
SecProCT: In Silico Prediction of Human Secretory Proteins Based on Capsule Network and Transformer


Abstract
:1. Introduction
2. Results
2.1. Evaluating the Performance of the Binary Classification
2.2. Ablation Study on Binary Classification
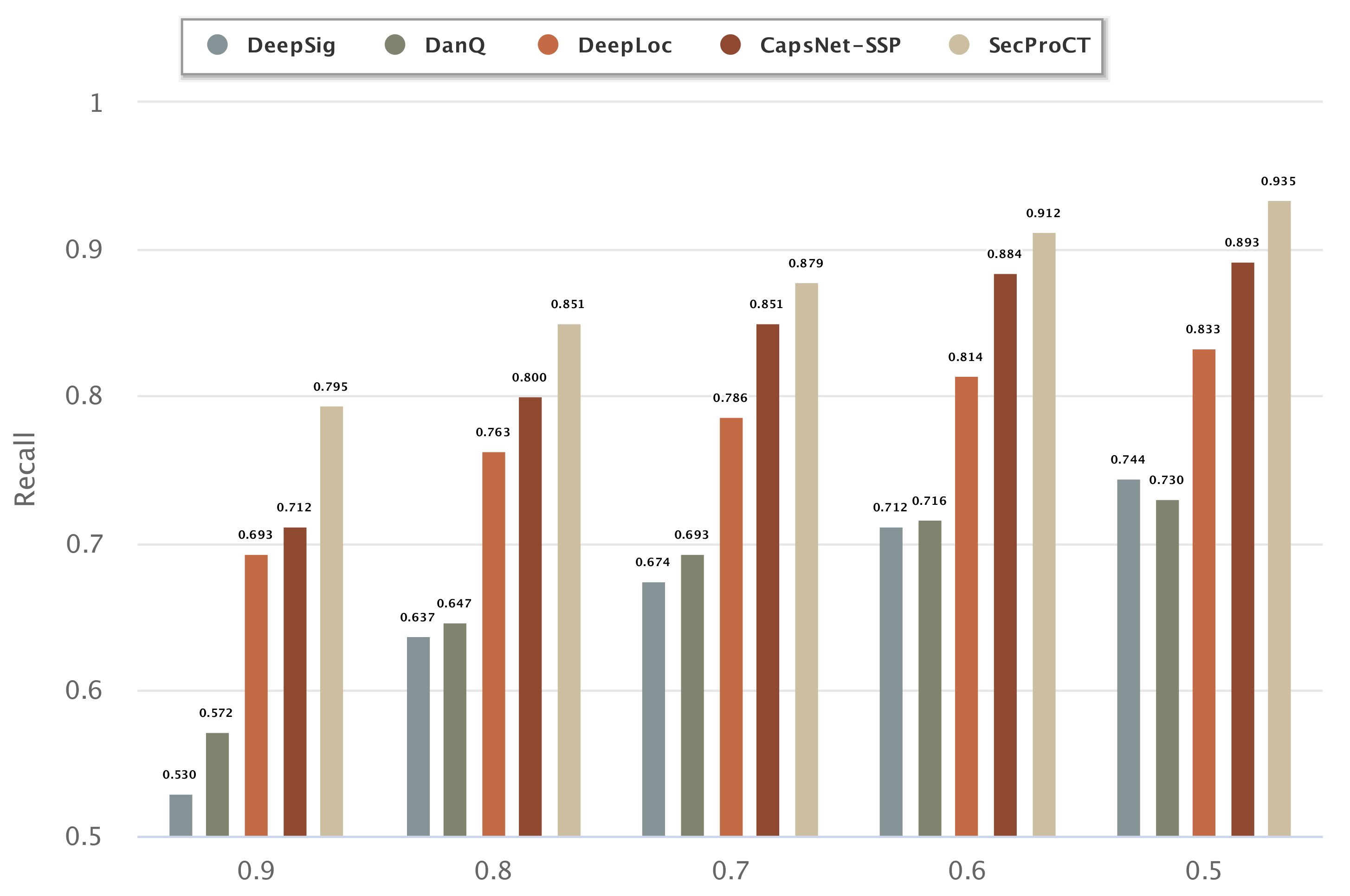
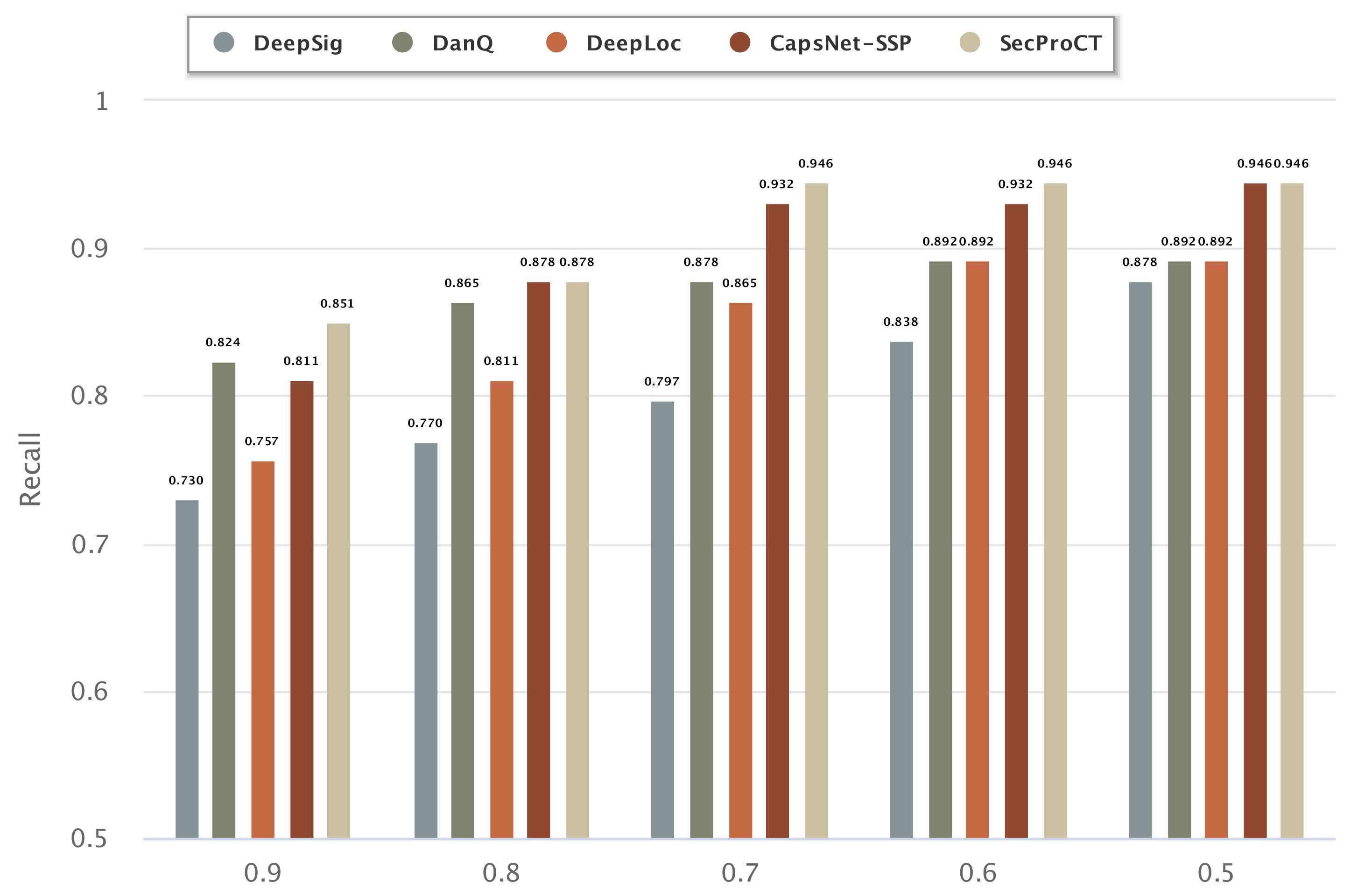
2.3. Evaluating the Performance of Human Secretory Protein Prediction
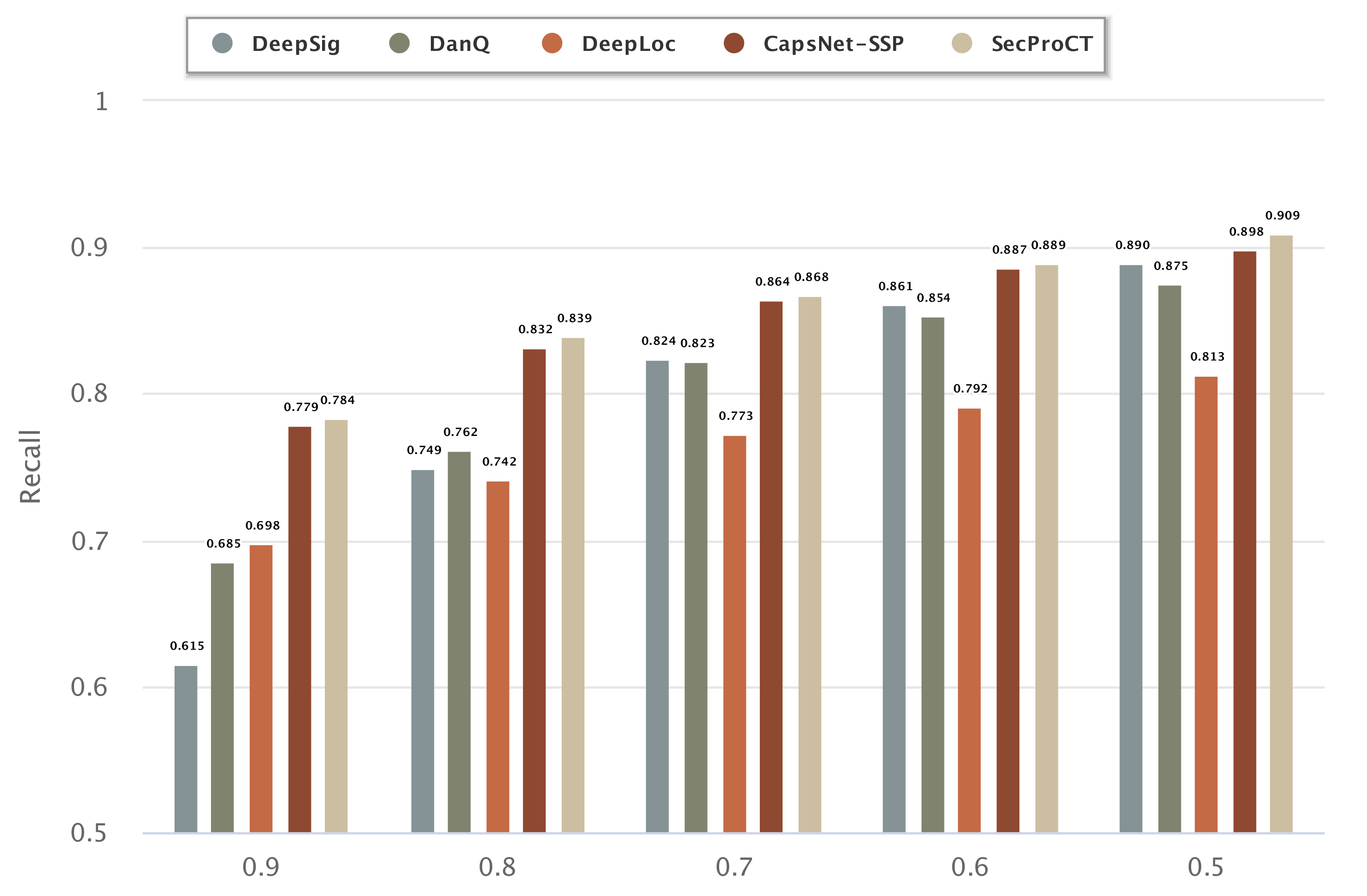
2.4. Evaluating the Performance of Cancer Biomarker Prediction
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Input Sequence Encoding
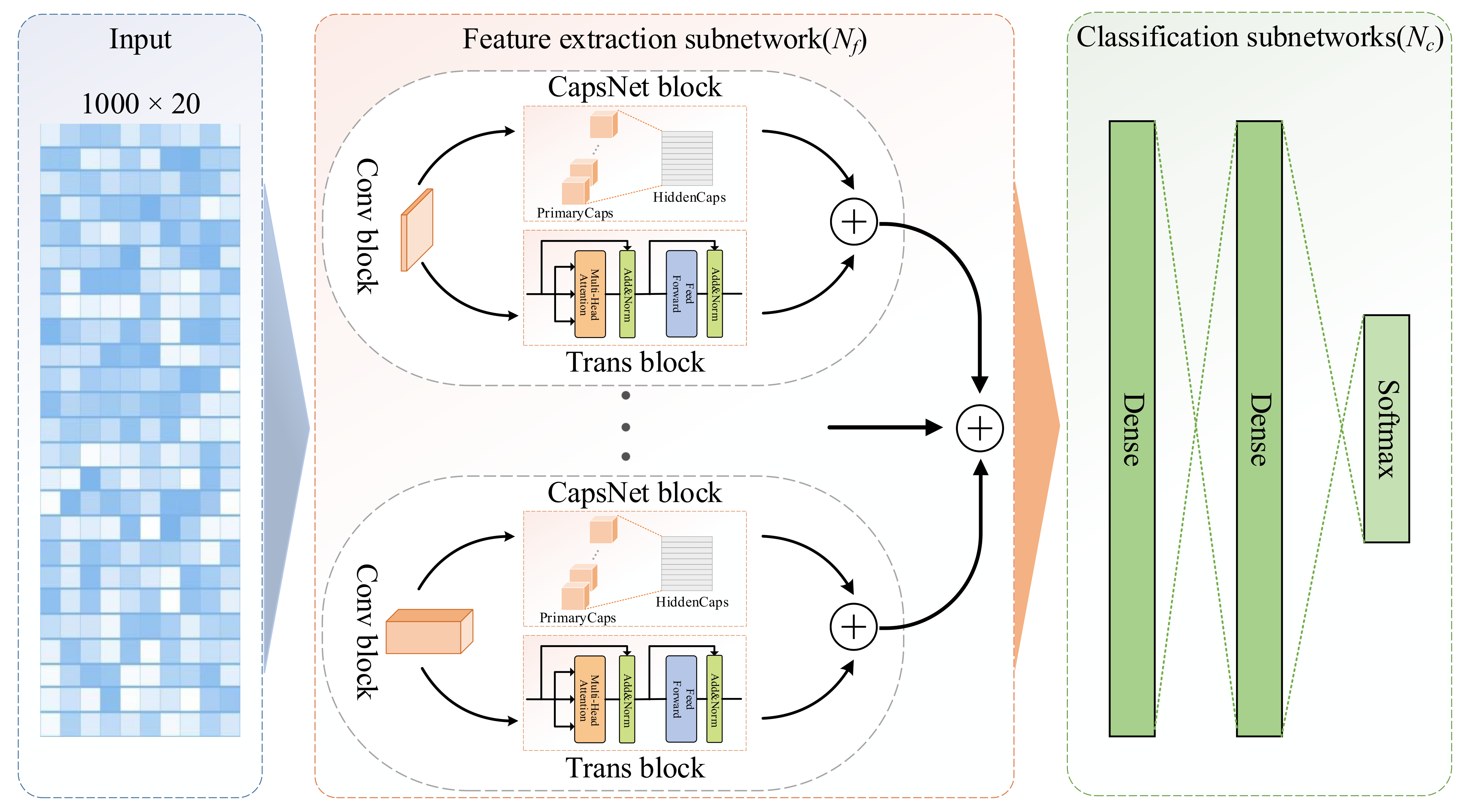
4.3. Architecture Design
4.4. Model Training
| Algorithm 1: Bagging Algorithm [49] |
| Input: : Training set with positive samples : Training set with negative samples T: Number of iterations n: The size of a random selection : Weak classifier
Bagged classifier: where |
4.5. Performance Measurements
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nagpal, M.; Singh, S.; Singh, P.; Chauhan, P.; Zaidi, M.A. Tumor markers: A diagnostic tool. Natl. J. Maxillofac. Surg. 2016, 7, 17–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loke, S.Y.; Lee, A.S.G. The future of blood-based biomarkers for the early detection of breast cancer. Eur. J. Cancer 2018, 92, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Pfaffe, T.; Cooper-White, J.; Beyerlein, P.; Kostner, K.; Punyadeera, C. Diagnostic potential of saliva: Current state and future applications. Clin. Chem. 2011, 57, 675–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castagnola, M.; Scarano, E.; Passali, G.C.; Messana, I.; Cabras, T.; Iavarone, F.; Di Cintio, G.; Fiorita, A.; De Corso, E.; Paludetti, G. Salivary biomarkers and proteomics: Future diagnostic and clinical utilities. Acta Otorhinolaryngol. Ital. 2017, 37, 94–101. [Google Scholar] [CrossRef]
