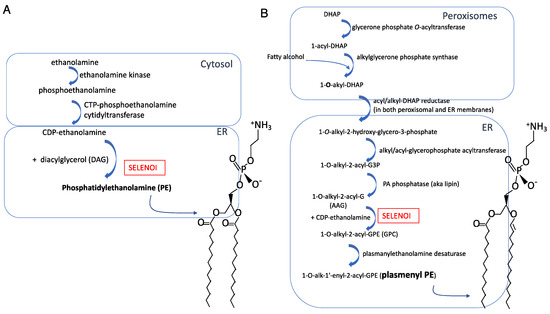
Figure 1.
Synthesis pathways and molecular structures for (A) PE and (B) plasmenyl PE.
As discussed above, SELENOI belongs to the family of selenoproteins and also to the family of phospholipid transferases. The latter enzymes are identified by a conserved CDP-alcohol phosphotransferase motif D(X)2DG(X)2AR(X)7-12G(X)3D(X)3D (X represents any amino acid and subscript numbers represent number of residues) commonly shared by all enzymes catalyzing the biosynthesis of phospholipids [20,36]. SELENOI is a predicted transmembrane protein that is fully embedded into the lipid bilayer, with its transmembrane helices traversing the membrane multiple times, as shown in Figure 2 [37]. Since most phospholipid transferases reside primarily in the (ER) membranes [38], SELENOI was initially thought to share the same localization. Recent studies showed that SELENOI is mainly localized in the Golgi apparatus [33], although these data involved overexpressed, tagged SELENOI in a cell line. Similar to other phospholipid transferases, the catalytic domain of the enzyme is predicted to be located within a pocket of the lipid bilayer that is accessible from the cytoplasm [37]. In redox selenoenzymes, selenocysteine residue is located within the catalytic domain in a prototypical C-X-X-U motif (X represents any amino acid), and replacement of selenocysteine with cysteine has been shown to reduce the catalytic activity of some of these enzymes [39,40]. In contrast, SELENOI does not contain a C-X-X-U motif and SELENOI’s selenocysteine is located near the C-terminus at amino acid position 387 that is predicted to reside in the cytosol, apart from active site in within the membrane pocket [37]. This raises the question if selenocysteine is necessary for the catalytic function of this enzyme? Overexpression of SELENOI cDNA in a bacterial expression system led to a truncated form of the protein due to the lack of recognition of eukaryotic SECIS elements, and this Sec-deficient protein retained in vitro Ept activity [20]. This suggests the selenocysteine residue is not directly involved in the catalytic function of SELENOI, which is further supported by the observation described above that CEPT1 (that lacks a selenocysteine residue) may exert Ept activity when compensating for a lack of SELENOI.
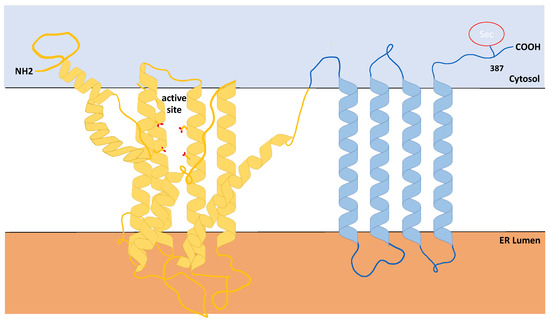
Figure 2.
Predicted structure of Selenoprotein I. Results from Phyre Alarm and other online prediction programs show that, similar to other phosphotransferases, SELENOI is predominantly comprised of hydrophobic amino acids (~90%) with the catalytic domain residing within a membrane pocket. Note the C-terminal Sec residue is located outside of the catalytic domain.
