
Nonsense Suppression as an Approach to Treat Lysosomal Storage Diseases

Abstract
:1. Premature Termination Codons Are Frequently the Cause of Disease
2. Mechanism of PTC Suppression
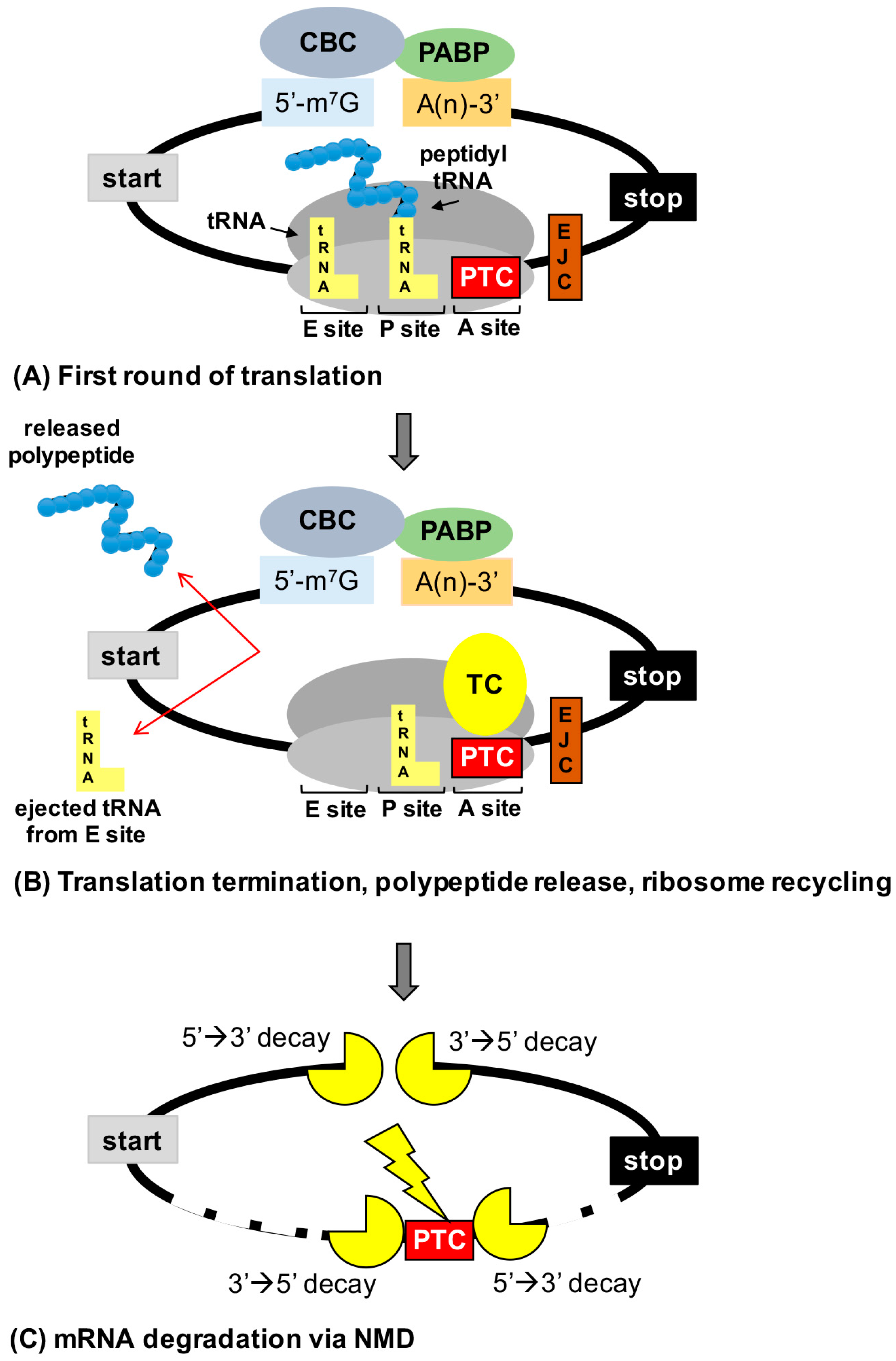
