Lysosomal Storage Diseases
A topical collection in Diseases (ISSN 2079-9721). This collection belongs to the section "Rare Syndrome".
Viewed by 122989
Share This Topical Collection
Editors
 Dr. Jose Sanchez-Alcazar
Dr. Jose Sanchez-Alcazar
Dr. Jose Sanchez-Alcazar
E-Mail
Website
Collection Editor
Centro Andaluz de Biología del Desarrollo (CABD-CSIC-Universidad Pablo de Olavide), 41013 Sevilla, Spain
Interests: rare diseases; mitochondrial diseases; lysosome diseases; neurodegeneration with brain iron accumulation; neurodevelopmental disorders; congenital myopathies; genetic susceptibility to cancer
Special Issues, Collections and Topics in MDPI journals
 Dr. Luis M. Jiménez Jiménez
Dr. Luis M. Jiménez Jiménez
Dr. Luis M. Jiménez Jiménez
E-Mail
Website
Collection Editor
Servicio de Fisiopatología Celular y Bioenergética, Departamento de Fisiología, Anatomía y Biología Celular, Universidad Pablo de Olavide, Sevilla, Spain
Interests: rare diseases; lysosome diseases; oxidative stress
Topical Collection Information
Dear Colleagues,
Lysosomal storage diseases (LSDs) comprise a group of over 50 genetically inherited disorders that are characterized by accumulation of undigested material inside the lysosome, most often caused by a deficiency of the enzyme normally responsible for catabolism of various molecules derived from cellular turnover. This condition results in accumulation of the undegraded substrate in various tissues and organs of the body causing these organs to function less efficiently, resulting in progressive deterioration in physical and/or mental state, and eventually cell degeneration.
This Topical Collection provides an open access opportunity for investigators to contribute with original research articles as well as review articles that will allow a better understanding of the biochemical and molecular physiopathology underlying LSDs, potential biomarkers of disease progression and new therapeutic strategies to treat these diseases, and evaluation of treatment efficiency.
Potential topics include, but are not limited to:
- Recent progress in LSDs research
- Diagnosis of LSDs
- Technological developments in newborn and population screening
- New pathophysiological mechanisms involved in LSDs
- Biomarkers in disease staging, monitoring, and efficiency of treatment
- Recent advances in treatment and drug delivery
Prof. Dr. Jose A. Sanchez-Alcazar
Dr. Luis M. Jiménez Jiménez
Collection Editors
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 250 words) can be sent to the Editorial Office for assessment.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Diseases is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 1800 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- lysosomal diseases
- physiopathology
- diagnosis
- biomarkers
- treatment
- screening
- autophagy
Published Papers (16 papers)
Open AccessArticle
The Effect of Fabry Disease Therapy on Bone Mineral Density
by
Tess Aitken, Mark K. Tiong, Andrew S. Talbot, Irene Ruderman and Kathleen M. Nicholls
Cited by 2 | Viewed by 2474
Abstract
Fabry disease (FD) is an X-linked lysosomal storage disorder, characterised by the cellular accumulation of globotriaosylceramide due to impaired alpha-galactosidase A enzyme activity. FD may manifest with multisystem pathology, including reduced bone mineral density (BMD). Registry data suggest that the introduction of Fabry-specific
[...] Read more.
Fabry disease (FD) is an X-linked lysosomal storage disorder, characterised by the cellular accumulation of globotriaosylceramide due to impaired alpha-galactosidase A enzyme activity. FD may manifest with multisystem pathology, including reduced bone mineral density (BMD). Registry data suggest that the introduction of Fabry-specific therapies (enzyme replacement therapy or chaperone therapy) has led to significant improvements in overall patient outcomes; however, there are limited data on the impact on bone density. The aim of this study was to describe the effect of Fabry-specific therapies on longitudinal changes in bone mineral density (BMD) in FD. We performed a retrospective observational study analysing bone densitometry (DXA) in patients with genetically confirmed FD. Patients were grouped based on the use of Fabry-specific therapies. The between-group longitudinal change in BMD Z-score was analysed using linear mixed effects models. A total of 88 FD patients were analysed (50 untreated; 38 treated). The mean age at first DXA was 38.5 years in the untreated group (84% female) and 43.7 years in the treated group (34% female). There was no significant longitudinal between-group difference in the BMD Z-score at the lumbar spine. However, the Z-score per year at the total hip (β = −0.105,
p < 0.001) and femoral neck (β = −0.081,
p = 0.001) was significantly lower over time in the treated than the untreated group. This may reflect those receiving therapy having a more severe underlying disease. Nevertheless, this suggests that Fabry-specific therapies do not reverse all disease mechanisms and that the additional management of BMD may be required in this patient population.
Full article
►▼
Show Figures
Open AccessCase Report
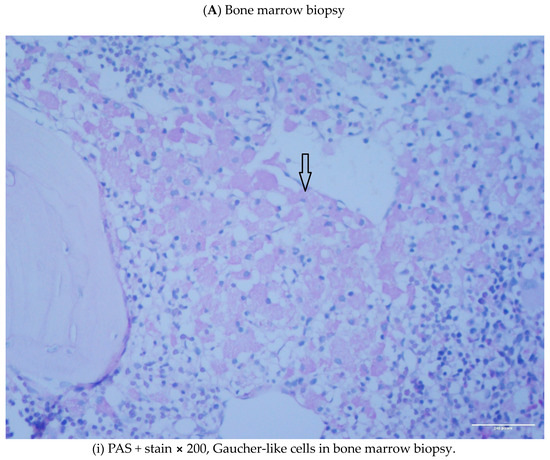
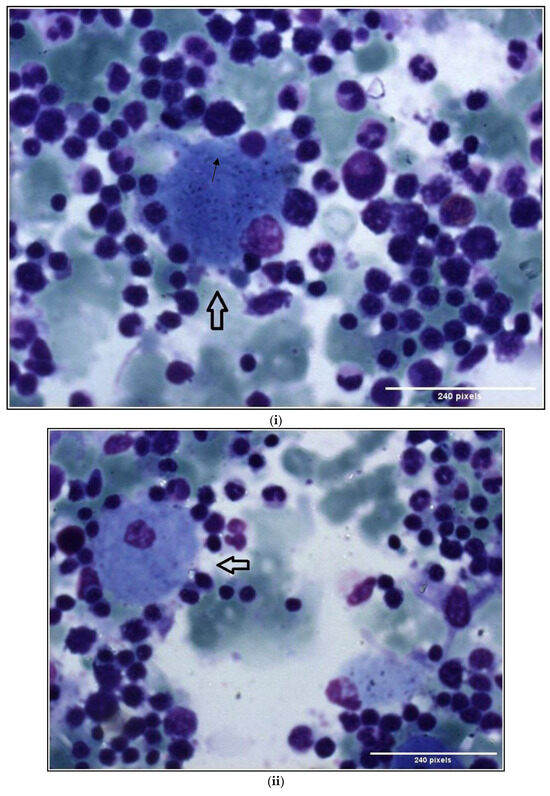
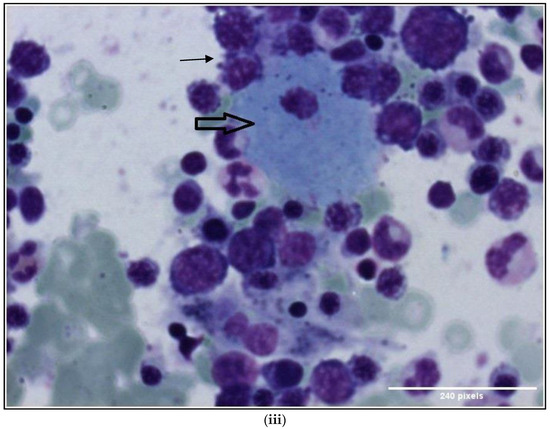
Gaucher-like Cells in Thalassemia Intermedia: Is It a Challenge?
by
Veroniki Komninaka, Pagona Flevari, Georgios Karkaletsis, Theodoros Androutsakos, Theofili Karkaletsi, Ioannis Ntanasis-Stathopoulos, Evaggelia-Eleni Ntelaki and Evangelos Terpos
Cited by 1 | Viewed by 4104
Abstract
We describe two cases of thalassemia intermedia (TI) patients with the presence of Gaucher-like cells in hematopoietic tissue biopsies, raising diagnostic dilemmas. The first is a 56-year-old female with bone lesions, splenomegaly, hypochromic microcytic anemia and Gaucher-like cells in the bone marrow, with
[...] Read more.
