
Toward a Coronavirus Knowledge Graph


 , ,
, , 
Abstract
:1. Introduction
2. Related Work
3. Datasets
3.1. Analytical Graph (AG)
3.2. CORD-19
4. Merging Different KGs
5. Cases
5.1. Ego-Centered Subgraph
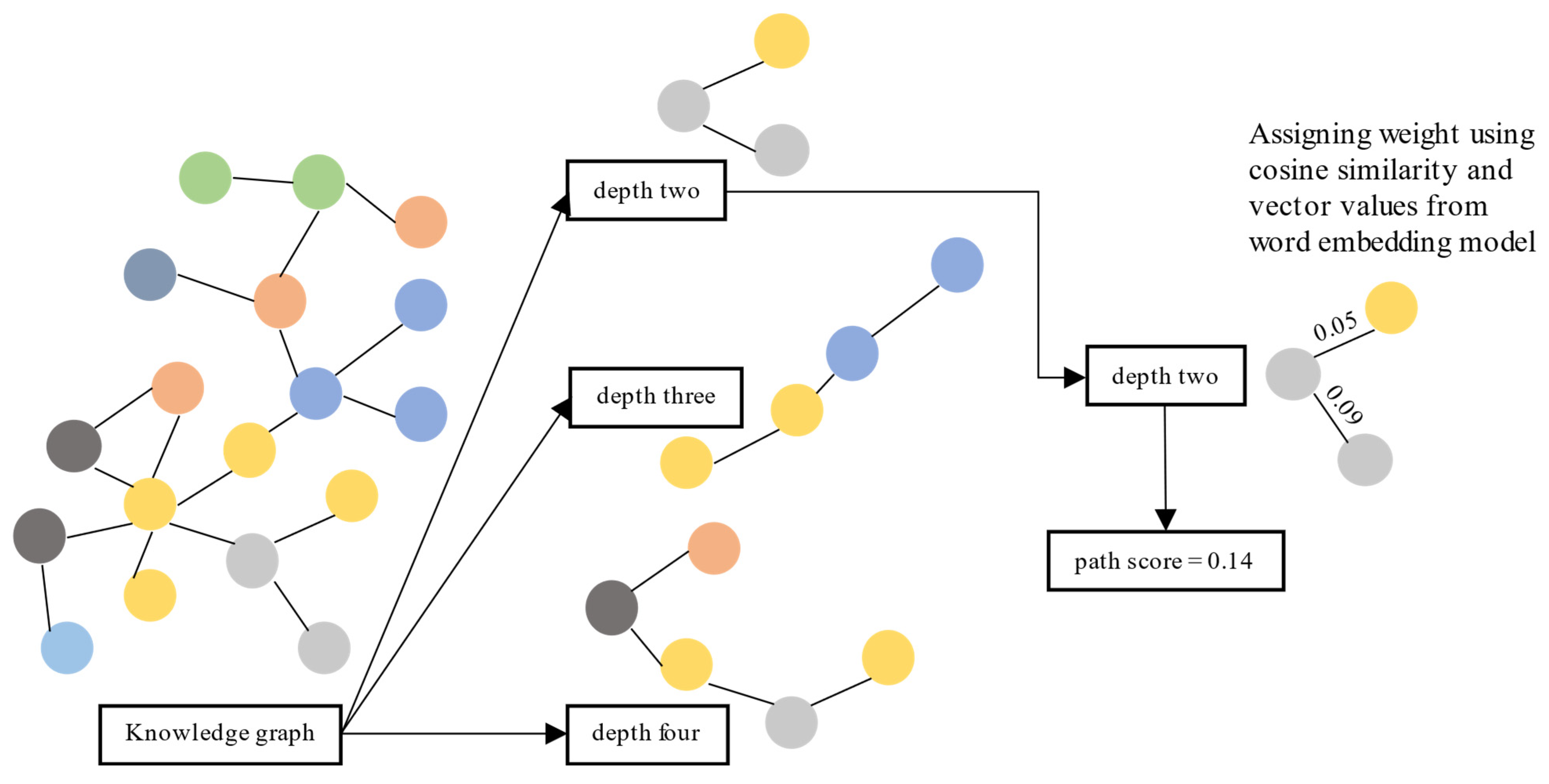
5.2. Path