4. Cellular Membranes and SELENOI KO
Published data show that SELENOI KO does not affect TCR induced signaling [18], which is somewhat surprising given that SELENOI deficiency decreases phospholipids involved in cellular membrane structure. In particular, the ratio of PE levels relative to PC levels plays a key role in membrane fluidity or rigidity [28,41]. Sufficient membrane rigidity is crucial in TCR signaling strength required to drive proliferation and differentiation [42]. Thus, one may expect a disruption of TCR signaling or, at a minimum, weaker TCR signaling with the lowered PE and plasmenyl PE levels in T cells that accompanies SELENOI KO. The lack of an effect of SELENOI KO on TCR signaling may suggest a compensatory process serves to maintain cellular membrane integrity in the absence of SELENOI. There is also a question regarding lipid rafts, which are fluctuating nanoscale assemblies of sphingolipid, cholesterol, and proteins that can be stabilized to coalesce, forming platforms that function in membrane signaling and trafficking [43]. Lipid rafts are enriched for plasmenyl PE [44], so it would reason that SELENOI deficiency in T cells that reduces plasmenyl PE should affect lipid raft organization. However, membrane raft organization does not appear differ between SELENOI KO versus WT control T cells as determined by fluorescent staining (Figure 3). This is consistent with the lack of effect of SELENOI KO on TCR signaling described above. Overall, there is insufficient evidence to date suggesting that membrane integrity or structure is affected by SELENOI KO in T cells.
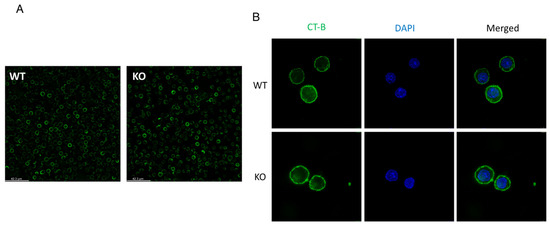
Figure 3.
Lipid raft staining is similar between SELENOI KO T cells and WT controls. Mouse T cells isolated from mice and activated through the TCR for 24 h were stained for lipid rafts using standard fluorescent cholera toxin subunit B protocols. (A) Fluorescent microscopy images from live cells at 20× and (B) confocal microscopy images of paraformaldehyde fixed cells at 63× reveal similar patterns of staining between KO and WT T cells.
5. Metabolic Reprogramming During T Cell Activation Requires SELENOI for Proliferation
In the non-activated or quiescent state, T cells mainly exhibit catabolic activity involving the break-down of nutrients to fuel cell survival. Upon T cell receptor (TCR) triggered activation, T cells transition to a state of anabolism in which nutrients are used to construct the molecular building blocks that are incorporated into cellular biomass to support proliferation [45,46]. In addition to small molecule precursors, energy is also needed for proliferation. This requirement is met during TCR-induced activation through increased glucose uptake via upregulated glucose transporters, which is accompanied by induced aerobic metabolism [47]. These changes are akin to shifts in cancer cell metabolism known as the Warburg effect. TCR-induced metabolic reprogramming is critical for generating sufficient energy and precursor molecules for subsequent rounds of mitosis, which promotes the T cell expansion phase that is so crucial for immune clearance of pathogens. The balance of catabolic and anabolic pathways in a cell determines how much adenosine triphosphate (ATP) is generated versus consumed, the availability of biosynthetic precursors, and the redox status of the cell [48]. Redox status may be controlled by antioxidant and redox regulating selenoproteins, with free thiols playing a key role [16]. However, SELENOI is an unconventional selenoprotein that is not involved in redox reactions. Since SELENOI is directly involved in two different anabolic pathways, one for PE synthesis and the other for plasmenyl PE synthesis, it follows that this selenoprotein is likely an integral part of the metabolic reprogramming during T cell activation.
To understand the role of SELENOI in T cell proliferation, our research group conducted loss-of-function studies in T cells isolated from different transgenic mouse models. In particular, an inducible knockdown (KD) mouse model along with a T cell specific knockout (KO) mouse model were compared to wild-type (WT) controls for TCR-induced proliferative capacity. SELENOI KD led to a ~22% decrease and KO to a ~56% decrease in proliferation compared to WT controls [18]. In vivo T cell expansion to vaccination was also decreased in T cell specific SELENOI KO mice compared to WT controls. Surprisingly, the TCR signaling was not affected by SELENOI deficiency and levels of PE and plasmenyl PE were only partially decreased. The latter observation may be explained by the fact that enzyme activity of a related phospholipid transferase may compensate for PE and plasmenyl PE when SELENOI is absent [33]. The most impressive result of SELENOI deficiency in activated T cells was a progressive accumulation of ATP, which was detected by the metabolic sensor AMPK and led to lower activation of this kinase. In fact, treating WT T cells with the AMPK inhibitor, dorsomorphin, reduced proliferation by similar levels as SELENOI KO. These data collectively suggest that SELENOI serves a critical function during T cell activation to maintain ethanolamine phospholipid synthesis and thereby keep a balanced metabolism within the cells as they undergo metabolic reprogramming. Removing SELENOI causes a ripple effect-these synthesis pathways are disrupted and this causes ATP to accumulate, which is sensed within the cells by AMPK and eventually proliferation is reduced.