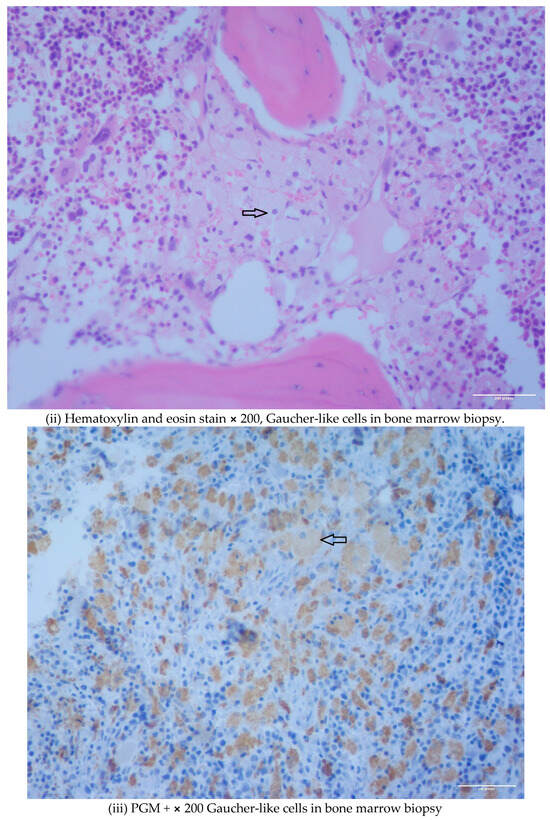
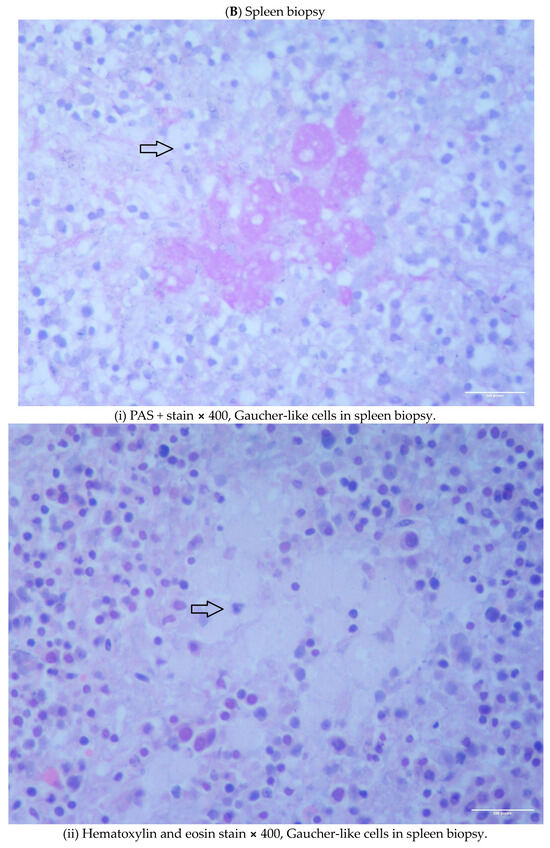
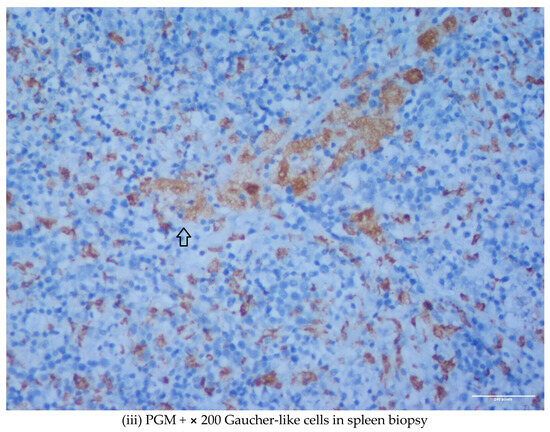
We describe two cases of thalassemia intermedia (TI) patients with the presence of Gaucher-like cells in hematopoietic tissue biopsies, raising diagnostic dilemmas. The first is a 56-year-old female with bone lesions, splenomegaly, hypochromic microcytic anemia and Gaucher-like cells in the bone marrow, with a final diagnosis of TI, and the second is a 69-year-old male with TI, monoclonal gammopathy of undetermined significance (MGUS) that accelerated to multiple myeloma (MM) requiring treatment, bone disease and Gaucher-like cells in the bone marrow and the spleen, and heterozygoty of Gaucher disease (GD). Gaucher-like cells are difficult to differentiate from true Gaucher cells, that are the hallmark of GD suspicion. These cells are usually reported in the lymphohematopoietic system. They have been described in myeloproliferative disorders, hematological malignancies, infectious diseases, hemoglobinopathies and other hemolytic anemias. The presence of Gaucher-like cells in patients with thalassemia major has been well documented, whereas there are limited references regarding cases with thalassemia intermedia. The identification of these cells in thalassemia probably reflects the high cell turnover. The bony complications in GD and TIare not yet fully explained in the literature, and this raises the question of whether Gaucher-like cells could play a pathogenetic role in the bone disease of thalassemia, as Gaucher cells are considered to play a similar role in bone complications of GD. Moreover, given the rarity and similarity of Gaucher and Gaucher-like cells, we would like to highlight that the presence of Gaucher-like cells in the bone marrow should not be overlooked, as they might be obscuring an underlying pathology, in order to ensure that hematologists, internists and hematopathologists will be promptly and accurately diagnosed.
Full article
►▼
Show Figures
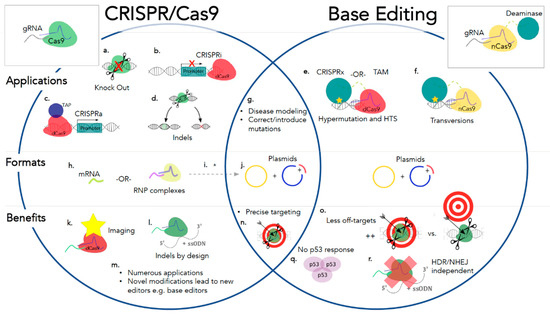
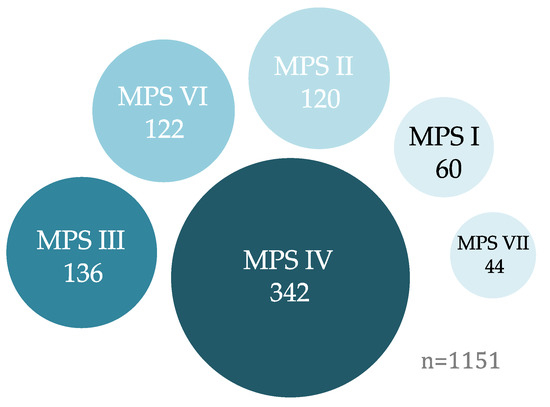
Open AccessReview
Cell and Gene Therapies for Mucopolysaccharidoses: Base Editing and Therapeutic Delivery to the CNS
by
Chloe L. Christensen, Rhea E. Ashmead and Francis Y. M. Choy
Cited by 12 | Viewed by 7386
Abstract
Although individually uncommon, rare diseases collectively account for a considerable proportion of disease impact worldwide. A group of rare genetic diseases called the mucopolysaccharidoses (MPSs) are characterized by accumulation of partially degraded glycosaminoglycans cellularly. MPS results in varied systemic symptoms and in some
[...] Read more.