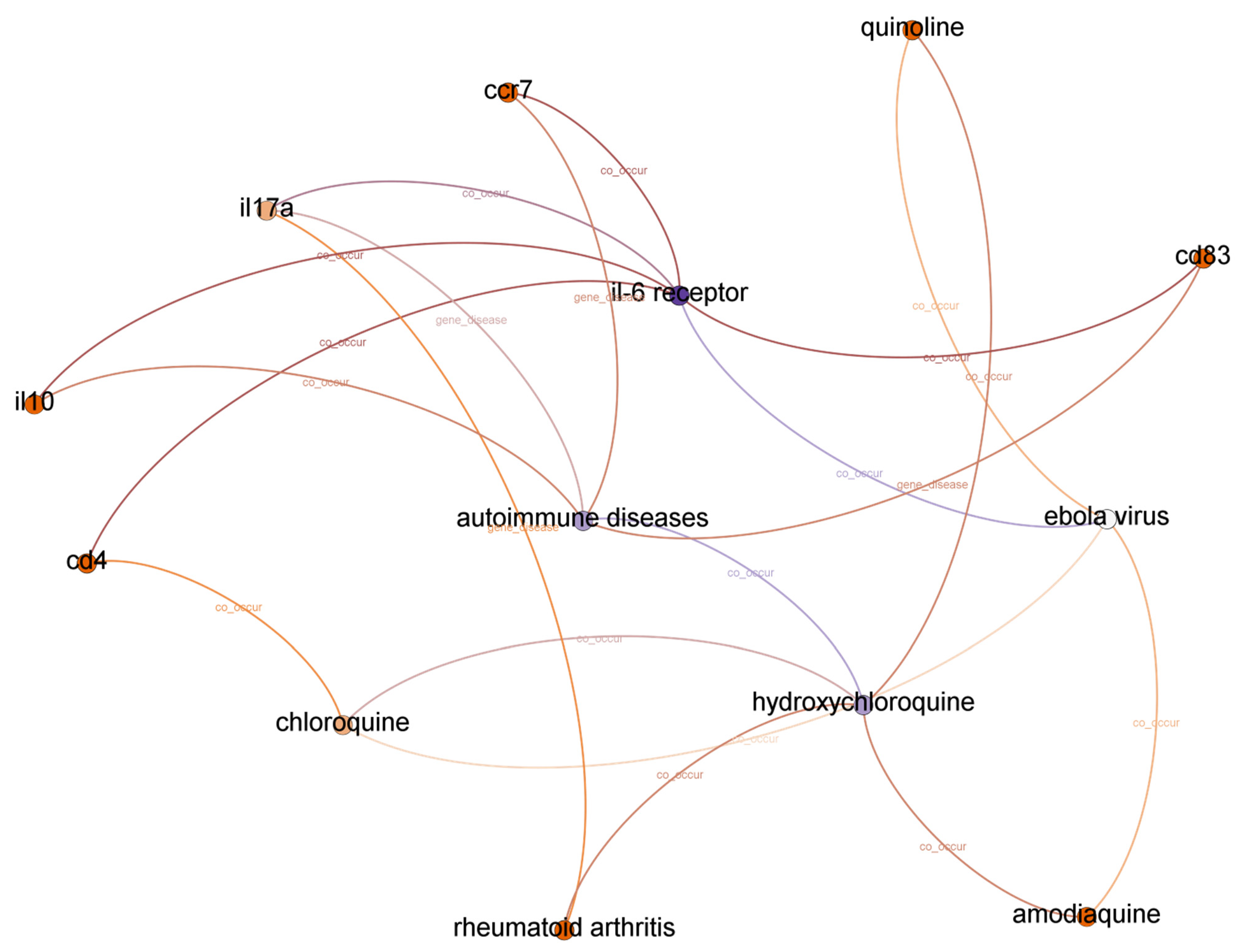
5.2.1. IL-6 Receptor and Hydroxychloroquine
5.2.2. STAT1 and Chloroquine
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Coronavirus Disease (COVID-19). Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 5 August 2020).