6. SELENOI and T Helper Cell Differentiation
As proliferation takes place, the fates of the daughter T cells differ through asymmetric cell division and differentiation [49]. In particular, naïve CD4+ T cells differentiate into one of several T helper cell lineages depending on signals from antigen presenting cells and cytokines present in the surrounding environment. In addition to these external stimuli, internal factors, such as cellular metabolism, can influence differentiation [50]. CD4+ T cell subsets express unique sets of cell surface markers and transcription factors, while secreting a defined array of cytokines that determines their functional properties [51]. It was recently demonstrated that CD4+ T cell differentiation was dependent on ethanolamine kinase 1, CTP-phosphoethanolamine cytidyltransferase, and SELENOI enzymes that comprise the CDP-ethanolamine pathway for de novo synthesis of PE and plasmenyl PE [52]. In particular, this pathway was important for T follicular helper (TFH) cell differentiation by promoting the surface expression and functional effects of the chemokine receptor, CXCR5. Unlike our SELENOI KO T cells that were decreased, but not lacking, PE and plasmenyl PE as described above, these studies largely focused on T cells lacking these phospholipids. A state of complete loss of PE and plasmenyl PE in T cells is unlikely given the essential roles these phospholipids play in development [53,54], but the importance of ethanolamine phospholipid synthesis in TFH cell formation does highlight how certain metabolic pathways promote T cell differentiation outcomes over others.
T helper type 17 (Th17) cells represent another subtype of T cells differentiating from naïve Th cells that promote inflammation. Th17 cells are required for immune responses to specific extracellular bacteria and fungi [55], but dysregulated Th17 responses may also contribute to pathogenesis in autoimmune diseases, such as rheumatoid arthritis and multiple sclerosis (MS) [56,57]. In contrast, CD4+ regulatory T (Treg) cells contribute to the suppression of immune responses and immune homeostasis, countering Th17 cells to protect against autoimmune disorders. Treg differentiation relies on the upregulation of the transcription factor FoxP3, and Treg cells function to secrete immunosuppressive cytokines TGF-β and IL-10 [58]. A major factor influencing Th17/Treg fates during T cell activation is the type of metabolic reprogramming that occurs after TCR engagement, which serves to meet increased demand for energy and metabolites [59].
Th17 cells take on a distinct metabolic signature that promotes differentiation into this subtype, heavily relying on finely tuned glycolysis, glutamine metabolism, and fatty acid synthesis for their differentiation and pro-inflammatory function [60,61,62]. Sustained mitochondrial oxidative phosphorylation has also been shown to regulate the fate decision between pathogenic Th17 and Treg cells [63]. These coordinated shifts in specific metabolic pathways that occur during T helper cell differentiation may be disrupted in a number of ways, and we recently identified SELENOI as a metabolic enzyme involved in regulating T helper cell differentiation. SELENOI mRNA and protein levels increased at early stages of Th17 differentiation, similar to levels of the master regulator that drives Th17 phenotypes, RORγt (manuscript submitted). Using T cell specific SELENOI KO mice, our studies have found that naïve CD4+ T cells were directed toward Treg and away from Th1 and Th17 subtypes when activated through the TCR. Moreover, T cell specific KO of SELENOI protected mice from a mouse model of MS, experimental autoimmune encephalitis (EAE), showing that SELENOI deficiency reduces Th17 pathology. These data may suggest that targeting SELENOI in T cells may represent a potential therapeutic approach to treating MS. Indeed, cell-based therapies have been proposed for restoring homeostasis in MS patients such as tolerogenic dendritic cells, Tregs, mesenchymal stem cells, and vaccination with T cells [64]. Interfering with SELENOI activity to skew Th17/Treg cells toward a tolerogenic phenotype certainly presents its challenges, but may provide a new cell-based therapeutic approach for this or other autoimmune disorders.