Although individually uncommon, rare diseases collectively account for a considerable proportion of disease impact worldwide. A group of rare genetic diseases called the mucopolysaccharidoses (MPSs) are characterized by accumulation of partially degraded glycosaminoglycans cellularly. MPS results in varied systemic symptoms and in some forms of the disease, neurodegeneration. Lack of treatment options for MPS with neurological involvement necessitates new avenues of therapeutic investigation. Cell and gene therapies provide putative alternatives and when coupled with genome editing technologies may provide long term or curative treatment. Clustered regularly interspaced short palindromic repeats (CRISPR)-based genome editing technology and, more recently, advances in genome editing research, have allowed for the addition of base editors to the repertoire of CRISPR-based editing tools. The latest versions of base editors are highly efficient on-targeting deoxyribonucleic acid (DNA) editors. Here, we describe a number of putative guide ribonucleic acid (RNA) designs for precision correction of known causative mutations for 10 of the MPSs. In this review, we discuss advances in base editing technologies and current techniques for delivery of cell and gene therapies to the site of global degeneration in patients with severe neurological forms of MPS, the central nervous system, including ultrasound-mediated blood-brain barrier disruption.
Full article
►▼
Show Figures
Open AccessFeature PaperReview
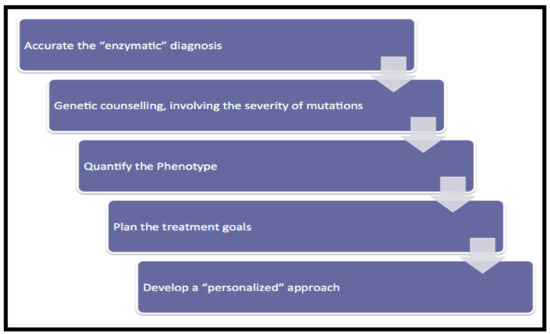
The Importance of a Multidisciplinary Approach in the Management of a Patient with Type I Gaucher Disease
by
Miguel-Ángel Torralba-Cabeza, Susana Olivera-González and José-Luis Sierra-Monzón
Cited by 9 | Viewed by 6596
Abstract
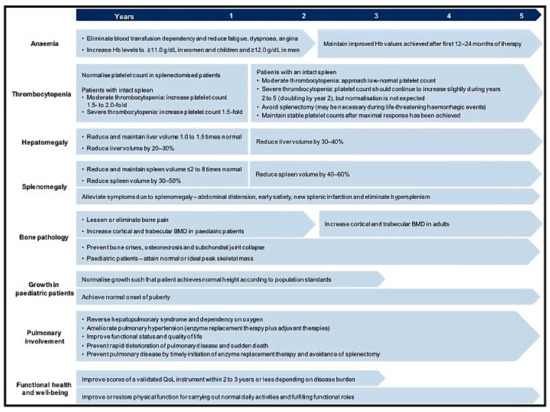
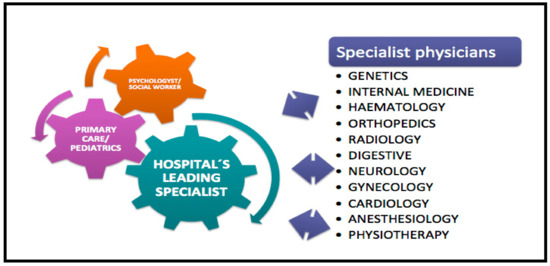
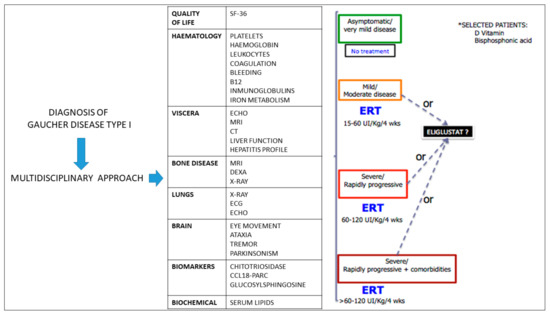
Managing the multisystemic symptoms of type I Gaucher Disease (GD) requires a multidisciplinary team approach that includes disease-specific treatments, as well as supportive care. This involves a range of medical specialists, general practitioners, supportive care providers, and patients. Phenotype classification and the setting
[...] Read more.
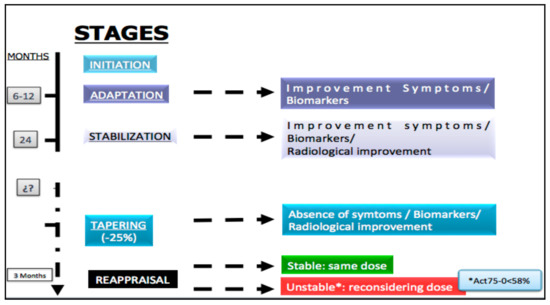
Managing the multisystemic symptoms of type I Gaucher Disease (GD) requires a multidisciplinary team approach that includes disease-specific treatments, as well as supportive care. This involves a range of medical specialists, general practitioners, supportive care providers, and patients. Phenotype classification and the setting of treatment goals are important for optimizing the management of type I GD, and for providing personalized care. The ability to classify disease severity using validated measurement tools allows the standardization of patient monitoring, and the measurement of disease progression and treatment response. Defining treatment goals is useful to provide a benchmark for assessing treatment response and managing the expectations of patients and their families. Although treatment goals will vary depending on disease severity, they include the stabilization, improvement or reversal (if possible) of clinical manifestations. Enzyme replacement therapy (ERT) is the standard care for patients with type I GD, but a novel substrate reduction therapy (SRT), Eliglustat, has demonstrated safety and efficacy in selected patients. To ensure that treatment goals are being achieved, regular and comprehensive follow up are necessary.
Full article
►▼
Show Figures
Open AccessCase Report
Role of Handheld In Vivo Reflectance Confocal Microscopy for the Diagnosis of Fabry Disease: A Case Report
by
Elisa Cinotti, Luca Provvidenziale, Michele Fimiani, Jean Luc Perrot, Frederic Cambazard and Pietro Rubegni
Cited by 1 | Viewed by 4750
Abstract
Fabry disease (FD) is a rare X-linked lysosomal storage disorder caused by the deficient activity of the lysosomal enzyme α-galactosidase that leads to a systemic accumulation of globotriaosylceramide. Handheld in vivo reflectance confocal microscopy (HH-RCM) is a useful modern technique in diagnosis and
[...] Read more.