- Chahrour, M.; Assi, S.; Bejjani, M.; Nasrallah, A.; Salhab, H.; Fares, M.Y.; Khachfe, H.H. A Bibliometric Analysis of COVID-19 Research Activity: A Call for Increased Output. Cureus 2020, 12, e7357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu Wang, L.; Lo, K.; Chandrasekhar, Y.; Reas, R.; Yang, J.; Eide, D.; Funk, K.; Kinney, R.; Liu, Z.; Merrill, W.; et al. CORD-19: The Covid-19 Open Research Dataset. arXiv 2020, arXiv:2004.10706v2. [Google Scholar]
- Wei, C.-H.; Kao, H.-Y.; Lu, Z.; Wei, C.-H.; Kao, H.-Y.; Lu, Z. PubTator: A web-based text mining tool for assisting biocuration. Nucleic Acids Res. 2013, 41, W518–W522. [Google Scholar] [CrossRef] [PubMed]
- Pyysalo, S.; Ginter, F.; Moen, H.; Salakoski, T.; Ananiadou, S. Distributional Semantics Resources for Biomedical Text. In Proceedings of the LBM, Tokyo, Japan, 12–13 December 2013; pp. 39–44. [Google Scholar]
- Domingo-Fernández, D.; Baksi, S.; Schultz, B.; Gadiya, Y.; Karki, R.; Raschka, T.; Ebeling, C.; Hofmann-Apitius, M.; Kodamullil, A.T. COVID-19 Knowledge Graph: A computable, multi-modal, cause-and-effect knowledge model of COVID-19 pathophysiology. Bioinformatics 2020, 37, 1332–1334. [Google Scholar] [CrossRef]
- Ge, Y.; Tian, T.; Huang, S.; Wan, F.; Li, J.; Li, S.; Yang, H.; Hong, L.; Wu, N.; Yuan, E.; et al. A data-driven drug repositioning framework discovered a potential therapeutic agent targeting COVID-19. bioRxiv 2020, 1–62. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Rawling, M.; Savory, E.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Kim, S.; Song, M.; Jeong, M.; Kim, D.; Kang, J.; Rousseau, J.F.; Li, X.; Xu, W.; Torvik, V.I.; et al. Building a PubMed knowledge graph. Sci. Data 2020, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bellomarini, L.; Benedetti, M.; Gentili, A.; Laurendi, R.; Magnanimi, D.; Muci, A.; Sallinger, E. COVID-19 and Company Knowledge Graphs: Assessing Golden Powers and Economic Impact of Selective Lockdown via AI Reasoning. arXiv 2004, arXiv:2004.10119. [Google Scholar]
- Bullock, J.; Luccioni, A.; Pham, K.H.; Lam, C.S.N.; Luengo-Oroz, M. Mapping the landscape of Artificial Intelligence applications against COVID-19. J. Artif. Intell. Res. 2020, 69, 807–845. [Google Scholar] [CrossRef]
- Sun, H.; Dhingra, B.; Zaheer, M.; Mazaitis, K.; Salakhutdinov, R.; Cohen, W. Open Domain Question Answering Using Early Fusion of Knowledge Bases and Text. In Proceedings of the 2018 Conference on Empirical Methods in Natural Language Processing, Brussels, Belgium, 31 October–4 November 2018; pp. 4231–4242. [Google Scholar]
- Chahal, M. Information Retrieval using Jaccard Similarity Coefficient. Int. J. Comput. Trends Technol. 2016, 36, 140–143. [Google Scholar] [CrossRef]
- van Eck, N.J.; Waltman, L. How to normalize cooccurrence data? An analysis of some well-known similarity measures. J. Am. Soc. Inf. Sci. Technol. 2009, 60, 1635–1651. [Google Scholar] [CrossRef] [Green Version]
- Der Brück, T.V.; Pouly, M. Text Similarity Estimation Based on Word Embeddings and Matrix Norms for Targeted Marketing. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Minneapolis, MA, USA, 2–7 June 2019. [Google Scholar] [CrossRef]
- Naili, M.; Chaibi, A.H.; Ben Ghezala, H.H. Comparative study of word embedding methods in topic segmentation. Procedia Comput. Sci. 2017, 112, 340–349. [Google Scholar] [CrossRef]
- Church, K.W. Word2Vec. Nat. Lang. Eng. 2016, 23, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Chambers, J.; Davies, M.; Gaulton, A.; Hersey, A.; Velankar, S.; Petryszak, R.; Hastings, J.; Bellis, L.J.; McGlinchey, S.; Overington, J.P. UniChem: A unified chemical structure cross-referencing and identifier tracking system. J. Cheminform. 2013, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Garcia Giron, C.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2016, 45, D158–D169. [CrossRef] [Green Version]
- The University of New Mexico. Target Central Resource Database. Available online: http://juniper.health.unm.edu/tcrd/ (accessed on 5 August 2020).
