
Deciphering the Role of Ferroptosis in the Pathogenesis of Peripheral Artery Disease Myopathy
 , , , , , ,
, , , , , , 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Muscle Biopsy
2.3. Myotube Cell Culture
2.4. RNA Isolation and qPCR
2.5. Protein Extraction and Western Blot
2.6. Statistical Analysis
3. Results
3.1. Patient Characteristics
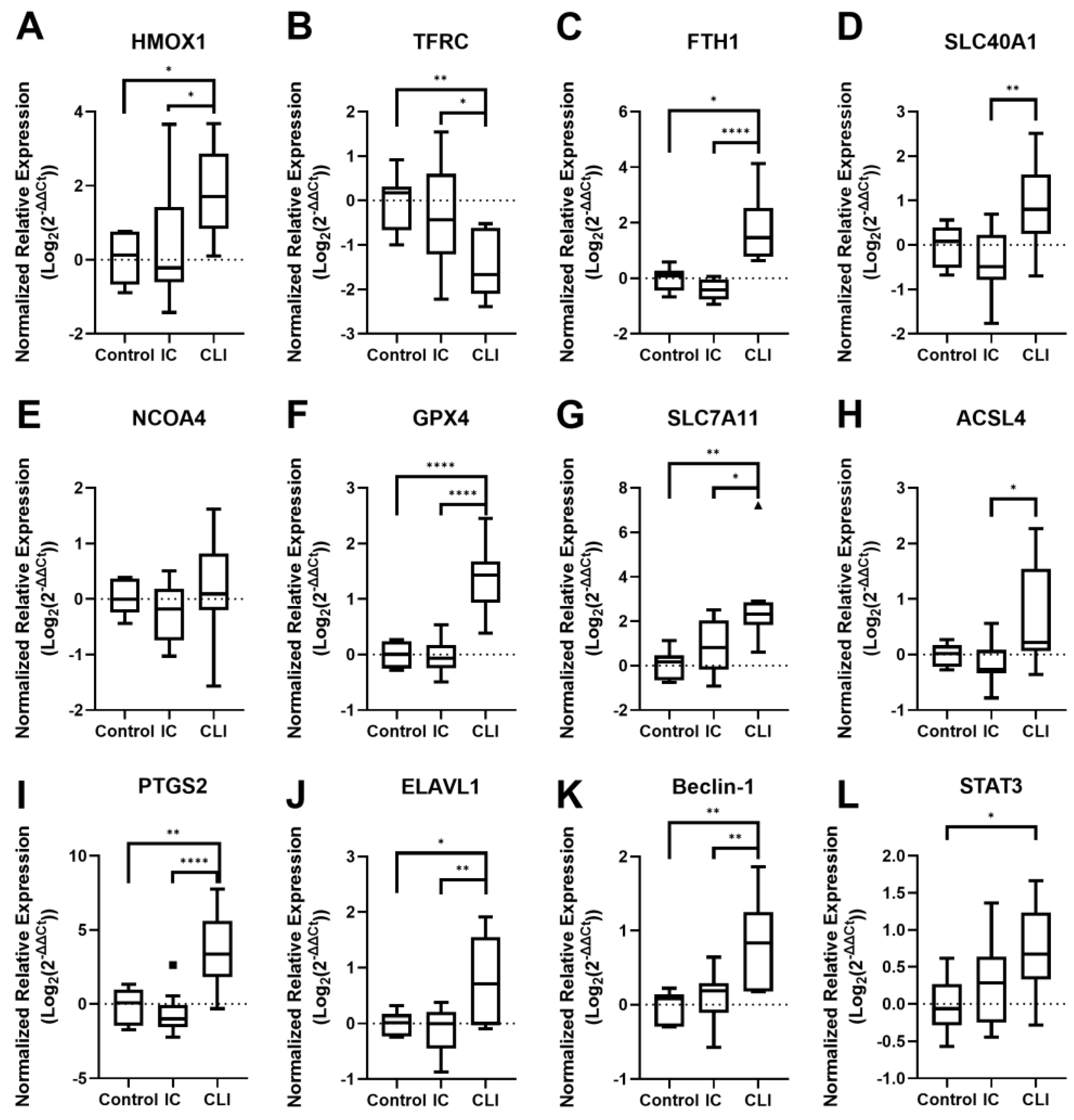
3.2. Ferroptosis Gene Expression
3.2.1. PAD Patient Muscle Tissue Gene Expression
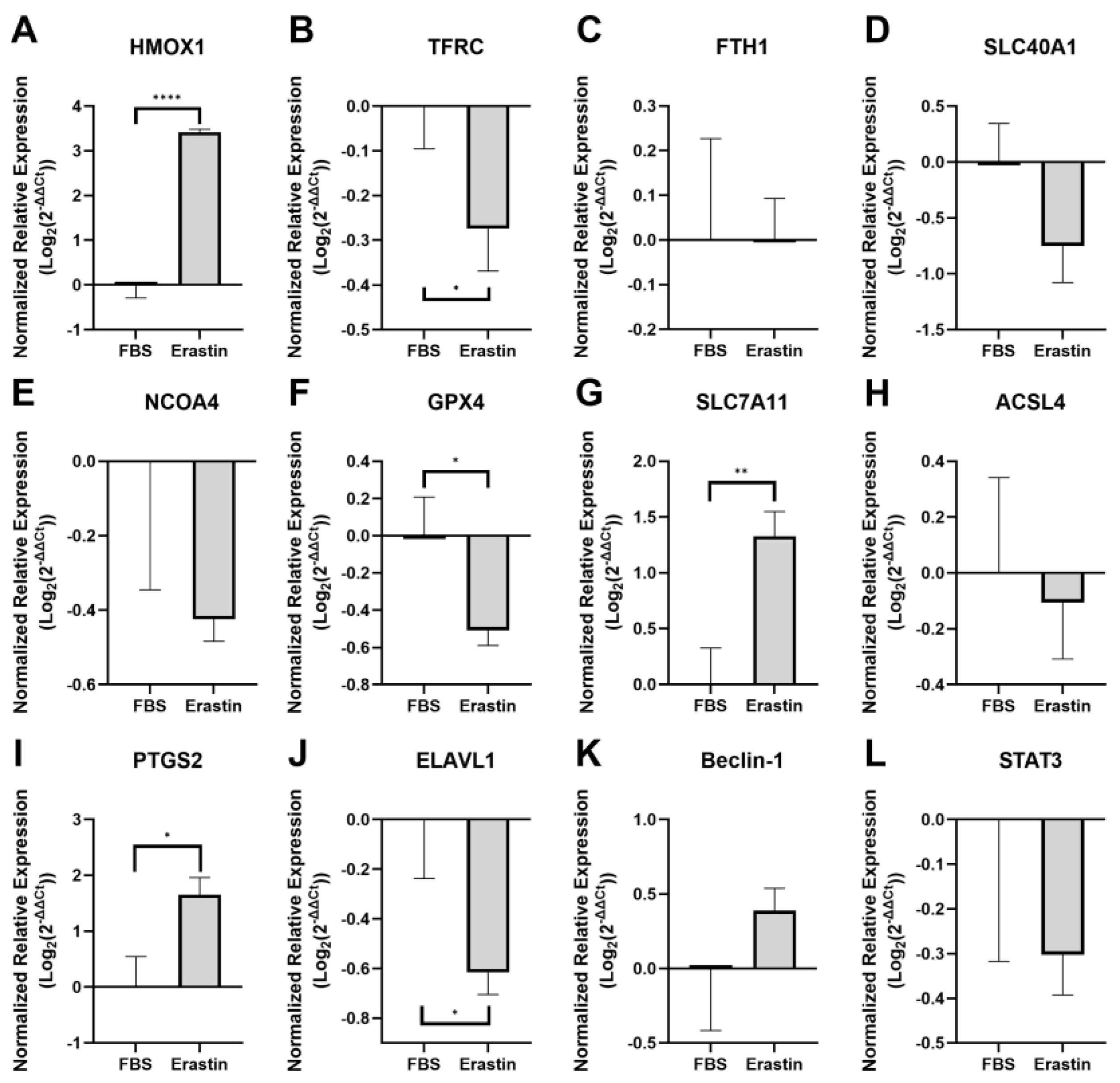
3.2.2. Myotube Gene Expression Following Ferroptosis Induction with Erastin
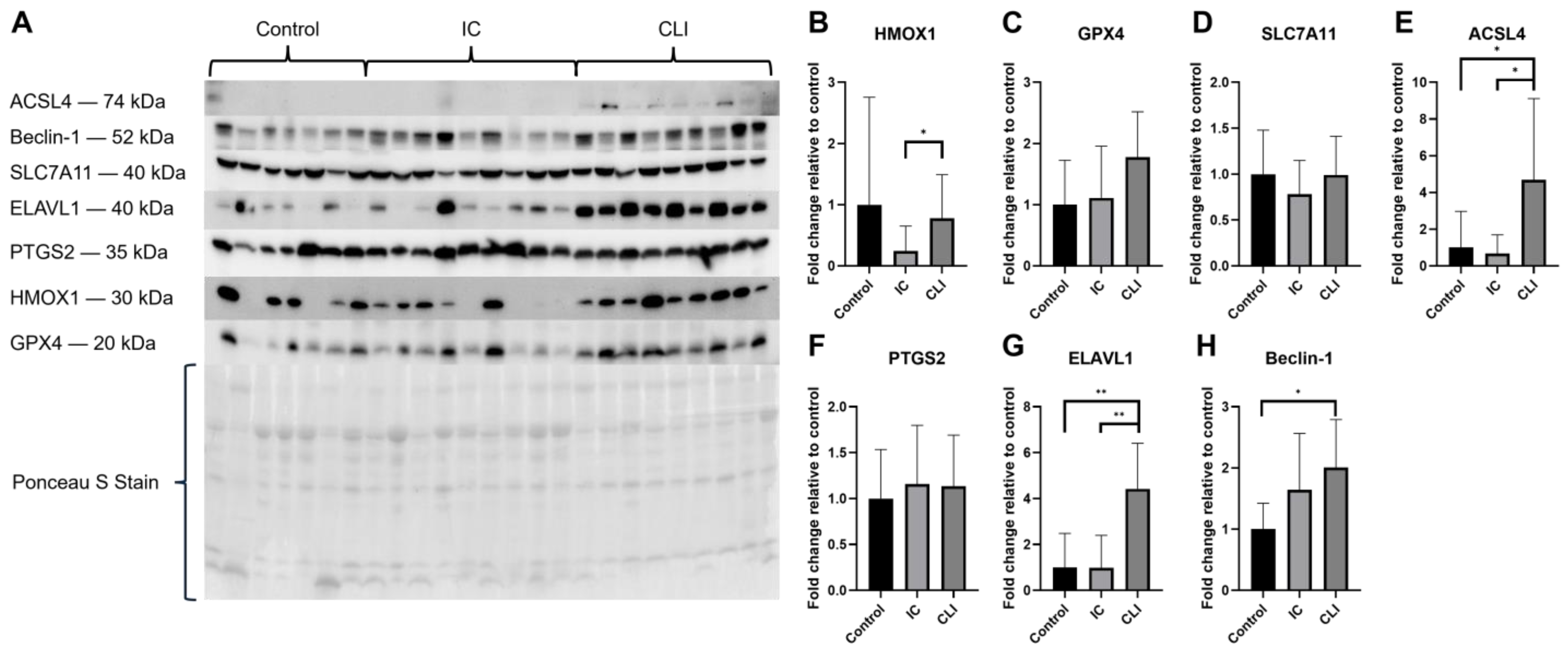
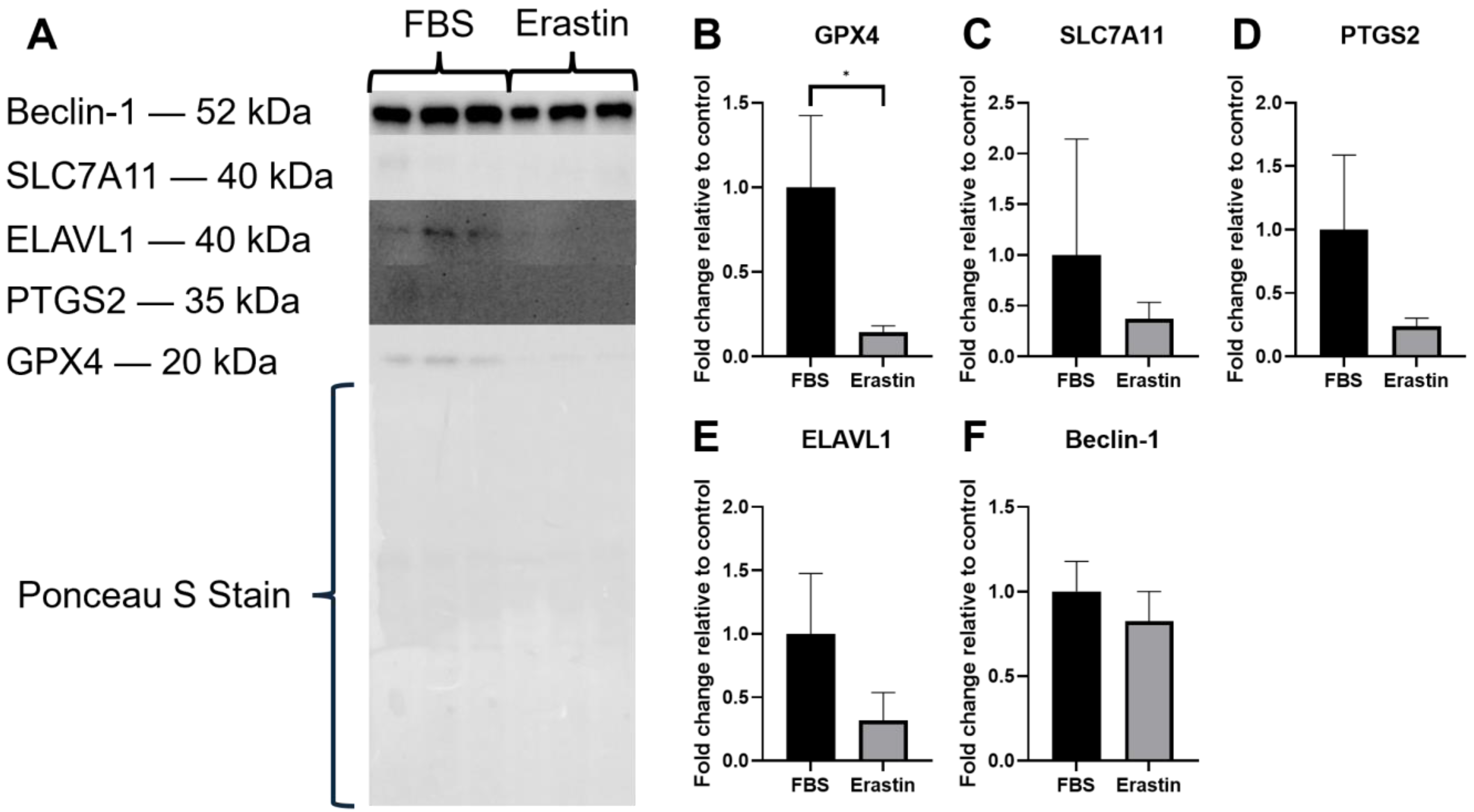
3.3. Protein Expression
3.3.1. PAD Patient Muscle Tissue Protein Expression
3.3.2. Erastin-Treated Myotubes Protein Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PAD | Peripheral artery disease |
| IC | Intermittent claudication |
| CLI | Critical limb ischemia |
| IRI | Ischemia/reperfusion injury |
| HMOX1 | Heme oxygenase 1 |
| TFRC | Transferrin receptor 1 |
| FTH1 | Ferritin heavy chain 1 |
| NCOA4 | Nuclear receptor coactivator 4 |
| FPN | Ferroportin |
| SLC40A1 | Solute carrier family 40 member 1 |
| GPX4 | Glutathione peroxidase 4 |
| SLC7A11 | Solute carrier family 7 member 11 |
| ACSL4 | Acyl-CoA synthetase long-chain family member 4 |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 |
| ELAVL1 | ELAV-like protein 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| ABI | Ankle brachial index |
| 4HNE | 4-Hydroxynonenal |
| LIP | Labile iron pool |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
References
- Criqui, M.H.; Matsushita, K.; Aboyans, V.; Hess, C.N.; Hicks, C.W.; Kwan, T.W.; McDermott, M.M.; Misra, S.; Ujueta, F.; Council on Arteriosclerosis; et al. Lower Extremity Peripheral Artery Disease: Contemporary Epidemiology, Management Gaps, and Future Directions: A Scientific Statement From the American Heart Association. Circulation 2021, 144, e171–e191. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.S.