Fabry disease (FD) is a rare X-linked lysosomal storage disorder caused by the deficient activity of the lysosomal enzyme α-galactosidase that leads to a systemic accumulation of globotriaosylceramide. Handheld in vivo reflectance confocal microscopy (HH-RCM) is a useful modern technique in diagnosis and follow-ups of many skin diseases. This noninvasive device provides high-resolution and high-contrast real-time images to study both the skin and the ocular surface structures that can help clinicians to confirm the diagnosis of FD. HH-RCM could be helpful even for the follow-ups of these patients, enabling us to monitor the effect of enzyme replacement therapy on corneal cells and keratinocytes.
Full article
►▼
Show Figures
Open AccessReview
Biomarkers and Imaging Findings of Anderson–Fabry Disease—What We Know Now
by
Idalina Beirão, Ana Cabrita, Márcia Torres, Fernando Silva, Patrício Aguiar, Francisco Laranjeira and Ana Marta Gomes
Cited by 19 | Viewed by 7679
Abstract
Anderson–Fabry disease (AFD) is an X-linked lysosomal storage disorder, caused by deficiency or absence of the alpha-galactosidase A activity, with a consequent glycosphingolipid accumulation. Biomarkers and imaging findings may be useful for diagnosis, identification of an organ involvement, therapy monitoring and prognosis. The
[...] Read more.
Anderson–Fabry disease (AFD) is an X-linked lysosomal storage disorder, caused by deficiency or absence of the alpha-galactosidase A activity, with a consequent glycosphingolipid accumulation. Biomarkers and imaging findings may be useful for diagnosis, identification of an organ involvement, therapy monitoring and prognosis. The aim of this article is to review the current available literature on biomarkers and imaging findings of AFD patients.
An extensive bibliographic review from PubMed, Medline and Clinical Key databases was performed by a group of experts from nephrology, neurology, genetics, cardiology and internal medicine, aiming for consensus. Lyso-GB3 is a valuable biomarker to establish the diagnosis. Proteinuria and creatinine are the most valuable to detect renal damage. Troponin I and high-sensitivity assays for cardiac troponin T can identify patients with cardiac lesions, but new techniques of cardiac imaging are essential to detect incipient damage. Specific cerebrovascular imaging findings are present in AFD patients. Techniques as metabolomics and proteomics have been developed in order to find an AFD fingerprint. Lyso-GB3 is important for evaluating the pathogenic mutations and monitoring the response to treatment. Many biomarkers can detect renal, cardiac and cerebrovascular involvement, but none of these have proved to be important to monitoring the response to treatment. Imaging features are preferred in order to find cardiac and cerebrovascular compromise in AFD patients.
Full article
Open AccessFeature PaperReview
The Spectrum of Neurological Manifestations Associated with Gaucher Disease
by
Tamanna Roshan Lal and Ellen Sidransky
Cited by 68 | Viewed by 12077
Abstract
Gaucher disease, the most common lysosomal storage disorder, is due to a deficiency in the enzyme glucocerebrosidase. This leads to the accumulation of its normal substrate, glucocerebroside, in tissue macrophages, affecting the hematological, visceral, bone and neurologic systems. Gaucher disease is classified into
[...] Read more.
Gaucher disease, the most common lysosomal storage disorder, is due to a deficiency in the enzyme glucocerebrosidase. This leads to the accumulation of its normal substrate, glucocerebroside, in tissue macrophages, affecting the hematological, visceral, bone and neurologic systems. Gaucher disease is classified into three broad phenotypes based upon the presence or absence of neurological involvement: type 1 (non-neuronopathic), type 2 (acute neuronopathic), and type 3 (subacute neuronopathic). Phenotypically, there is a wide spectrum of visceral and neurological manifestations. Enzyme replacement is effective in managing the visceral disease; however, treating the neurological manifestations has proved to be more challenging. This review discusses the various neurological manifestations encountered in Gaucher disease, and provides a brief overview regarding the treatment and ongoing research challenges.
Full article
Open AccessReview
Lysosomal Storage Disorders and Malignancy
by
Gregory M. Pastores and Derralynn A. Hughes
Cited by 13 | Viewed by 7034
Abstract
Lysosomal storage disorders (LSDs) are infrequent to rare conditions caused by mutations that lead to a disruption in the usual sequential degradation of macromolecules or their transit within the cell. Gaucher disease (GD), a lipidosis, is among the most common LSD, with an
[...] Read more.
Lysosomal storage disorders (LSDs) are infrequent to rare conditions caused by mutations that lead to a disruption in the usual sequential degradation of macromolecules or their transit within the cell. Gaucher disease (GD), a lipidosis, is among the most common LSD, with an estimated incidence of 1 in 40,000 among the Caucasian, non-Jewish population. Studies have indicated an increased frequency of polyclonal and monoclonal gammopathy among patients with GD. It has been shown that two major sphingolipids that accumulate in GD, namely, β-glucosylceramide 22:0 (βGL1-22) and glucosylsphingosine (LGL1), can be recognized by a distinct subset of CD1d-restricted human and murine type II natural killer T (NKT) cells. Investigations undertaken in an affected mouse model revealed βGL1-22- and LGL1-specific NKT cells were present and constitutively promoted the expression of a T-follicular helper (TFH) phenotype; injection of these lipids led to downstream induction of germinal center B cells, hypergammaglobulinemia, and the production of antilipid antibodies. Subsequent studies have found clonal immunoglobulin in 33% of sporadic human monoclonal gammopathies is also specific for the lysolipids LGL1 and lysophosphatidylcholine (LPC). Furthermore, substrate reduction ameliorated GD-associated gammopathy in mice. It had been hypothesized that chronic antigenic stimulation by the abnormal lipid storage and associated immune dysregulation may be the underlying mechanism for the increased incidence of monoclonal and polyclonal gammopathies, as well as an increased incidence of multiple myeloma in patients with GD. Current observations support this proposition and illustrate the value of investigations into rare diseases, which as ‘experiments of nature’ may provide insights into conditions found in the general population that continue to remain incompletely understood.
Full article
Open AccessReview
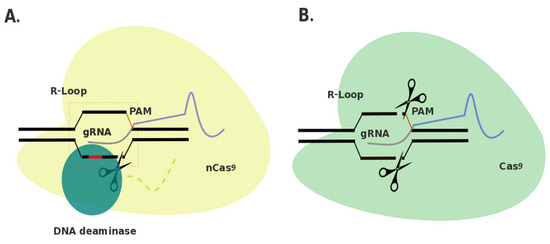
A Prospective Treatment Option for Lysosomal Storage Diseases: CRISPR/Cas9 Gene Editing Technology for Mutation Correction in Induced Pluripotent Stem Cells
by
Chloe L. Christensen and Francis Y. M. Choy
Cited by 9 | Viewed by 10050
Abstract
Ease of design, relatively low cost and a multitude of gene-altering capabilities have all led to the adoption of the sophisticated and yet simple gene editing system: clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9). The CRISPR/Cas9 system holds promise for the
[...] Read more.
Ease of design, relatively low cost and a multitude of gene-altering capabilities have all led to the adoption of the sophisticated and yet simple gene editing system: clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9). The CRISPR/Cas9 system holds promise for the correction of deleterious mutations by taking advantage of the homology directed repair pathway and by supplying a correction template to the affected patient’s cells. Currently, this technique is being applied in vitro in human-induced pluripotent stem cells (iPSCs) to correct a variety of severe genetic diseases, but has not as of yet been used in iPSCs derived from patients affected with a lysosomal storage disease (LSD). If adopted into clinical practice, corrected iPSCs derived from cells that originate from the patient themselves could be used for therapeutic amelioration of LSD symptoms without the risks associated with allogeneic stem cell transplantation. CRISPR/Cas9 editing in a patient’s cells would overcome the costly, lifelong process associated with currently available treatment methods, including enzyme replacement and substrate reduction therapies. In this review, the overall utility of the CRISPR/Cas9 gene editing technique for treatment of genetic diseases, the potential for the treatment of LSDs and methods currently employed to increase the efficiency of this re-engineered biological system will be discussed.