- McDonald, A.G.; Boyce, S.; Tipton, K.F. ExplorEnz: The primary source of the IUBMB enzyme list. Nucleic Acids Res. 2009, 37, D593–D597. [Google Scholar] [CrossRef] [PubMed]
- Mungall, C.J.; Torniai, C.; Gkoutos, G.V.; Lewis, S.E.; Haendel, M.A. Uberon, an integrative multi-species anatomy ontology. Genome Biol. 2012, 13. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; I Furlong, L. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodenreider, O. The Unified Medical Language System (UMLS): Integrating biomedical terminology. Nucleic Acids Res. 2004, 32, 267D–270D. [Google Scholar] [CrossRef] [Green Version]
- Janssens, Y.; Bronselaer, A.; Wynendaele, E.; De Tré, G.; De Spiegeleer, B. Disbiome: A database describing microbiome alterations in different disease states. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Gaudet, P.; Michel, P.-A.; Zahn, M.; Britan, A.; Cusin, I.; Domagalski, M.; Duek, P.D.; Gateau, A.; Gleizes, A.; Hinard, V.; et al. The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res. 2016, 45, D177–D182. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2017, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2015, 44, D1075–D1079. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Tatonetti, N.; Ye, P.P.; Daneshjou, R.; Altman, R.B.; Tatonetti, N.; Ye, P.P.; Daneshjou, R.; Altman, R.B. Data-Driven Prediction of Drug Effects and Interactions. Sci. Transl. Med. 2012, 4, 125ra31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M.; von Mering, C.; Campillos, M.; Jensen, L.J.; Bork, P. STITCH: Interaction networks of chemicals and proteins. Nucleic Acids Res. 2007, 36, D684–D688. [Google Scholar] [CrossRef]
- Wei, Y.; Yu, W.; Jianqiao, T.; Xiaoli, X.; Yuesheng, Z.; Suqi, Y. Visualization analysis on treatment of coronavirus based on knowledge graph. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2020, 32, 279–286. [Google Scholar] [CrossRef]
- Vrandečić, D.; Krötzsch, M. Wikidata: A free collaborative knowledgebase. Commun. ACM 2014, 57, 78–85. [Google Scholar] [CrossRef]
- South, A.M.; Diz, D.I.; Chappell, M.C. COVID-19, ACE2, and the cardiovascular consequences. Am. J. Physiol. Circ. Physiol. 2020, 318, H1084–H1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zisman, L.S. ACE and ACE2: A tale of two enzymes. Eur. Heart J. 2005, 26, 322–324. [Google Scholar] [CrossRef]
- Wishart, D.S. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Baize, S.; Leroy, E.M.; Georges, A.J.; Georges-Courbot, M.-C.; Capron, M.; Bedjabaga, I.; Lansoud-Soukate, J.; Mavoungou, E. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 2002, 128, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Hober, D.; Blondiaux, J. Addressing Therapeutic Options for Ebola Virus Infection in Current and Future Outbreaks. Antimicrob. Agents Chemother. 2015, 59, 5892–5902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sawalha, A.H.; Lu, Q. COVID-19 and autoimmune diseases. Curr. Opin. Rheumatol. 2020, 33, 155–162. [Google Scholar] [CrossRef]
- Hussein, M.I.H.; Albashir, A.A.D.; Elawad, O.A.M.A.; Homeida, A. Malaria and COVID-19: Unmasking their ties. Malar. J. 2020, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, T.A.; Oliveira, J.B.; Torgerson, T.R. Congenital immune dysregulation disorders. In Pediatric Allergy: Principles and Practice; Elsevier: Amsterdam, The Netherlands, 2016; pp. 124–132. [Google Scholar]
- Mu, J.; Fang, Y.; Yang, Q.; Shu, T.; Wang, A.; Huang, M.; Jin, L.; Deng, F.; Qiu, Y.; Zhou, X. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhu, X.; Ji, X.; Quanquin, N.; Deng, Y.-Q.; Tian, M.; Aliyari, R.; Zuo, X.; Yuan, L.; Afridi, S.K.; et al. Chloroquine, a FDA-approved Drug, Prevents Zika Virus Infection and its Associated Congenital Microcephaly in Mice. EBioMedicine 2017, 24, 189–194. [Google Scholar] [CrossRef]