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Barone Gibbs, B.; Beaton, A.Z.; Boehme, A.K.; et al. 2024 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation 2024, 149, e347–e913. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Rudan, D.; Zhu, Y.; Fowkes, F.J.I.; Rahimi, K.; Fowkes, F.G.R.; Rudan, I. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: An updated systematic review and analysis. Lancet Glob. Health 2019, 7, e1020–e1030. [Google Scholar] [CrossRef]
- Steven, S.; Daiber, A.; Dopheide, J.F.; Munzel, T.; Espinola-Klein, C. Peripheral artery disease, redox signaling, oxidative stress—Basic and clinical aspects. Redox Biol. 2017, 12, 787–797. [Google Scholar] [CrossRef]
- Koutakis, P.; Weiss, D.J.; Miserlis, D.; Shostrom, V.K.; Papoutsi, E.; Ha, D.M.; Carpenter, L.A.; McComb, R.D.; Casale, G.P.; Pipinos, I.I. Oxidative damage in the gastrocnemius of patients with peripheral artery disease is myofiber type selective. Redox Biol. 2014, 2, 921–928. [Google Scholar] [CrossRef]
- Koutakis, P.; Ismaeel, A.; Farmer, P.; Purcell, S.; Smith, R.S.; Eidson, J.L.; Bohannon, W.T. Oxidative stress and antioxidant treatment in patients with peripheral artery disease. Physiol. Rep. 2018, 6, e13650. [Google Scholar] [CrossRef]
- Koutakis, P.; Hernandez, H.; Miserlis, D.; Thompson, J.R.; Papoutsi, E.; Mietus, C.J.; Haynatzki, G.; Kim, J.K.; Casale, G.P.; Pipinos, I.I. Oxidative damage in the gastrocnemius predicts long-term survival in patients with peripheral artery disease. NPJ Aging 2024, 10, 21. [Google Scholar] [CrossRef]
- Signorelli, S.S.; Katsiki, N. Oxidative Stress and Inflammation: Their Role in the Pathogenesis of Peripheral Artery Disease with or Without Type 2 Diabetes Mellitus. Curr. Vasc. Pharmacol. 2018, 16, 547–554. [Google Scholar] [CrossRef]
- Ismaeel, A.; McDermott, M.M.; Joshi, J.K.; Sturgis, J.C.; Zhang, D.; Ho, K.J.; Sufit, R.; Ferrucci, L.; Peterson, C.A.; Kosmac, K. Cocoa flavanols, Nrf2 activation, and oxidative stress in peripheral artery disease: Mechanistic findings in muscle based on outcomes from a randomized trial. Am. J. Physiol. Cell Physiol. 2024, 326, C589–C605. [Google Scholar] [CrossRef]