7. Conclusions
Overall, insights into roles for SELENOI in T cell functions have been made using SELENOI loss-of-function models (Table 1). Upregulated SELENOI triggered by TCR engagement contributes to both proliferation and differentiation, which may be crucial for a variety of immune responses (Figure 4). The emerging field of metabolic reprogramming during T cell activation is providing new insights into factors and pathways that regulate optimal responses to pathogens and tumors. SELENOI has recently been identified as an anabolic enzyme involved in metabolic reprogramming, and disruption of its activity in mouse models has been shown to effectively decrease EAE. Ethanolamine phospholipid synthesis is particularly important for metabolic reprogramming in pluripotent stem cells and proliferating tumor cells [31,65], and our recent work showed a crucial role for ethanolamine phospholipid synthesis during T cell activation. How this may be translated into new therapies remains uncertain, but targeting metabolism may be incorporated into cell-based therapies involving T cells. For example, chimeric antigen receptor (CAR) expressing T cells have become an effective approach for treating some cancers, and advances have been made to improve the metabolic fitness and efficacy of CAR T cells [66]. This may provide a framework for developing new strategies in treating diseases, including autoimmune disorders, focused on regulating T cell metabolism and providing optimal immunity.

Table 1.
A summary of roles for SELENOI in T cell functions.
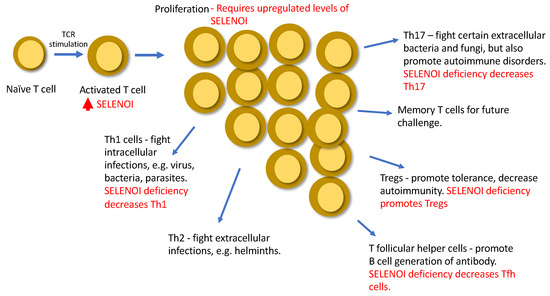
Figure 4.
A summary of roles of SELENOI in T cells.
Funding
This research was supported by NIH grant R01AI147496.
Institutional Review Board Statement
No human studies were included. Animal protocols were approved by the University of Hawaii Institutional Animal Care and Use Committee.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Reeves, M.A.; Hoffmann, P.R. The human selenoproteome: Recent insights into functions and regulation. Cell. Mol. Life Sci. 2009, 66, 2457–2478. [Google Scholar] [CrossRef]
- Schweizer, U.; Fradejas-Villar, N. Why 21? The significance of selenoproteins for human health revealed by inborn errors of metabolism. FASEB J. 2016, 30, 3669–3681. [Google Scholar] [CrossRef] [PubMed]
- Seyedali, A.; Berry, M.J. Nonsense-mediated decay factors are involved in the regulation of selenoprotein mRNA levels during selenium deficiency. RNA 2014, 20, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-C.; Yeh, C.-W.; Chen, Y.-F.; Lee, T.-T.; Hsieh, P.-Y.; Rusnac, D.V.; Lin, S.-Y.; Elledge, S.J.; Zheng, N.; Yen, H.-C.S. C-Terminal End-Directed Protein Elimination by CRL2 Ubiquitin Ligases. Mol. Cell 2018, 70, 602e3–613e3. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E. Regulation of Selenium Metabolism and Transport. Annu. Rev. Nutr. 2015, 35, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.W.; Hashimoto, A.C.; Shafer, L.A.; Dow, S.; Berry, M.J.; Hoffmann, P.R. Dietary Selenium Modulates Activation and Differentiation of CD4+ T Cells in Mice through a Mechanism Involving Cellular Free Thiols. J. Nutr. 2010, 140, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Broome, C.S.; McArdle, F.; Kyle, J.A.M.; Andrews, F.; Lowe, N.; Hart, C.A.; Arthur, J.R.; Jackson, M. An increase in selenium intake improves immune function and poliovirus handling in adults with marginal selenium status. Am. J. Clin. Nutr. 2004, 80, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.R.; Berry, M.J. The influence of selenium on immune responses. Mol. Nutr. Food Res. 2008, 52, 1273–1280. [Google Scholar] [CrossRef]