- Robinson, J.L.; Feizi, A.; Uhlen, M.; Nielsen, J. A Systematic Investigation of the Malignant Functions and Diagnostic Potential of the Cancer Secretome. Cell Rep. 2019, 26, 2622–2635.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Huang, L.; Shao, D.; Wang, Y.; Cui, X.; Li, Y.; Chen, Q.; Cui, J. Human body-fluid proteome: Quantitative profiling and computational prediction. Brief. Bioinform. 2021, 22, 315–333. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Du, W.; Zhou, C.; Zhou, Y.; Cao, Z.; Tian, Y.; Wang, Y. A computational method for prediction of saliva-secretory proteins and its application to identification of head and neck cancer biomarkers for salivary diagnosis. IEEE Trans. Nanobiosci. 2015, 14, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zeng, S.; Xu, C.; Qiu, W.; Liang, Y.; Joshi, T.; Xu, D. MusiteDeep: A deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 2017, 33, 3909–3916. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Liu, Q.; Puett, D.; Xu, Y. Computational prediction of human proteins that can be secreted into the bloodstream. Bioinformatics 2008, 24, 2370–2375. [Google Scholar] [CrossRef]
- Liu, Q.; Cui, J.; Yang, Q.; Xu, Y. In-silico prediction of blood-secretory human proteins using a ranking algorithm. BMC Bioinform. 2010, 11, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liang, Y.; Wang, Y.; Cui, J.; Liu, M.; Du, W.; Xu, Y. Computational prediction of human salivary proteins from blood circulation and application to diagnostic biomarker identification. PLoS ONE 2013, 8, e80211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chai, H.; Guo, S.; Guo, H.; Li, Y. High-Throughput Identification of Mammalian Secreted Proteins Using Species-Specific Scheme and Application to Human Proteome. Molecules 2018, 23, 1448. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Y.; Ma, Z. In silico Prediction of Human Secretory Proteins in Plasma Based on Discrete Firefly Optimization and Application to Cancer Biomarkers Identification. Front. Genet. 2019, 10, 542. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; UniProt, C. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Casadio, R. DeepSig: Deep learning improves signal peptide detection in proteins. Bioinformatics 2018, 34, 1690–1696. [Google Scholar] [CrossRef] [Green Version]
- Quang, D.; Xie, X. DanQ: A hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016, 44, e107. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Sonderby, C.K.; Sonderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Du, W.; Sun, Y.; Li, G.; Cao, H.; Pang, R.; Li, Y. CapsNet-SSP: Multilane capsule network for predicting human saliva-secretory proteins. BMC Bioinform. 2020, 21, 237. [Google Scholar] [CrossRef]
- Emilsson, V.; Ilkov, M.; Lamb, J.R.; Finkel, N.; Gudmundsson, E.F.; Pitts, R.; Hoover, H.; Gudmundsdottir, V.; Horman, S.R.; Aspelund, T.; et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 2018, 361, 769–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Apweiler, R.; Balgley, B.M.; Boontheung, P.; Bundy, J.L.; Cargile, B.J.; Cole, S.; Fang, X.; Gonzalez-Begne, M.; Griffin, T.J.; et al. Systematic comparison of the human saliva and plasma proteomes. Proteom. Clin. Appl. 2009, 3, 116–134. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Rhodus, N.L.; Griffin, R.J.; Carlis, J.V.; Griffin, T.J. A catalogue of human saliva proteins identified by free flow electrophoresis-based peptide separation and tandem mass spectrometry. Mol. Cell Proteom. 