- Ismaeel, A.; Papoutsi, E.; Miserlis, D.; Lavado, R.; Haynatzki, G.; Casale, G.P.; Bohannon, W.T.; Smith, R.S.; Eidson, J.L.; Brumberg, R.; et al. The Nitric Oxide System in Peripheral Artery Disease: Connection with Oxidative Stress and Biopterins. Antioxidants 2020, 9, 590. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.M.; Carpenter, L.C.; Koutakis, P.; Swanson, S.A.; Zhu, Z.; Hanna, M.; DeSpiegelaere, H.K.; Pipinos, I.I.; Casale, G.P. Transforming growth factor-beta 1 produced by vascular smooth muscle cells predicts fibrosis in the gastrocnemius of patients with peripheral artery disease. J. Transl. Med. 2016, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Casale, G.P.; Thompson, J.R.; Carpenter, L.C.; Kim, J.; Lackner, T.J.; Mietus, C.J.; Ha, D.M.; Myers, S.A.; Brunette, K.E.; Li, S.; et al. Cytokine signature of inflammation mediated by autoreactive Th-cells, in calf muscle of claudicating patients with Fontaine stage II peripheral artery disease. Transl. Res. 2021, 228, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, S.S.; Anzaldi, M.; Fiore, V. Inflammation in peripheral arterial disease (PAD). Curr. Pharm. Des. 2012, 18, 4350–4357. [Google Scholar] [CrossRef]
- Gardner, A.W.; Parker, D.E.; Montgomery, P.S.; Blevins, S.M.; Teague, A.M.; Casanegra, A.I. Monitored daily ambulatory activity, inflammation, and oxidative stress in patients with claudication. Angiology 2014, 65, 491–496. [Google Scholar] [CrossRef]
- McDermott, M.M.; Ferrucci, L.; Gonzalez-Freire, M.; Kosmac, K.; Leeuwenburgh, C.; Peterson, C.A.; Saini, S.; Sufit, R. Skeletal Muscle Pathology in Peripheral Artery Disease: A Brief Review. Arter. Thromb. Vasc. Biol. 2020, 40, 2577–2585. [Google Scholar] [CrossRef]
- Koutakis, P.; Myers, S.A.; Cluff, K.; Ha, D.M.; Haynatzki, G.; Mccomb, R.D.; Uchida, K.; Miserlis, D.; Papoutsi, E.; Johanning, J.M.; et al. Abnormal myofiber morphology and limb dysfunction in claudication. J. Surg. Res. 2015, 196, 172–179. [Google Scholar] [CrossRef]