- Ivory, K.; Prieto, E.; Spinks, C.; Armah, C.N.; Goldson, A.J.; Dainty, J.R.; Nicoletti, C. Selenium supplementation has beneficial and detrimental effects on immunity to influenza vaccine in older adults. Clin. Nutr. 2015, 36, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.; Hansbro, P.M. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Cui, W.; Kaech, S.M. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol. Rev. 2010, 236, 151–166. [Google Scholar] [CrossRef]
- Fredericks, G.J.; Hoffmann, F.W.; Rose, A.H.; Osterheld, H.J.; Hess, F.M.; Mercier, F.; Hoffmann, P.R. Stable expression and function of the inositol 1,4,5-triphosphate receptor requires palmitoylation by a DHHC6/selenoprotein K complex. Proc. Natl. Acad. Sci. USA 2014, 111, 16478–16483. [Google Scholar] [CrossRef]
- Carlson, B.A.; Yoo, M.-H.; Shrimali, R.K.; Irons, R.; Gladyshev, V.N.; Hatfield, D.L.; Park, J.M. Role of selenium-containing proteins in T-cell and macrophage function. Proc. Nutr. Soc. 2010, 69, 300–310. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Irons, R.D.; Carlson, B.A.; Sano, Y.; Gladyshev, V.N.; Park, J.M.; Hatfield, D.L. Selenoproteins Mediate T Cell Immunity through an Antioxidant Mechanism. J. Biol. Chem. 2008, 283, 20181–20185. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Stadtman, T.C.; Hatfield, D.L.; Jeang, K.-T. Levels of major selenoproteins in T cells decrease during HIV infection and low molecular mass selenium compounds increase. Proc. Natl. Acad. Sci. USA 1999, 96, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Hoffmann, F.W.; Marciel, M.P.; Page, K.E.; Williams-Aduja, M.A.; Akana, E.N.; Gojanovich, G.S.; Gerschenson, M.; Urschitz, J.; Moisyadi, S.; et al. Upregulated ethanolamine phospholipid synthesis via selenoprotein I is required for effective metabolic reprogramming during T cell activation. Mol. Metab. 2021, 47, 101170. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of Mammalian Selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Horibata, Y.; Hirabayashi, Y. Identification and characterization of human ethanolaminephosphotransferase1. J. Lipid Res. 2007, 48, 503–508. [Google Scholar] [CrossRef]
- Horibata, Y.; Elpeleg, O.; Eran, A.; Hirabayashi, Y.; Savitzki, D.; Tal, G.; Mandel, H.; Sugimoto, H. Ethanolamine phosphotransferase 1 (selenoprotein I) is critical for the neural development and maintenance of plasmalogen in human. J. Lipid Res. 2018, 59, 1015–1026. [Google Scholar] [CrossRef]
- Ahmed, M.Y.; Al-Khayat, A.; Al-Murshedi, F.; Al-Futaisi, A.; Chioza, B.A.; Fernandez-Murray, J.P.; Self, J.E.; Salter, C.G.; Harlalka, G.V.; Rawlins, L.E.; et al. A mutation ofEPT1 (SELENOI)underlies a new disorder of Kennedy pathway phospholipid biosynthesis. Brain 2017, 140, 547–554. [Google Scholar] [CrossRef]
- Devaux, P.F. Static and dynamic lipid asymmetry in cell membranes. Biochemistry 1991, 30, 1163–1173. [Google Scholar] [CrossRef]
- Murate, M.; Abe, M.; Kasahara, K.; Iwabuchi, K.; Umeda, M.; Kobayashi, T. Transbilayer distribution of lipids at nano scale. J. Cell Sci. 2015, 128, 1627–1638. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2014, 16, 1–18. [Google Scholar] [CrossRef]
- Gibellini, F.; Smith, T.K. The Kennedy pathway-De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef]
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. 2012, 1822, 1442–1452. [Google Scholar] [CrossRef]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Noyon, C.; Ruysschaert, J.-M.; Van Antwerpen, P.; Govaerts, C. Phosphatidylethanolamine Is a Key Regulator of Membrane Fluidity in Eukaryotic Cells. J. Biol. Chem. 2016, 291, 3658–3667. [Google Scholar] [CrossRef]