2005, 4, 1826–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denny, P.; Hagen, F.K.; Hardt, M.; Liao, L.; Yan, W.; Arellanno, M.; Bassilian, S.; Bedi, G.S.; Boontheung, P.; Cociorva, D.; et al. The proteomes of human parotid and submandibular/sublingual gland salivas collected as the ductal secretions. J. Proteome Res. 2008, 7, 1994–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Yang, L.; Wang, W.; Song, T.; Lee, C.S.; DeVoe, D.L.; Balgley, B.M. Comparison of electrokinetics-based multidimensional separations coupled with electrospray ionization-tandem mass spectrometry for characterization of human salivary proteins. Anal. Chem. 2007, 79, 5785–5792. [Google Scholar] [CrossRef]
- Bandhakavi, S.; Stone, M.D.; Onsongo, G.; Van Riper, S.K.; Griffin, T.J. A dynamic range compression and three-dimensional peptide fractionation analysis platform expands proteome coverage and the diagnostic potential of whole saliva. J. Proteome Res. 2009, 8, 5590–5600. [Google Scholar] [CrossRef] [Green Version]
- Sivadasan, P.; Kumar Gupta, M.; Sathe, G.J.; Balakrishnan, L.; Palit, P.; Gowda, H.; Suresh, A.; Abraham Kuriakose, M.; Sirdeshmukh, R. Data from human salivary proteome—A resource of potential biomarkers for oral cancer. Data Brief 2015, 4, 374–378. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.B.; Sharma, S.; Mohamedali, A.; Mahboob, S.; Redmond, W.J.; Pascovici, D.; Wu, J.X.; Zaw, T.; Adhikari, S.; Vaibhav, V.; et al. Potential early clinical stage colorectal cancer diagnosis using a proteomics blood test panel. Clin. Proteom. 2019, 16, 34. [Google Scholar] [CrossRef]
- Ahn, J.M.; Sung, H.J.; Yoon, Y.H.; Kim, B.G.; Yang, W.S.; Lee, C.; Park, H.M.; Kim, B.J.; Kim, B.G.; Lee, S.Y.; et al. Integrated glycoproteomics demonstrates fucosylated serum paraoxonase 1 alterations in small cell lung cancer. Mol. Cell Proteom. 2014, 13, 30–48. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.W.; Chang, K.P.; Hsu, C.W.; Chang, I.Y.; Liu, H.P.; Chen, Y.T.; Wu, C.C. Identification of Salivary Biomarkers for Oral Cancer Detection with Untargeted and Targeted Quantitative Proteomics Approaches. Mol. Cell Proteom. 