- Prasad, K.; Khatoon, F.; Rashid, S.; Ali, N.; AlAsmari, A.; Ahmed, M.Z.; Alqahtani, A.S.; Alqahtani, M.; Kumar, V. Targeting hub genes and pathways of innate immune response in COVID-19: A network biology perspective. Int. J. Biol. Macromol. 2020, 163, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wei, J.; Zou, L.; Jiang, T.; Wang, G.; Chen, L.; Huang, L.; Meng, F.; Wang, N.; Zhou, X.; et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): A multicenter, single-blind, randomized controlled trial. J. Allergy Clin. Immunol. 2020, 146, 137–146.e3. [Google Scholar] [CrossRef]
- Arora, S.; Singh, P.; Dohare, R.; Jha, R.; Syed, M.A. Unravelling host-pathogen interactions: ceRNA network in SARS-CoV-2 infection (COVID-19). Gene 2020, 762, 145057. [Google Scholar] [CrossRef]
- Di Maria, E.; Latini, A.; Borgiani, P.; Novelli, G. Genetic variants of the human host influencing the coronavirus-associated phenotypes (SARS, MERS and COVID-19): Rapid systematic review and field synopsis. Hum. Genom. 2020, 14, 1–19. [Google Scholar] [CrossRef]
- Colalto, C. Volatile molecules for COVID-19: A possible pharmacological strategy? Drug Dev. Res. 2020, 81, 950–968. [Google Scholar] [CrossRef] [PubMed]
- Campioli, C.C.; Cevallos, E.C.; Assi, M.; Patel, R.; Binnicker, M.J.; O’Horo, J.C. Clinical predictors and timing of cessation of viral RNA shedding in patients with COVID-19. J. Clin. Virol. 2020, 130, 104577. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Li, X.; Huang, B.; Su, H.; Li, Y.; Luo, D.J.; Chen, S.; Ma, L.; Wang, S.H.; Nie, X.; et al. Pathological changes of fatal coronavirus disease 2019 (COVID-19) in the lungs: Report of 10 cases by post-mortem needle autopsy. Chin. J. Pathol. 2020, 49, 568–575. [Google Scholar] [CrossRef]
- Sisó-Almirall, A.; Kostov, B.; Mas-Heredia, M.; Vilanova-Rotllan, S.; Sequeira-Aymar, E.; Corrales, M.S.; Sant-Arderiu, E.; Cayuelas-Redondo, L.; Martínez-Pérez, A.; García-Plana, N.; et al. Prognostic factors in Spanish COVID-19 patients: A case series from Barcelona. PLoS ONE 2020, 15, e0237960. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.Y.; Mullakary, R.M.; Iyer, V.N. Successful treatment of coronavirus disease 2019 in a patient with asthma. Allergy Asthma Proc. 2020, 41, 296–300. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Number of Entities |
|---|---|
| Compound | 588,820 |
| Phenotype | 96,924 |
| Gene | 19,946 |
| Biological process | 12,313 |
| Enzyme class | 8077 |
| Gene Ontology (GO) | 6002 |
| Pathway | 2205 |
| Organism | 1419 |
| Tissue | 94 |
| Type | Number of Relationships |
|---|---|
| COMPOUND_GENE | 1,331,963 |
| GENE_DISEASE | 648,348 |
| COMPOUND_ADVERSE_EFFECT | 453,684 |
| GENE_GENE | 381,389 |
| IS_A_PHENOTYPE | 247,563 |
| GENE_BIOLOGICALPROCESS | 177,898 |
| GENE_CELLULARCOMPONENT | 117,323 |
| GENE_MOLECULARFUNCTION | 92,316 |
| GENE_TISSUE | 48,900 |
| PATHWAY_GENE | 40,632 |
| COMPOUND_INDICATION | 33,868 |
| INSTANCE_OF | 21,936 |
| IS_A_EC | 8069 |
| CHANGES_WITH | 6952 |
| REPURPOSED_INDICATION | 6609 |
| PATHWAY_COMPOUND | 5869 |
| PATHWAY_CELLULARCOMPONENT | 4608 |
| PART_OF | 3705 |
| GENE_EC | 2331 |
| PATHWAY_CONTAINS_PATHWAY | 2245 |
| CANONICAL_TARGET | 2080 |
| POSITIVELY_REGULATES | 1439 |
| NEGATIVELY_REGULATES | 1278 |
| REGULATES | 1199 |
| HAS_PART | 338 |
| OCCURS_IN | 111 |
| GENE_GO | 1 |
| Type | Number of Entities |
|---|---|
| Disease | 16,487 |
| Chemical | 8677 |
| Gene | 7080 |
| Species | 5596 |
| Protein mutation | 703 |
| Single nucleotide polymorphisms (SNPs) | 162 |
| DNA mutation | 155 |
| Cell line | 68 |
| Genus | 15 |
| Strain | 2 |
| Disease Name | Synonym |
|---|---|
| pneumocystis Carinii infection | pneumocystis infections Wegener’s granulomatosis |
| acute lymphoblastic leukemia | acute lymphocytic leukemia |
| adult respiratory distress syndrome | respiratory distress syndrome, adult malignant gliomas |