- Silin, V.I.; Hoogerheide, D.P. pH dependent electrical properties of the inner- and outer- leaflets of biomimetic cell membranes. J. Colloid Interface Sci. 2021, 594, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Rubio, J.; Astudillo, A.M.; Casas, J.; Balboa, M.A.; Balsinde, J. Regulation of Phagocytosis in Macrophages by Membrane Ethanolamine Plasmalogens. Front. Immunol. 2018, 9, 1723. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, K.; Xing, G.; Li, L.; Ma, B.; Hu, Z.; Duan, L.; Liu, X. Phospholipid remodeling is critical for stem cell pluripotency by facilitating mesenchymal-to-epithelial transition. Sci. Adv. 2019, 5, eaax7525. [Google Scholar] [CrossRef]
- Lebrero, P.; Astudillo, A.M.; Rubio, J.M.; Fernandez-Caballero, L.; Kokotos, G.; Balboa, M.A.; Balsinde, J. Cellular Plasmalogen Content Does Not Influence Arachidonic Acid Levels or Distribution in Macrophages: A Role for Cytosolic Phospholipase A2gamma in Phospholipid Remodeling. Cells 2019, 8, 799. [Google Scholar] [CrossRef]
- Horibata, Y.; Ando, H.; Sugimoto, H. Locations and contributions of the phosphotransferases EPT1 and CEPT1 to the biosynthesis of ethanolamine phospholipids. J. Lipid Res. 2020, 61, 1221–1231. [Google Scholar] [CrossRef]
- Vance, J.E. Historical perspective: Phosphatidylserine and phosphatidylethanolamine from the 1800s to the present. J. Lipid Res. 2018, 59, 923–944. [Google Scholar] [CrossRef]
- Bleijerveld, O.B.; Brouwers, J.F.; Vaandrager, A.B.; Helms, J.B.; Houweling, M. The CDP-ethanolamine Pathway and Phosphatidylserine Decarboxylation Generate Different Phosphatidylethanolamine Molecular Species. J. Biol. Chem. 2007, 282, 28362–28372. [Google Scholar] [CrossRef] [PubMed]
- McMaster, C.R.; Bell, R.M. CDP-ethanolamine:1,2-diacylglycerol ethanolaminephosphotransferase. Biochim. Biophys. Acta 1997, 1348, 117–123. [Google Scholar] [CrossRef]
- Liu, J.; Rozovsky, S. Membrane-Bound Selenoproteins. Antioxidants Redox Signal. 2015, 23, 795–813. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E.; Vance, D.E. Does rat liver Golgi have the capacity to synthesize phospholipids for lipoprotein secretion? J Biol Chem 1988, 263, 5898–5909. [Google Scholar] [CrossRef]
- Kuiper, G.G.J.M.; Klootwijk, W.; Visser, T.J. Substitution of Cysteine for Selenocysteine in the Catalytic Center of Type III Iodothyronine Deiodinase Reduces Catalytic Efficiency and Alters Substrate Preference. Endocrinology 2003, 144, 2505–2513. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Fomenko, D.E.; Yoon, Y.-E.; Gladyshev, V.N. Catalytic Advantages Provided by Selenocysteine in Methionine-S-Sulfoxide Reductases†. Biochemistry 2006, 45, 13697–13704. [Google Scholar] [CrossRef][Green Version]
- Vance, J.E.; Tasseva, G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta 2013, 1831, 543–554. [Google Scholar] [CrossRef]
- He, H.-T.; Bongrand, P. Membrane dynamics shape TCR-generated signaling. Front. Immunol. 2012, 3, 90. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid Rafts as a Membrane-Organizing Principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J.; Han, X.; Chung, K.-N.; Gross, R.W. Lipid Rafts Are Enriched in Arachidonic Acid and Plasmenylethanolamine and Their Composition Is Independent of Caveolin-1 Expression: A Quantitative Electrospray Ionization/Mass Spectrometric Analysis. Biochemistry 2002, 41, 2075–2088. [Google Scholar] [CrossRef]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- MacIver, N.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Herman, C.E.; MacIver, N.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef]