2019, 18, 1796–1806. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, L.; Zhou, H.; Lee, J.M.; Garon, E.B.; Wong, D.T. Proteomic analysis of human saliva from lung cancer patients using two-dimensional difference gel electrophoresis and mass spectrometry. Mol. Cell Proteom. 2012, 11, M111.012112. [Google Scholar] [CrossRef] [Green Version]
- Porto-Mascarenhas, E.C.; Assad, D.X.; Chardin, H.; Gozal, D.; De Luca Canto, G.; Acevedo, A.C.; Guerra, E.N. Salivary biomarkers in the diagnosis of breast cancer: A review. Crit. Rev. Oncol. Hematol. 2017, 110, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Y.; Yin, Y.; Gao, G.; Li, S.; Jiang, Y.; Gu, X.; Luo, J. SPD–a web-based secreted protein database. Nucleic Acids Res. 2005, 33, D169–D173. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, J.; Lynn Fink, J.; Karunaratne, S.; Hanson, K.; Hamilton, N.A.; Teasdale, R.D. LOCATE: A mammalian protein subcellular localization database. Nucleic Acids Res. 2008, 36, D230–D233. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Meinken, J.; Walker, G.; Cooper, C.R.; Min, X.J. MetazSecKB: The human and animal secretome and subcellular proteome knowledgebase. Database 2015, 2015, bav077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.J.; Peng, M.; Li, H.; Liu, B.S.; Wang, C.; Wu, J.R.; Li, Y.X.; Zeng, R. Sys-BodyFluid: A systematical database for human body fluid proteome research. Nucleic Acids Res. 2009, 37, D907–D912. [Google Scholar] [CrossRef]
- Omenn, G.S. The HUPO Human Plasma Proteome Project. Proteom. Clin. Appl. 2007, 1, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Loo, J.A.; Wong, D.T. Human saliva proteome analysis and disease biomarker discovery. Expert Rev. Proteom. 2007, 4, 531–538. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; Lopez de la Paz, M.