| bunyavirus infection | Bunyaviridae infections |
| breast cancer | breast carcinoma |
| thyroid cancer | thyroid neoplasm |
| Gene Name | Synonym |
|---|---|
| msg1 | cited1 |
| pla2s | pla2g2a |
| amyloid precursor protein | app caveolin 1 |
| bcl-w | bcl2l2 |
| ro52 | trim21 |
| timp | timp1 |
| dead box helicase 5 | ddx5 |
| aconitase 2 | aco2 |
| Type | Number of Entities |
|---|---|
| Compound | 588,820 |
| Phenotype | 94,251 |
| Gene | 21,761 |
| Biological process | 12,120 |
| Enzyme class | 8077 |
| GO | 5737 |
| Chemical | 4817 |
| Species | 3060 |
| Disease | 2565 |
| Pathway | 2201 |
| Organism | 1419 |
| Protein mutation | 678 |
| SNP | 162 |
| DNA mutation | 148 |
| Tissue | 94 |
| Cell line | 39 |
| Genus | 15 |
| Strain | 2 |
| Depth | Path | Score |
|---|---|---|
| 2 | Il-6_receptor--co_occur--ebola virus--co_occur--hydroxychloroquine | 0.271000 |
| 3 | il-6 receptor--co_occur--ebola virus--co_occur--chloroquine--co_occur--hydroxychloroquine | 0.983559 |
| 3 | il-6 receptor--co_occur--ebola virus--co_occur--quinoline--co_occur--hydroxychloroquine | 0.967936 |
| 3 | il-6 receptor--co_occur--ebola virus--co_occur--amodiaquine--co_occur--hydroxychloroquine | 0.964461 |
| 3 | il-6 receptor--co_occur--cd4--co_occur--chloroquine--co_occur--hydroxychloroquine | 0.902787 |
| 3 | il-6 receptor--co_occur--il17a--gene_disease--rheumatoid arthritis--co_occur--hydroxychloroquine | 0.895744 |
| 3 | il-6 receptor--co_occur--il17a--gene_disease--autoimmune diseases--co_occur--hydroxychloroquine | 0.887945 |
| 3 | il-6 receptor--co_occur--il10--gene_disease--autoimmune diseases--co_occur--hydroxychloroquine | 0.884826 |
| 3 | il-6 receptor--co_occur--il10--co_occur--autoimmune diseases--co_occur--hydroxychloroquine | 0.884826 |
| 3 | il-6 receptor--co_occur--cd83--gene_disease--autoimmune diseases--co_occur--hydroxychloroquine | 0.882492 |
| 3 | il-6 receptor--co_occur--ccr7--gene_disease--autoimmune diseases--co_occur--hydroxychloroquine | 0.860170 |
| 4 | il-6 receptor--co_occur--cd83--gene_gene--cd86--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.275014 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccr1--gene_disease--malaria--co_occur--hydroxychloroquine | 1.262865 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccr3--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.247009 |
| 4 | il-6 receptor--co_occur--ccr2--co_occur--cx3cr1--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.229658 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cxcr3--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.225782 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccr5--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.214904 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cxcr5--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.198998 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccr6--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.185817 |
| 4 | il-6 receptor--co_occur--gsto1--gene_gene--prdx2--gene_disease--malaria--co_occur--hydroxychloroquine | 1.177270 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccr3--gene_disease--malaria--co_occur--hydroxychloroquine | 1.169309 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cxcr3--gene_disease--malaria--co_occur--hydroxychloroquine | 1.151890 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccl7--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.149486 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cxcr1--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.149216 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cxcl10--gene_disease--malaria--co_occur--hydroxychloroquine | 1.144627 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccl22--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.138721 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cxcr2--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.136466 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccr7--gene_disease--malaria--co_occur--hydroxychloroquine | 1.126956 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cxcl8--gene_disease--malaria--co_occur--hydroxychloroquine | 1.123747 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccl20--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.119462 |