- Van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef]
- Chang, J.T.; Palanivel, V.R.; Kinjyo, I.; Schambach, F.; Intlekofer, A.M.; Banerjee, A.; Longworth, S.A.; Vinup, K.E.; Mrass, P.; Oliaro, J.; et al. Asymmetric T Lymphocyte Division in the Initiation of Adaptive Immune Responses. Science 2007, 315, 1687–1691. [Google Scholar] [CrossRef]
- Chisolm, D.A.; Weinmann, A.S. Connections Between Metabolism and Epigenetics in Programming Cellular Differentiation. Annu. Rev. Immunol. 2018, 36, 221–246. [Google Scholar] [CrossRef]
- Pawlak, M.; Ho, A.W.; Kuchroo, V.K. Cytokines and transcription factors in the differentiation of CD4+ T helper cell subsets and induction of tissue inflammation and autoimmunity. Curr. Opin. Immunol. 2020, 67, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Guy, C.S.; Chapman, N.M.; Palacios, G.; Wei, J.; Zhou, P.; Long, L.; Wang, Y.-D.; Qian, C.; Dhungana, Y.; et al. Metabolic control of TFH cells and humoral immunity by phosphatidylethanolamine. Nature 2021, 595, 724–729. [Google Scholar] [CrossRef]
- Wang, L.; Magdaleno, S.; Tabas, I.; Jackowski, S. Early Embryonic Lethality in Mice with Targeted Deletion of the CTP:Phosphocholine Cytidylyltransferase α Gene (Pcyt1a). Mol. Cell. Biol. 2005, 25, 3357–3363. [Google Scholar] [CrossRef]
- Avery, J.C.; Yamazaki, Y.; Hoffmann, F.W.; Folgelgren, B.; Hoffmann, P.R. Selenoprotein I is essential for murine embryogenesis. Arch. Biochem. Biophys. 2020, 689, 108444. [Google Scholar] [CrossRef]
- E Harrington, L.; Hatton, R.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23–IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; Yang, X.-P.; Hirahara, K.; O’Shea, J.J. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 2011, 32, 395–401. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Shen, H.; Shi, L.Z. Metabolic regulation of TH17 cells. Mol. Immunol. 2019, 109, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Araujo, L.; Khim, P.; Mkhikian, H.; Mortales, C.-L.; Demetriou, M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. eLife 2017, 6, e21330. [Google Scholar] [CrossRef]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bahre, H.; et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 2014, 20, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR Kinase Differentially Regulates Effector and Regulatory T Cell Lineage Commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Benavides, G.A.; Geng, J.; Koralov, S.; Hu, H.; Darley-Usmar, V.M.; Harrington, L.E. Mitochondrial Oxidative Phosphorylation Regulates the Fate Decision between Pathogenic Th17 and Regulatory T Cells. Cell Rep. 2020, 30, 1898–1909. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, M.J.; Presas-Rodríguez, S.; Teniente-Serra, A.; González-Larreategui, I.; Quirant-Sánchez, B.; Fondelli, F.; Djedovic, N.; Iwaszkiewicz-Grześ, D.; Chwojnicki, K.; Miljković, D.; et al. Paving the way towards an effective treatment for multiple sclerosis: Advances in cell therapy. Cell. Mol. Immunol. 2021, 18, 1353–1374. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Rong, X.; Palladino, E.N.; Wang, J.; Fogelman, A.M.; Martín, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.; del Bufalo, F.; de Angelis, B.; Quintarelli, C.; Caruana, I.; de Billy, E. Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy. Cells 2020, 10, 14. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).