- Koutakis, P.; Miserlis, D.; Myers, S.A.; Kim, J.K.; Zhu, Z.; Papoutsi, E.; Swanson, S.A.; Haynatzki, G.; Ha, D.M.; Carpenter, L.A.; et al. Abnormal accumulation of desmin in gastrocnemius myofibers of patients with peripheral artery disease: Associations with altered myofiber morphology and density, mitochondrial dysfunction and impaired limb function. J. Histochem. Cytochem. 2015, 63, 256–269. [Google Scholar] [CrossRef]
- Sartipy, F.; Sigvant, B.; Lundin, F.; Wahlberg, E. Ten Year Mortality in Different Peripheral Arterial Disease Stages: A Population Based Observational Study on Outcome. Eur. J. Vasc. Endovasc. Surg. 2018, 55, 529–536. [Google Scholar] [CrossRef]
- Mueller, T.; Hinterreiter, F.; Luft, C.; Poelz, W.; Haltmayer, M.; Dieplinger, B. Mortality rates and mortality predictors in patients with symptomatic peripheral artery disease stratified according to age and diabetes. J. Vasc. Surg. 2014, 59, 1291–1299. [Google Scholar] [CrossRef]
- McDermott, M.M.; Liu, K.; Tian, L.; Guralnik, J.M.; Criqui, M.H.; Liao, Y.; Ferrucci, L. Calf muscle characteristics, strength measures, and mortality in peripheral arterial disease: A longitudinal study. J. Am. Coll. Cardiol. 2012, 59, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 2020, 160, 92–102. [Google Scholar] [CrossRef]
- Li, W.; Leng, Y.; Xiong, Y.; Xia, Z. Ferroptosis Is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury Through Endoplasmic Reticulum Stress. DNA Cell Biol. 2020, 39, 210–225. [Google Scholar] [CrossRef]
- Meng, Z.; Liang, H.; Zhao, J.; Gao, J.; Liu, C.; Ma, X.; Liu, J.; Liang, B.; Jiao, X.; Cao, J.; et al. HMOX1 upregulation promotes ferroptosis in diabetic atherosclerosis. Life Sci. 2021, 284, 119935. [Google Scholar] [CrossRef]
- Eshima, H.; Shahtout, J.L.; Siripoksup, P.; Pearson, M.J.; Mahmassani, Z.S.; Ferrara, P.J.; Lyons, A.W.; Maschek, J.A.; Peterlin, A.D.; Verkerke, A.R.P.; et al. Lipid hydroperoxides promote sarcopenia through carbonyl stress. Elife 2023, 12, e85289. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Li, C.; Deng, X.; Xie, X.; Liu, Y.; Friedmann Angeli, J.P.; Lai, L. Activation of Glutathione Peroxidase 4 as a Novel Anti-inflammatory Strategy. Front. Pharmacol. 2018, 9, 1120. [Google Scholar] [CrossRef]