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Magnan, C.N.; Baldi, P. SSpro/ACCpro 5: Almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 2014, 30, 2592–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabour, S.; Frosst, N.; Hinton, G.E. Dynamic routing between capsules. arXiv 2017, arXiv:1710.09829. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention is all you need. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Li, Y.; Yuan, Y. Convergence analysis of two-layer neural networks with relu activation. arXiv 2017, arXiv:1705.09886. [Google Scholar]
- Caruana, R. Learning many related tasks at the same time with backpropagation. In Advances in Neural Information Processing Systems; Massachusetts Institute of Technology Press: Cambridge, MA, USA, 1995; pp. 657–664. [Google Scholar]
- Ng, H.W.; Nguyen, V.D.; Vonikakis, V.; Winkler, S. Deep learning for emotion recognition on small datasets using transfer learning. In Proceedings of the 2015 ACM on International Conference on Multimodal Interaction, Seattle, WA, USA, 9–13 November 2015; pp. 443–449. [Google Scholar]
- Galar, M.; Fernandez, A.; Barrenechea, E.; Bustince, H.; Herrera, F. A review on ensembles for the class imbalance problem: Bagging-, boosting-, and hybrid-based approaches. IEEE Trans. Syst. Man Cybern. Part C 2011, 42, 463–484. [Google Scholar] [CrossRef]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Rosasco, L.; Caponnetto, A. On early stopping in gradient descent learning. Construct. Approx. 2007, 26, 289–315. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2017, arXiv:cs.LG/1412.6980. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Body Fluid | Algorithm | Advantage | Disadvantage | Ref |
|---|---|---|---|---|---|
| Cui et al. (2008) | Blood | SVM | 1 | 5 | [10] |
| Liu et al. (2010) | Blood | Ranking | 2 | 5 | [11] |