Full article
►▼
Show Figures
Open AccessArticle
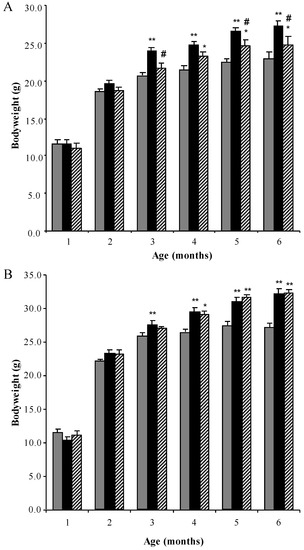
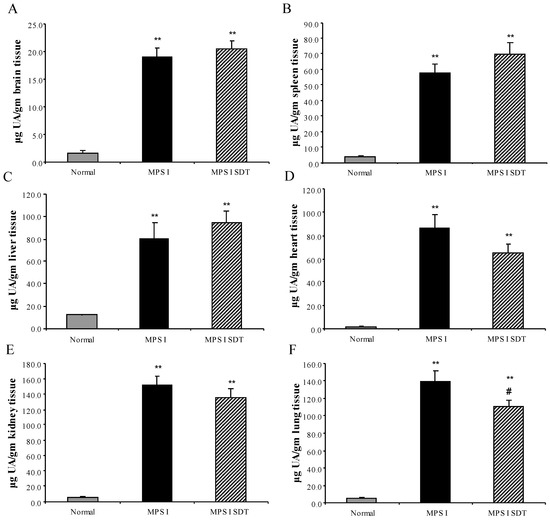
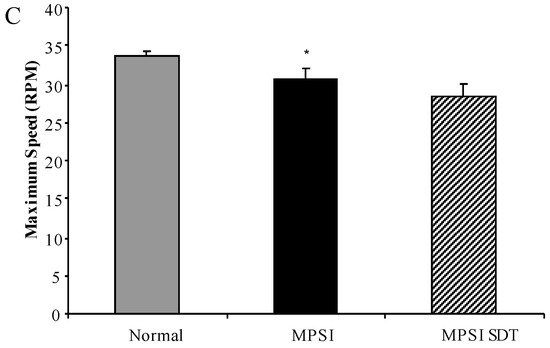
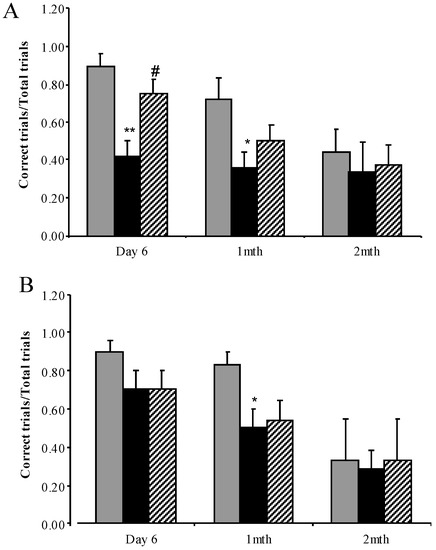
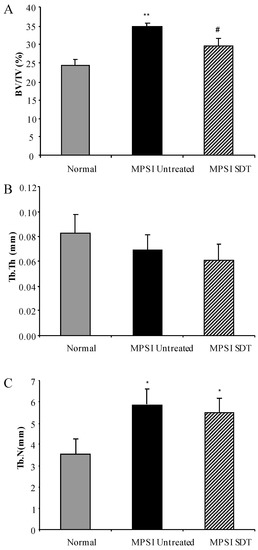
Substrate Deprivation Therapy to Reduce Glycosaminoglycan Synthesis Improves Aspects of Neurological and Skeletal Pathology in MPS I Mice
by
Ainslie L. K. Derrick-Roberts, Matilda R. Jackson, Carmen E. Pyragius and Sharon Byers
Cited by 16 | Viewed by 6712
Abstract
Mucopolysaccharidosis type I (MPS I) is the most common form of the MPS group of genetic diseases. MPS I results from a deficiency in the lysosomal enzyme α-
l-iduronidase, leading to accumulation of undegraded heparan and dermatan sulphate glycosaminoglycan (GAG) chains in
[...] Read more.
Mucopolysaccharidosis type I (MPS I) is the most common form of the MPS group of genetic diseases. MPS I results from a deficiency in the lysosomal enzyme α-
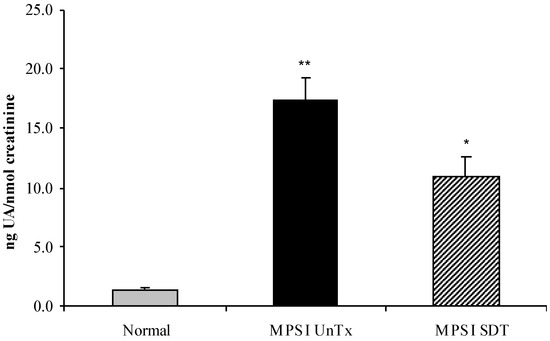
l-iduronidase, leading to accumulation of undegraded heparan and dermatan sulphate glycosaminoglycan (GAG) chains in patient cells. MPS children suffer from multiple organ failure and die in their teens to early twenties. In particular, MPS I children also suffer from profound mental retardation and skeletal disease that restricts growth and movement. Neither brain nor skeletal disease is adequately treated by current therapy approaches. To overcome these barriers to effective therapy we have developed and tested a treatment called substrate deprivation therapy (SDT). MPS I knockout mice were treated with weekly intravenous injections of 1 mg/kg rhodamine B for six months to assess the efficacy of SDT. Mice were assessed using biochemistry, micro-CT and a battery of behaviour tests to determine the outcome of treatment. A reduction in female bodyweight gain was observed with the treatment as well as a decrease in lung GAG. Behavioural studies showed slight improvements in inverted grid and significant improvements in learning ability for female MPS I mice treated with rhodamine B. Skeletal disease also improved with a reduction in bone mineral volume observed. Overall, rhodamine B is safe to administer to MPS I knockout mice where it had an effect on improving aspects of neurological and skeletal disease symptoms and may therefore provide a potential therapy or adjunct therapy for MPS I patients.
Full article
►▼
Show Figures
Open AccessReview
Biomarkers in Lysosomal Storage Diseases
by
Joaquin Bobillo Lobato, Maria Jiménez Hidalgo and Luis M. Jiménez Jiménez
Cited by 21 | Viewed by 7714
Abstract
A biomarker is generally an analyte that indicates the presence and/or extent of a biological process, which is in itself usually directly linked to the clinical manifestations and outcome of a particular disease. The biomarkers in the field of lysosomal storage diseases (LSDs)
[...] Read more.
A biomarker is generally an analyte that indicates the presence and/or extent of a biological process, which is in itself usually directly linked to the clinical manifestations and outcome of a particular disease. The biomarkers in the field of lysosomal storage diseases (LSDs) have particular relevance where spectacular therapeutic initiatives have been achieved, most notably with the introduction of enzyme replacement therapy (ERT). There are two main types of biomarkers. The first group is comprised of those molecules whose accumulation is directly enhanced as a result of defective lysosomal function. These molecules represent the storage of the principal macro-molecular substrate(s) of a specific enzyme or protein, whose function is deficient in the given disease. In the second group of biomarkers, the relationship between the lysosomal defect and the biomarker is indirect. In this group, the biomarker reflects the effects of the primary lysosomal defect on cell, tissue, or organ functions. There is no “gold standard” among biomarkers used to diagnosis and/or monitor LSDs, but there are a number that exist that can be used to reasonably assess and monitor the state of certain organs or functions. A number of biomarkers have been proposed for the analysis of the most important LSDs. In this review, we will summarize the most promising biomarkers in major LSDs and discuss why these are the most promising candidates for screening systems.