- Tran, L.; Xie, B.; Assaf, E.; Ferrari, R.; Pipinos, I.I.; Casale, G.P.; Mota Alvidrez, R.I.; Watkins, S.; Sachdev, U. Transcriptomic Profiling Identifies Ferroptosis-Related Gene Signatures in Ischemic Muscle Satellite Cells Affected by Peripheral Artery Disease-Brief Report. Arter. Thromb. Vasc. Biol. 2023, 43, 2023–2029. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, G.A.; Narkowicz, C.; Lord, R.; Howard, B.J.; Chung, S. Effect of celecoxib on cyclooxygenase-2 expression and possible variants in a patient with Barrett’s esophagus. Dis. Esophagus 2007, 20, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, M.R.; Bicknell, L.S. The molecular genetics of nELAVL in brain development and disease. Eur. J. Hum. Genet. 2023, 31, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Pabis, M.; Popowicz, G.M.; Stehle, R.; Fernández-Ramos, D.; Asami, S.; Warner, L.; García-Mauriño, S.M.; Schlundt, A.; Martínez-Chantar, M.L.; Díaz-Moreno, I.; et al. HuR biological function involves RRM3-mediated dimerization and RNA binding by all three RRMs. Nucleic Acids Res. 2019, 47, 1011–1029. [Google Scholar] [CrossRef]
- Wu, D.; Hu, Q.; Wang, Y.; Jin, M.; Tao, Z.; Wan, J. Identification of HMOX1 as a Critical Ferroptosis-Related Gene in Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 833642. [Google Scholar] [CrossRef]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [CrossRef]
- Kobak, K.A.; Franczuk, P.; Schubert, J.; Dzięgała, M.; Kasztura, M.; Tkaczyszyn, M.; Drozd, M.; Kosiorek, A.; Kiczak, L.; Bania, J.; et al. Primary Human Cardiomyocytes and Cardiofibroblasts Treated with Sera from Myocarditis Patients Exhibit an Increased Iron Demand and Complex Changes in the Gene Expression. Cells 2021, 10, 818. [Google Scholar] [CrossRef]
- Malorni, W.; Testa, U.; Rainaldi, G.; Tritarelli, E.; Peschle, C. Oxidative stress leads to a rapid alteration of transferrin receptor intravesicular trafficking. Exp. Cell Res. 1998, 241, 102–116. [Google Scholar] [CrossRef]
- Lee, J.; Hyun, D.-H. The Interplay between Intracellular Iron Homeostasis and Neuroinflammation in Neurodegenerative Diseases. Antioxidants 2023, 12, 918. [Google Scholar] [CrossRef]