| Wang et al. (2013) | Saliva | SVM | 4 | 5 | [12] |
| Sun et al. (2015) | Saliva | SVM | 1 | 5 | [8] |
| Zhang et al. (2018) | Body fluids | SVM | 3 | 5 | [13] |
| Zhang et al. (2019) | Blood | DFO * | 1 | 5 | [14] |
| Methods | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| KNN | 0.879 | 0.848 | 0.911 | 0.759 | 0.873 |
| Decision Tree | 0.858 | 0.852 | 0.864 | 0.716 | 0.838 |
| Random Forest | 0.866 | 0.818 | 0.916 | 0.736 | 0.922 |
| AdaBoost | 0.859 | 0.838 | 0.881 | 0.719 | 0.912 |
| SVM | 0.893 | 0.877 | 0.910 | 0.787 | 0.925 |
| SecProCT | 0.917 | 0.906 | 0.930 | 0.835 | 0.967 |
| Methods | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| KNN | 0.778 | 0.649 | 0.907 | 0.575 | 0.809 |
| Decision Tree | 0.772 | 0.692 | 0.851 | 0.550 | 0.740 |
| Random Forest | 0.781 | 0.804 | 0.758 | 0.563 | 0.836 |
| AdaBoost | 0.792 | 0.703 | 0.881 | 0.593 | 0.847 |
| SVM | 0.781 | 0.784 | 0.778 | 0.562 | 0.857 |
| SecProCT | 0.909 | 0.898 | 0.919 | 0.817 | 0.956 |
| Methods | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| DeepSig | 0.893 | 0.906 | 0.880 | 0.786 | 0.939 |
| DanQ | 0.850 | 0.783 | 0.920 | 0.708 | 0.927 |
| DeepLoc | 0.898 | 0.877 | 0.920 | 0.797 | 0.940 |
| CapsNet-SSP | 0.893 | 0.897 | 0.890 | 0.786 | 0.942 |
| SecProCT | 0.917 | 0.906 | 0.930 | 0.835 | 0.967 |
| Methods | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| DeepSig | 0.792 | 0.745 | 0.838 | 0.586 | 0.867 |
| DanQ | 0.802 | 0.745 | 0.859 | 0.608 | 0.886 |
| DeepLoc | 0.843 | 0.755 | 0.929 | 0.695 | 0.891 |
| CapsNet-SSP | 0.888 | 0.847 | 0.929 | 0.779 | 0.948 |
| SecProCT | 0.909 | 0.898 | 0.919 | 0.817 | 0.956 |
| Methods | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| SecretomeP | 0.762 | 0.632 | 0.787 | 0.340 | 0.764 |
| SRTpred | 0.782 | 0.678 | 0.802 | 0.392 | 0.770 |
| iMSP-U | 0.829 | 0.631 | 0.866 | 0.443 | 0.821 |