| 4 | il-6 receptor--co_occur--ccr2--co_occur--ccl2--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.119136 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccl2--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.119136 |
| 4 | il-6 receptor--co_occur--gsto1--gene_gene--gstk1--gene_disease--malaria--co_occur--hydroxychloroquine | 1.119127 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccl22--gene_disease--malaria--co_occur--hydroxychloroquine | 1.117001 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--ccl2--gene_disease--malaria--co_occur--hydroxychloroquine | 1.116415 |
| 4 | il-6 receptor--co_occur--ccr2--co_occur--ccl2--gene_disease--malaria--co_occur--hydroxychloroquine | 1.116415 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cx3cl1--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.092402 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cxcl12--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.092225 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cxcl10--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.086009 |
| 4 | il-6 receptor--co_occur--ccr2--gene_gene--cxcr6--gene_disease--hiv infections--co_occur--hydroxychloroquine | 1.081086 |
| Depth | Path | Score |
|---|---|---|
| 2 | stat1--co_occur--weight loss--co_occur--chloroquine | 0.700930 |
| 2 | stat1--co_occur--mice--co_occur--chloroquine | 0.593730 |
| 2 | stat1--co_occur--mnv--co_occur--chloroquine | 0.559861 |
| 3 | stat1--gene_gene--oasl--co_occur--eif2ak2--co_occur--chloroquine | 1.402100 |
| 3 | stat1--gene_gene--oasl--co_occur--eif2ak2--co_occur--chloroquine | 1.402100 |
| 3 | stat1--gene_gene--oas2--co_occur--oas1--co_occur--chloroquine | 1.347831 |
| 3 | stat1--gene_gene--oas2--co_occur--oas1--co_occur--chloroquine | 1.347831 |
| 3 | stat1--gene_gene--mx2--co_occur--eif2ak2--co_occur--chloroquine | 1.345132 |
| 3 | stat1--co_occur--mx2--co_occur--eif2ak2--co_occur--chloroquine | 1.345132 |
| 3 | stat1--gene_gene--mx2--co_occur--eif2ak2--co_occur--chloroquine | 1.345132 |
| 3 | stat1--gene_gene--mx2--gene_gene--eif2ak2--co_occur--chloroquine | 1.345132 |
| 3 | stat1--co_occur--mx2--gene_gene--eif2ak2--co_occur--chloroquine | 1.345132 |
| 3 | stat1--gene_gene--mx2--gene_gene--eif2ak2--co_occur--chloroquine | 1.345132 |
| 3 | stat1--co_occur--isg15--gene_gene--eif2ak2--co_occur--chloroquine | 1.267028 |
| 3 | stat1--gene_gene--isg15--gene_gene--eif2ak2--co_occur--chloroquine | 1.267028 |
| 3 | stat1--gene_gene--isg15--gene_gene--eif2ak2--co_occur--chloroquine | 1.267028 |
| 3 | stat1--co_occur--mx1--gene_gene--oas1--co_occur--chloroquine | 1.248431 |
| 3 | stat1--gene_gene--mx1--gene_gene--oas1--co_occur--chloroquine | 1.248431 |
| 3 | stat1--co_occur--mx1--co_occur--oas1--co_occur--chloroquine | 1.248431 |
| 3 | stat1--gene_gene--mx1--co_occur--oas1--co_occur--chloroquine | 1.248431 |
| 3 | stat1--gene_gene--mx1--co_occur--oas1--co_occur--chloroquine | 1.248431 |
| 3 | stat1--gene_gene--mx1--gene_gene--oas1--co_occur--chloroquine | 1.248431 |
| 3 | stat1--co_occur--jak1--co_occur--eif2ak2--co_occur--chloroquine | 1.242361 |
| 3 | stat1--gene_gene--jak1--gene_gene--eif2ak2--co_occur--chloroquine | 1.242361 |
| 3 | stat1--gene_gene--jak1--co_occur--eif2ak2--co_occur--chloroquine | 1.242361 |
| 3 | stat1--co_occur--jak1--gene_gene--eif2ak2--co_occur--chloroquine | 1.242361 |
| 3 | stat1--gene_gene--jak1--co_occur--eif2ak2--co_occur--chloroquine | 1.242361 |