- Chen, A.C.; Donovan, A.; Ned-Sykes, R.; Andrews, N.C. Noncanonical role of transferrin receptor 1 is essential for intestinal homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 11714–11719. [Google Scholar] [CrossRef]
- Ding, H.; Chen, S.; Pan, X.; Dai, X.; Pan, G.; Li, Z.; Mai, X.; Tian, Y.; Zhang, S.; Liu, B.; et al. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J. Cachexia Sarcopenia Muscle 2021, 12, 746–768. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.M.; Hoff, F.; Ferrucci, L.; Pearce, W.H.; Guralnik, J.M.; Tian, L.; Liu, K.; Schneider, J.R.; Sharma, L.; Tan, J.; et al. Lower extremity ischemia, calf skeletal muscle characteristics, and functional impairment in peripheral arterial disease. J. Am. Geriatr. Soc. 2007, 55, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Sugai, T.; Watanabe, T.; Otaki, Y.; Goto, J.; Watanabe, K.; Toshima, T.; Takahashi, T.; Yokoyama, M.; Tamura, H.; Nishiyama, S.; et al. Decreased Psoas Muscle Computed Tomography Value Predicts Poor Outcome in Peripheral Artery Disease. Circ. J. 2018, 82, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.; Miserlis, D.; Papoutsi, E.; Steiner, J.L.; Gordon, B.; Haynatzki, G.; Pacher, P.; Koutakis, P. Chronic alcohol consumption exacerbates ischemia-associated skeletal muscle mitochondrial dysfunction in a murine model of peripheral artery disease. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167584. [Google Scholar] [CrossRef]
- Fletcher, E.; Miserlis, D.; Sorokolet, K.; Wilburn, D.; Bradley, C.; Papoutsi, E.; Wilkinson, T.; Ring, A.; Ferrer, L.; Haynatzki, G.; et al. Diet-induced obesity augments ischemic myopathy and functional decline in a murine model of peripheral artery disease. Transl. Res. J. Lab. Clin. Med. 2023, 260, 17–31. [Google Scholar] [CrossRef]
- Meex, R.C.R.; Blaak, E.E.; van Loon, L.J.C. Lipotoxicity plays a key role in the development of both insulin resistance and muscle atrophy in patients with type 2 diabetes. Obes. Rev. 2019, 20, 1205–1217. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Coornaert, I.; Breynaert, A.; Hermans, N.; De Meyer, G.R.Y.; Martinet, W. GPX4 overexpression does not alter atherosclerotic plaque development in ApoE knock-out mice. Vasc. Biol. 2024, 6, e230020. [Google Scholar] [CrossRef]