Full article
Open AccessArticle
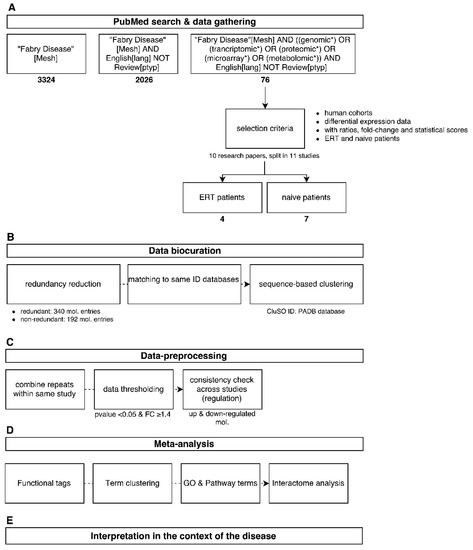
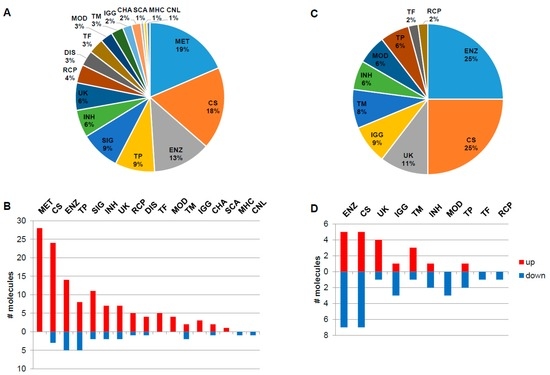
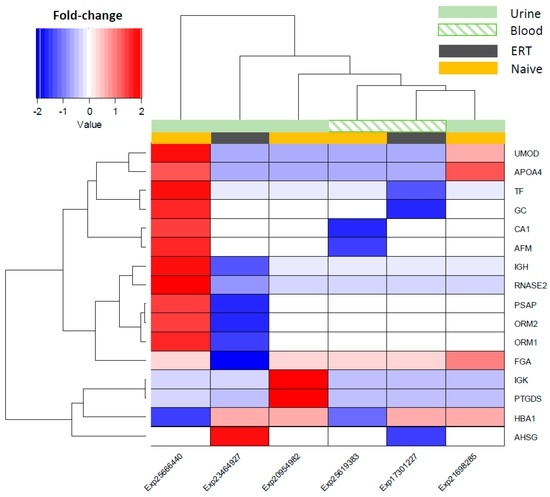
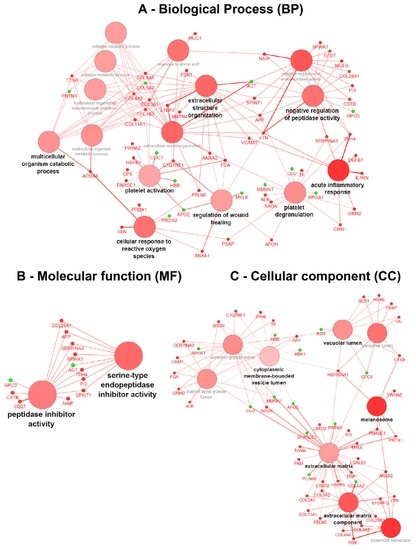
Integrative Systems Biology Investigation of Fabry Disease
by
Marco Fernandes and Holger Husi
Cited by 10 | Viewed by 9203
Abstract
Fabry disease (FD) is a rare X-linked recessive genetic disorder caused by a deficient activity of the lysosomal enzyme alpha-galactosidase A (GLA) and is characterised by intra-lysosomal accumulation of globotriaosylceramide (Gb3). We performed a meta-analysis of peer-reviewed publications including high-throughput omics technologies including
[...] Read more.
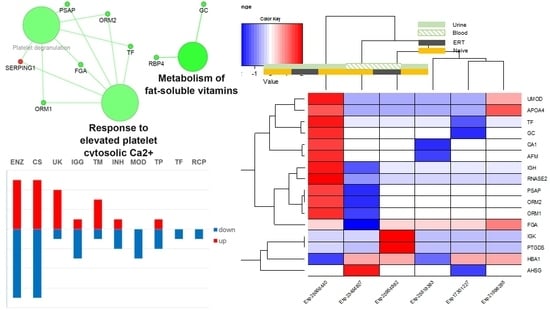
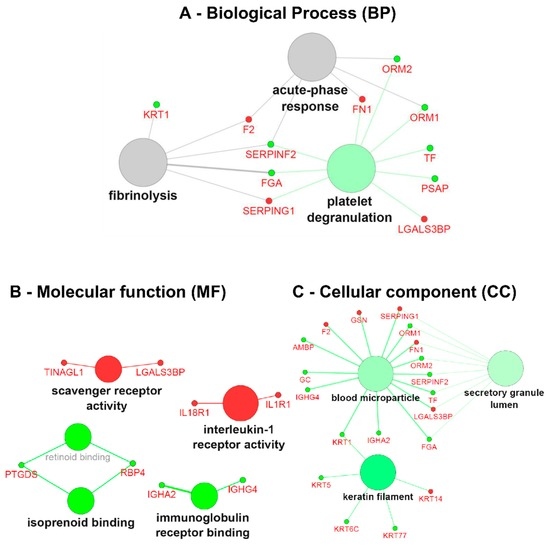
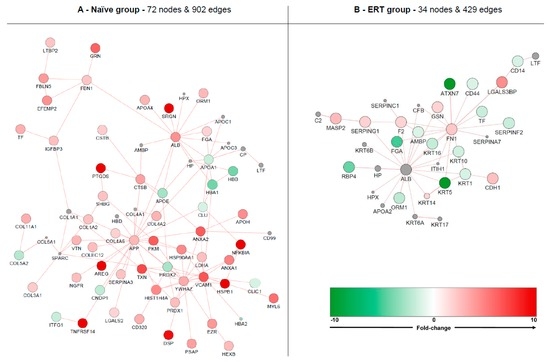
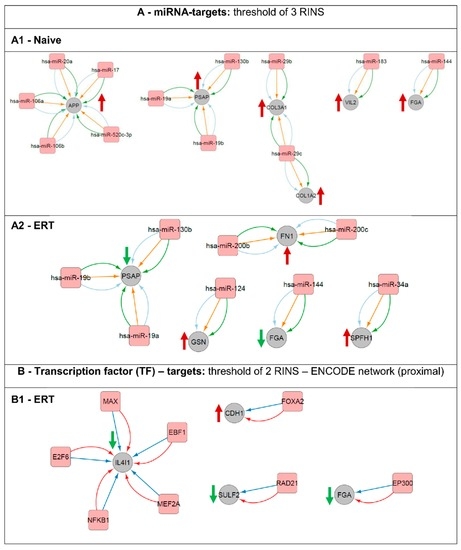
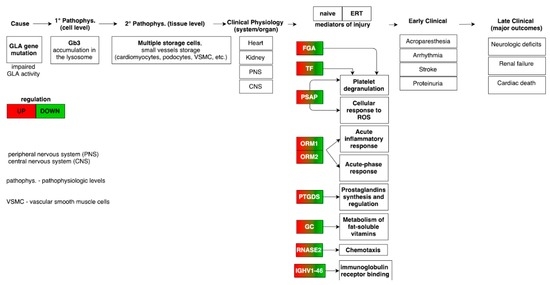
Fabry disease (FD) is a rare X-linked recessive genetic disorder caused by a deficient activity of the lysosomal enzyme alpha-galactosidase A (GLA) and is characterised by intra-lysosomal accumulation of globotriaosylceramide (Gb3). We performed a meta-analysis of peer-reviewed publications including high-throughput omics technologies including naïve patients and those undergoing enzyme replacement therapy (ERT). This study describes FD on a systems level using a systems biology approach, in which molecular data sourced from multi-omics studies is extracted from the literature and integrated as a whole in order to reveal the biochemical processes and molecular pathways potentially affected by the dysregulation of differentially expressed molecules. In this way new insights are provided that describe the pathophysiology of this rare disease. Using gene ontology and pathway term clustering, FD displays the involvement of major biological processes such as the acute inflammatory response, regulation of wound healing, extracellular matrix (ECM) remodelling, regulation of peptidase activity, and cellular response to reactive oxygen species (ROS). Differential expression of acute-phase response proteins in the groups of naïve (up-regulation of ORM1, ORM2, ITIH4, SERPINA3 and FGA) and ERT (down-regulation of FGA, ORM1 and ORM2) patients could be potential hallmarks for distinction of these two patient groups.