| iMSP-H | 0.850 | 0.538 | 0.908 | 0.441 | 0.817 |
| SCRIP | 0.858 | 0.716 | 0.884 | 0.537 | 0.865 |
| SecProCT | 0.873 | 0.809 | 0.884 | 0.596 | 0.920 |
| Architectures | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| Without Conv block | 0.882 | 0.853 | 0.912 | 0.604 | 0.915 |
| Without CapsNet block | 0.916 | 0.903 | 0.928 | 0.674 | 0.947 |
| Without Trans block | 0.905 | 0.889 | 0.921 | 0.647 | 0.938 |
| All | 0.921 | 0.911 | 0.931 | 0.686 | 0.950 |
| Architectures | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| Without Conv block | 0.875 | 0.848 | 0.902 | 0.671 | 0.909 |
| Without CapsNet block | 0.891 | 0.857 | 0.925 | 0.722 | 0.918 |
| Without Trans block | 0.888 | 0.828 | 0.948 | 0.750 | 0.915 |
| All | 0.892 | 0.834 | 0.950 | 0.759 | 0.920 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, W.; Zhao, X.; Sun, Y.; Zheng, L.; Li, Y.; Zhang, Y. SecProCT: In Silico Prediction of Human Secretory Proteins Based on Capsule Network and Transformer. Int. J. Mol. Sci. 2021, 22, 9054. https://doi.org/10.3390/ijms22169054
Du W, Zhao X, Sun Y, Zheng L, Li Y, Zhang Y. SecProCT: In Silico Prediction of Human Secretory Proteins Based on Capsule Network and Transformer. International Journal of Molecular Sciences. 2021; 22(16):9054. https://doi.org/10.3390/ijms22169054
Chicago/Turabian StyleDu, Wei, Xuan Zhao, Yu Sun, Lei Zheng, Ying Li, and Yu Zhang. 2021. "SecProCT: In Silico Prediction of Human Secretory Proteins Based on Capsule Network and Transformer" International Journal of Molecular Sciences 22, no. 16: 9054. https://doi.org/10.3390/ijms22169054
APA StyleDu, W., Zhao, X., Sun, Y., Zheng, L., Li, Y., & Zhang, Y. (2021). SecProCT: In Silico Prediction of Human Secretory Proteins Based on Capsule Network and Transformer. International Journal of Molecular Sciences, 22(16), 9054. https://doi.org/10.3390/ijms22169054