- Kanaan, M.N.; Pileggi, C.A.; Karam, C.Y.; Kennedy, L.S.; Fong-McMaster, C.; Cuperlovic-Culf, M.; Harper, M.E. Cystine/glutamate antiporter xCT controls skeletal muscle glutathione redox, bioenergetics and differentiation. Redox Biol. 2024, 73, 103213. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Klionsky, D.J.; Kroemer, G.; Lotze, M.T.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X. Curr. Biol. 2018, 28, 2388–2399.e2385. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Ouyang, S.; Li, H.; Lou, L.; Huang, Q.; Zhang, Z.; Mo, J.; Li, M.; Lu, J.; Zhu, K.; Chu, Y.; et al. Inhibition of STAT3-ferroptosis negative regulatory axis suppresses tumor growth and alleviates chemoresistance in gastric cancer. Redox Biol. 2022, 52, 102317. [Google Scholar] [CrossRef]
- Zhang, W.; Gong, M.; Mo, J.; Zhang, S.; Zhu, Z.; Wang, X.; Zhang, B.; Qian, W.; Wu, Z.; Ma, Q.; et al. Thiostrepton induces ferroptosis in pancreatic cancer cells through STAT3/GPX4 signalling. Cell Death Dis. 2022, 13, 630. [Google Scholar] [CrossRef]
- Galbraith, R.A.; Sassa, S.; Kappas, A. Heme binding to murine erythroleukemia cells. Evidence for a heme receptor. J. Biol. Chem. 1985, 260, 12198–12202. [Google Scholar] [CrossRef]
- Belcher, J.D.; Beckman, J.D.; Balla, G.; Balla, J.; Vercellotti, G. Heme degradation and vascular injury. Antioxid. Redox Signal 2010, 12, 233–248. [Google Scholar] [CrossRef]
- Gáll, T.; Balla, G.; Balla, J. Heme, Heme Oxygenase, and Endoplasmic Reticulum Stress-A New Insight into the Pathophysiology of Vascular Diseases. Int. J. Mol. Sci. 2019, 20, 3675. [Google Scholar] [CrossRef]
- Scrivner, O.; Fletcher, E.; Hoffmann, C.; Li, F.; Wilkinson, T.; Miserlis, D.; Smith, R.S.; Bohannon, W.T.; Sutliff, R.; Jordan, W.D.; et al. Myoglobinemia, Peripheral Arterial Disease, and Patient Mortality. J. Am. Coll. Surg. 2023, 236, 588–598. [Google Scholar] [CrossRef]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | IC | CLI | p-Value | |