Full article
►▼
Show Figures
Open AccessReview
Genetic Substrate Reduction Therapy: A Promising Approach for Lysosomal Storage Disorders
by
Maria Francisca Coutinho, Juliana Inês Santos, Liliana Matos and Sandra Alves
Cited by 23 | Viewed by 8977
Abstract
Lysosomal storage diseases are a group of rare genetic disorders characterized by the accumulation of storage molecules in late endosomes/lysosomes. Most of them result from mutations in genes encoding for the catabolic enzymes that ensure intralysosomal digestion. Conventional therapeutic options include enzyme replacement
[...] Read more.
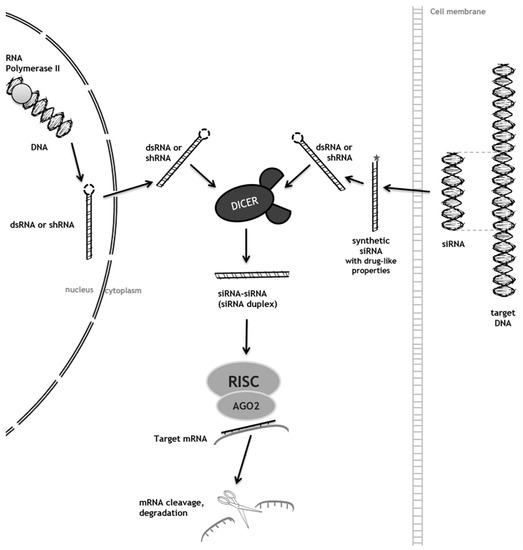
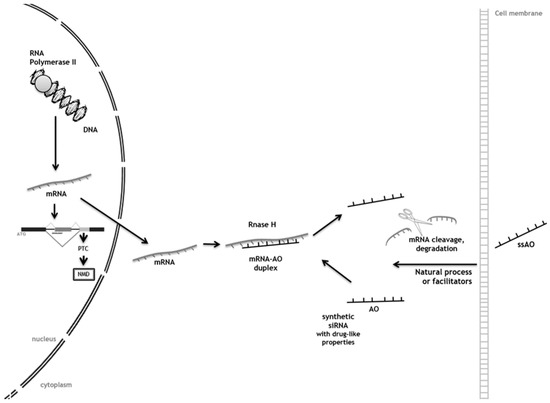
Lysosomal storage diseases are a group of rare genetic disorders characterized by the accumulation of storage molecules in late endosomes/lysosomes. Most of them result from mutations in genes encoding for the catabolic enzymes that ensure intralysosomal digestion. Conventional therapeutic options include enzyme replacement therapy, an approach targeting the functional loss of the enzyme by injection of a recombinant one. Even though this is successful for some diseases, it is mostly effective for peripheral manifestations and has no impact on neuropathology. The development of alternative therapeutic approaches is, therefore, mandatory, and striking innovations including the clinical development of pharmacological chaperones and gene therapy are currently under evaluation. Most of them, however, have the same underlying rationale: an attempt to provide or enhance the activity of the missing enzyme to re-establish substrate metabolism to a level that is consistent with a lack of progression and/or return to health. Here, we will focus on the one approach which has a different underlying principle: substrate reduction therapy (SRT), whose uniqueness relies on the fact that it acts upstream of the enzymatic defect, decreasing storage by downregulating its biosynthetic pathway. Special attention will be given to the most recent advances in the field, introducing the concept of genetic SRT (gSRT), which is based on the use of RNA-degrading technologies (RNA interference and single stranded antisense oligonucleotides) to promote efficient substrate reduction by decreasing its synthesis rate.
Full article
►▼
Show Figures
Open AccessReview
Nonsense Suppression as an Approach to Treat Lysosomal Storage Diseases
by
Kim M. Keeling
Cited by 23 | Viewed by 7948
Abstract
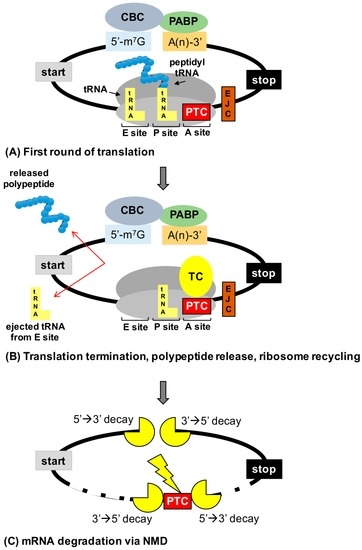
In-frame premature termination codons (PTCs) (also referred to as nonsense mutations) comprise ~10% of all disease-associated gene lesions. PTCs reduce gene expression in two ways. First, PTCs prematurely terminate translation of an mRNA, leading to the production of a truncated polypeptide that often
[...] Read more.
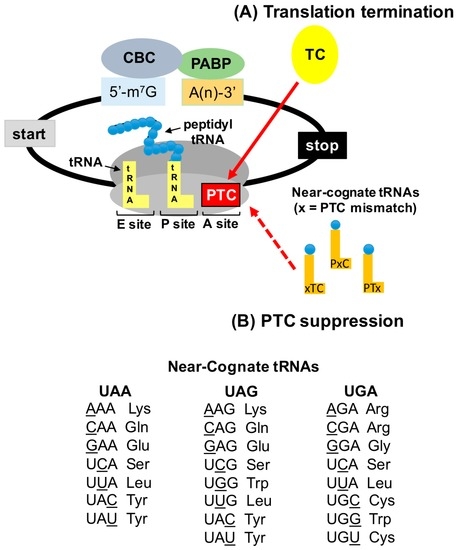
In-frame premature termination codons (PTCs) (also referred to as nonsense mutations) comprise ~10% of all disease-associated gene lesions. PTCs reduce gene expression in two ways. First, PTCs prematurely terminate translation of an mRNA, leading to the production of a truncated polypeptide that often lacks normal function and/or is unstable. Second, PTCs trigger degradation of an mRNA by activating nonsense-mediated mRNA decay (NMD), a cellular pathway that recognizes and degrades mRNAs containing a PTC. Thus, translation termination and NMD are putative therapeutic targets for the development of treatments for genetic diseases caused by PTCs. Over the past decade, significant progress has been made in the identification of compounds with the ability to suppress translation termination of PTCs (also referred to as readthrough). More recently, NMD inhibitors have also been explored as a way to enhance the efficiency of PTC suppression. Due to their relatively low threshold for correction, lysosomal storage diseases are a particularly relevant group of diseases to investigate the feasibility of nonsense suppression as a therapeutic approach. In this review, the current status of PTC suppression and NMD inhibition as potential treatments for lysosomal storage diseases will be discussed.