| 3 | stat1--gene_gene--jak1--gene_gene--eif2ak2--co_occur--chloroquine | 1.242361 |
| 3 | stat1--gene_gene--mx1--gene_gene--eif2ak2--co_occur--chloroquine | 1.233766 |
| 3 | stat1--co_occur--mx1--co_occur--eif2ak2--co_occur--chloroquine | 1.233766 |
| 3 | stat1--gene_gene--mx1--co_occur--eif2ak2--co_occur--chloroquine | 1.233766 |
| 3 | stat1--gene_gene--mx1--gene_gene--eif2ak2--co_occur--chloroquine | 1.233766 |
| 3 | stat1--co_occur--mx1--gene_gene--eif2ak2--co_occur--chloroquine | 1.233766 |
| 3 | stat1--gene_gene--mx1--co_occur--eif2ak2--co_occur--chloroquine | 1.233766 |
| 3 | stat1--gene_disease--jc virus infection--co_occur--myalgia--co_occur--arthralgia--co_occur--chloroquine | 1.402100 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--dengue shock syndrome--co_occur--arthralgia--co_occur--chloroquine | 1.015049 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--oas2--co_occur--oas1--co_occur--chloroquine | 0.973442 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--hyperglycemia--co_occur--metformin--co_occur--chloroquine | 0.966955 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--mx2--gene_gene--eif2ak2--co_occur--chloroquine | 0.906810 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--mx2--co_occur--eif2ak2--co_occur--chloroquine | 0.878100 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--phenazopyridine--co_occur--monensin sodium--co_occur--chloroquine | 0.878100 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--alphavirus infections--co_occur--arthralgia--co_occur--chloroquine | 0.876126 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--a226v--co_occur--arthralgia--co_occur--chloroquine | 0.793003 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--empyema--co_occur--pneumonia--co_occur--chloroquine | 0.787724 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--pleural effusion--co_occur--pneumonia--co_occur--chloroquine | 0.777438 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--pneumococcal pneumonia--co_occur--pneumonia--co_occur--chloroquine | 0.739655 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--ly96--gene_gene--eif2ak2--co_occur--chloroquine | 0.736294 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--usp18--gene_gene--eif2ak2--co_occur--chloroquine | 0.710234 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--hypoxia--co_occur--arthralgia--co_occur--chloroquine | 0.708574 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--isg15--gene_gene--eif2ak2--co_occur--chloroquine | 0.707835 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--bronchiectasis--co_occur--bronchiolitis--co_occur--chloroquine | 0.702962 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--asthma--co_occur--bronchiolitis--co_occur--chloroquine | 0.697532 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--dhf--co_occur--arthralgia--co_occur--chloroquine | 0.694675 |
| 4 | stat1--gene_disease--jc virus infection--co_occur--socs1--co_occur--eif2ak2--co_occur--chloroquine | 0.692914 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Bu, Y.; Jiang, P.; Shi, X.; Lun, B.; Chen, C.; Syafiandini, A.F.; Ding, Y.; Song, M. Toward a Coronavirus Knowledge Graph. Genes 2021, 12, 998. https://doi.org/10.3390/genes12070998
Zhang P, Bu Y, Jiang P, Shi X, Lun B, Chen C, Syafiandini AF, Ding Y, Song M. Toward a Coronavirus Knowledge Graph. Genes. 2021; 12(7):998. https://doi.org/10.3390/genes12070998
Chicago/Turabian StyleZhang, Peng, Yi Bu, Peng Jiang, Xiaowen Shi, Bing Lun, Chongyan Chen, Arida Ferti Syafiandini, Ying Ding, and Min Song. 2021. "Toward a Coronavirus Knowledge Graph" Genes 12, no. 7: 998. https://doi.org/10.3390/genes12070998
APA StyleZhang, P., Bu, Y., Jiang, P., Shi, X., Lun, B., Chen, C., Syafiandini, A. F., Ding, Y., & Song, M. (2021). Toward a Coronavirus Knowledge Graph. Genes, 12(7), 998. https://doi.org/10.3390/genes12070998