|---|---|---|---|---|
| Age, y | 69.1 ± 3.4 | 67.1 ± 8.1 | 63.3 ± 5.6 | 0.208 |
| Sex, No. (%) | 0.018 | |||
| Male | 7 (100) | 10 (100) | 6 (60) | |
| Race, No. (%) | <0.001 | |||
| White | 7 (100) | 10 (100) | 1 (10) | |
| Black | 0 (0) | 0 (0) | 3 (30) | |
| Hispanic | 0 (0) | 0 (0) | 6 (60) | |
| Comorbidities, No. (%) | ||||
| Coronary Artery Disease | 0 (0) | 3 (30) | 3 (30) | 0.259 |
| Hypertension | 7 (100) | 7 (70) | 6 (60) | 0.168 |
| Dyslipidemia | 5 (71) | 8 (80) | 6 (60) | 0.617 |
| Diabetes | 1 (14) | 2 (20) | 9 (90) | 0.001 |
| Smoking status, No. (%) | <0.001 | |||
| Never smoked | 3 (43) | 0 (0) | 8 (80) | |
| Current smokers | 1 (14) | 8 (80) | 1 (10) | |
| Former smokers | 3 (43) | 2 (20) | 1 (10) | |
| ABI | 1.10 ± 0.18 | 0.69 ± 0.26 * | 0.18 ± 0.21 *,# | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilkinson, T.; Fletcher, E.; Ring, A.; Bradley, C.; Papoutsi, E.; Miserlis, D.; Smith, R.S.; Bohannon, W.T.; Pipinos, I.I.; Koutakis, P. Deciphering the Role of Ferroptosis in the Pathogenesis of Peripheral Artery Disease Myopathy. Biology 2025, 14, 537. https://doi.org/10.3390/biology14050537
Wilkinson T, Fletcher E, Ring A, Bradley C, Papoutsi E, Miserlis D, Smith RS, Bohannon WT, Pipinos II, Koutakis P. Deciphering the Role of Ferroptosis in the Pathogenesis of Peripheral Artery Disease Myopathy. Biology. 2025; 14(5):537. https://doi.org/10.3390/biology14050537
Chicago/Turabian StyleWilkinson, Trevor, Emma Fletcher, Andrew Ring, Cassandra Bradley, Evlampia Papoutsi, Dimitrios Miserlis, Robert S. Smith, William T. Bohannon, Iraklis I. Pipinos, and Panagiotis Koutakis. 2025. "Deciphering the Role of Ferroptosis in the Pathogenesis of Peripheral Artery Disease Myopathy" Biology 14, no. 5: 537. https://doi.org/10.3390/biology14050537
APA StyleWilkinson, T., Fletcher, E., Ring, A., Bradley, C., Papoutsi, E., Miserlis, D., Smith, R. S., Bohannon, W. T., Pipinos, I. I., & Koutakis, P. (2025). Deciphering the Role of Ferroptosis in the Pathogenesis of Peripheral Artery Disease Myopathy. Biology, 14(5), 537. https://doi.org/10.3390/biology14050537