Full article
►▼
Show Figures
Open AccessReview
Mitochondrial Dysfunction in Lysosomal Storage Disorders
by
Mario De la Mata, David Cotán, Marina Villanueva-Paz, Isabel De Lavera, Mónica Álvarez-Córdoba, Raquel Luzón-Hidalgo, Juan M. Suárez-Rivero, Gustavo Tiscornia and Manuel Oropesa-Ávila
Cited by 52 | Viewed by 9214
Abstract
Lysosomal storage diseases (LSDs) describe a heterogeneous group of rare inherited metabolic disorders that result from the absence or loss of function of lysosomal hydrolases or transporters, resulting in the progressive accumulation of undigested material in lysosomes. The accumulation of substances affects the
[...] Read more.
Lysosomal storage diseases (LSDs) describe a heterogeneous group of rare inherited metabolic disorders that result from the absence or loss of function of lysosomal hydrolases or transporters, resulting in the progressive accumulation of undigested material in lysosomes. The accumulation of substances affects the function of lysosomes and other organelles, resulting in secondary alterations such as impairment of autophagy, mitochondrial dysfunction, inflammation and apoptosis. LSDs frequently involve the central nervous system (CNS), where neuronal dysfunction or loss results in progressive neurodegeneration and premature death. Many LSDs exhibit signs of mitochondrial dysfunction, which include mitochondrial morphological changes, decreased mitochondrial membrane potential (ΔΨm), diminished ATP production and increased generation of reactive oxygen species (ROS). Furthermore, reduced autophagic flux may lead to the persistence of dysfunctional mitochondria. Gaucher disease (GD), the LSD with the highest prevalence, is caused by mutations in the GBA1 gene that results in defective and insufficient activity of the enzyme β-glucocerebrosidase (GCase). Decreased catalytic activity and/or instability of GCase leads to accumulation of glucosylceramide (GlcCer) and glucosylsphingosine (GlcSph) in the lysosomes of macrophage cells and visceral organs. Mitochondrial dysfunction has been reported to occur in numerous cellular and mouse models of GD. The aim of this manuscript is to review the current knowledge and implications of mitochondrial dysfunction in LSDs.
Full article
Open AccessArticle
Role of Diffusion Tensor Imaging in Prognostication and Treatment Monitoring in Niemann-Pick Disease Type C1
by
Meghann W. Lau, Ryan W. Lee, Robin Miyamoto, Eun Sol Jung, Nicole Yanjanin Farhat, Shoko Yoshida, Susumu Mori, Andrea Gropman, Eva H. Baker and Forbes D. Porter
Cited by 5 | Viewed by 6427
Abstract
Niemann-Pick Disease, type C1 (NPC1) is a rapidly progressive neurodegenerative disorder characterized by cholesterol sequestration within late endosomes and lysosomes, for which no reliable imaging marker exists for prognostication and management. Cerebellar volume deficits are found to correlate with disease severity and diffusion
[...] Read more.
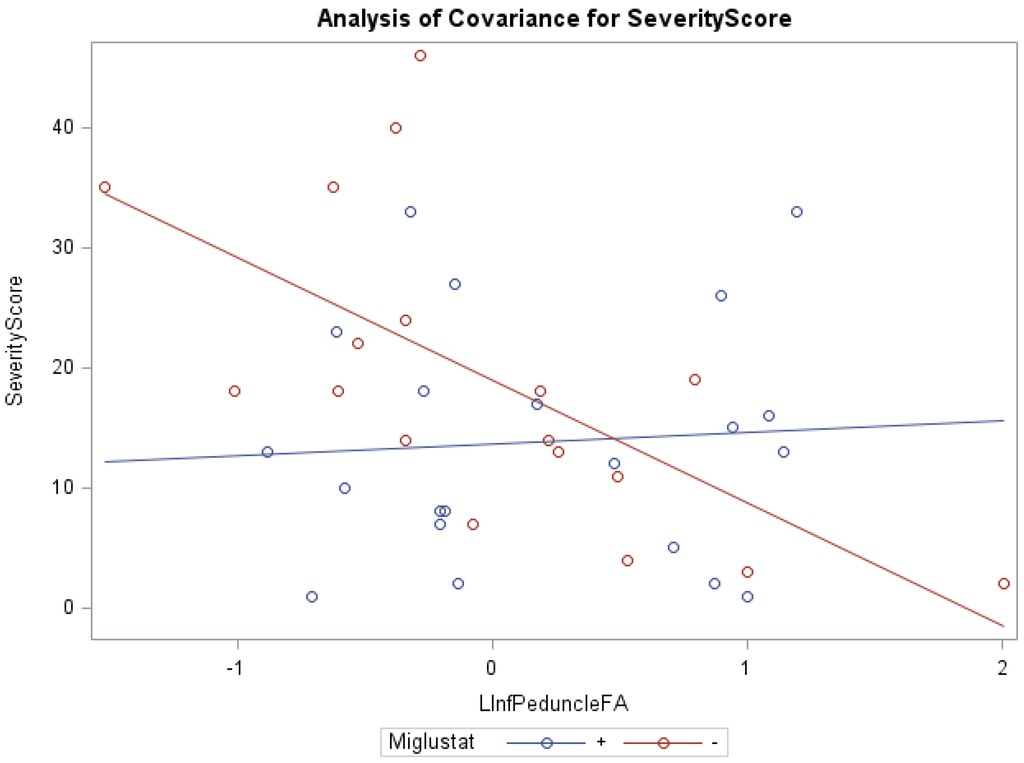
Niemann-Pick Disease, type C1 (NPC1) is a rapidly progressive neurodegenerative disorder characterized by cholesterol sequestration within late endosomes and lysosomes, for which no reliable imaging marker exists for prognostication and management. Cerebellar volume deficits are found to correlate with disease severity and diffusion tensor imaging (DTI) of the corpus callosum and brainstem, which has shown that microstructural disorganization is associated with NPC1 severity. This study investigates the utility of cerebellar DTI in clinical severity assessment. We hypothesize that cerebellar volume, fractional anisotropy (FA) and mean diffusivity (MD) negatively correlate with NIH NPC neurological severity score (NNSS) and motor severity subscores. Magnetic resonance imaging (MRI) was obtained for thirty-nine NPC1 subjects, ages 1–21.9 years (mean = 11.1, SD = 6.1). Using an atlas-based automated approach, the cerebellum of each patient was measured for FA, MD and volume. Additionally, each patient was given an NNSS. Decreased cerebellar FA and volume, and elevated MD correlate with higher NNSS. The cognition subscore and motor subscores for eye movement, ambulation, speech, swallowing, and fine motor skills were also statistically significant. Microstructural disorganization negatively correlated with motor severity in subjects. Additionally, Miglustat therapy correlated with lower severity scores across ranges of FA, MD and volume in all regions except the inferior peduncle, where a paradoxical effect was observed at high FA values. These findings suggest that DTI is a promising prognostication tool.
Full article
►▼
Show Figures





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}