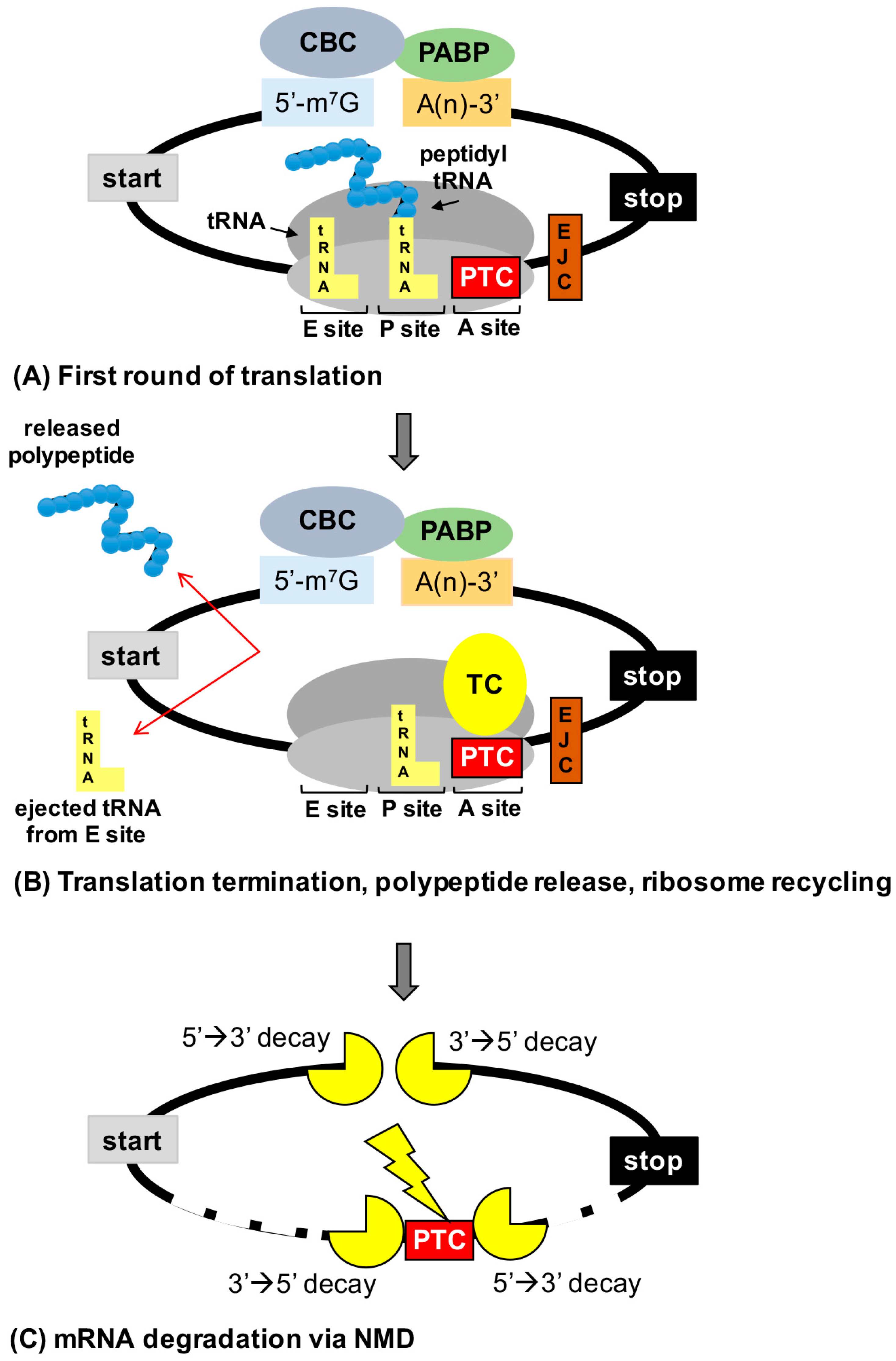
2.1. Overview of Translation
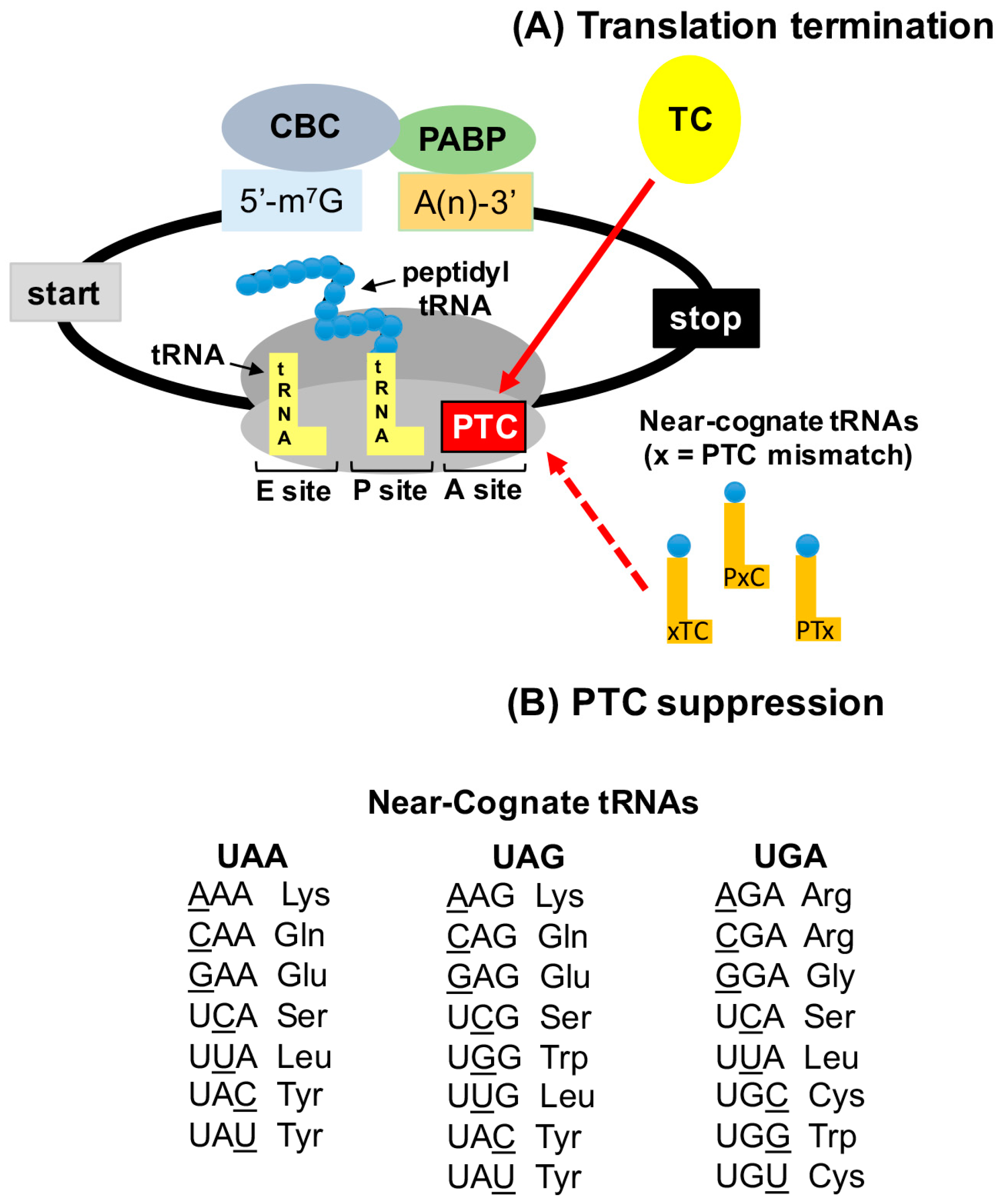
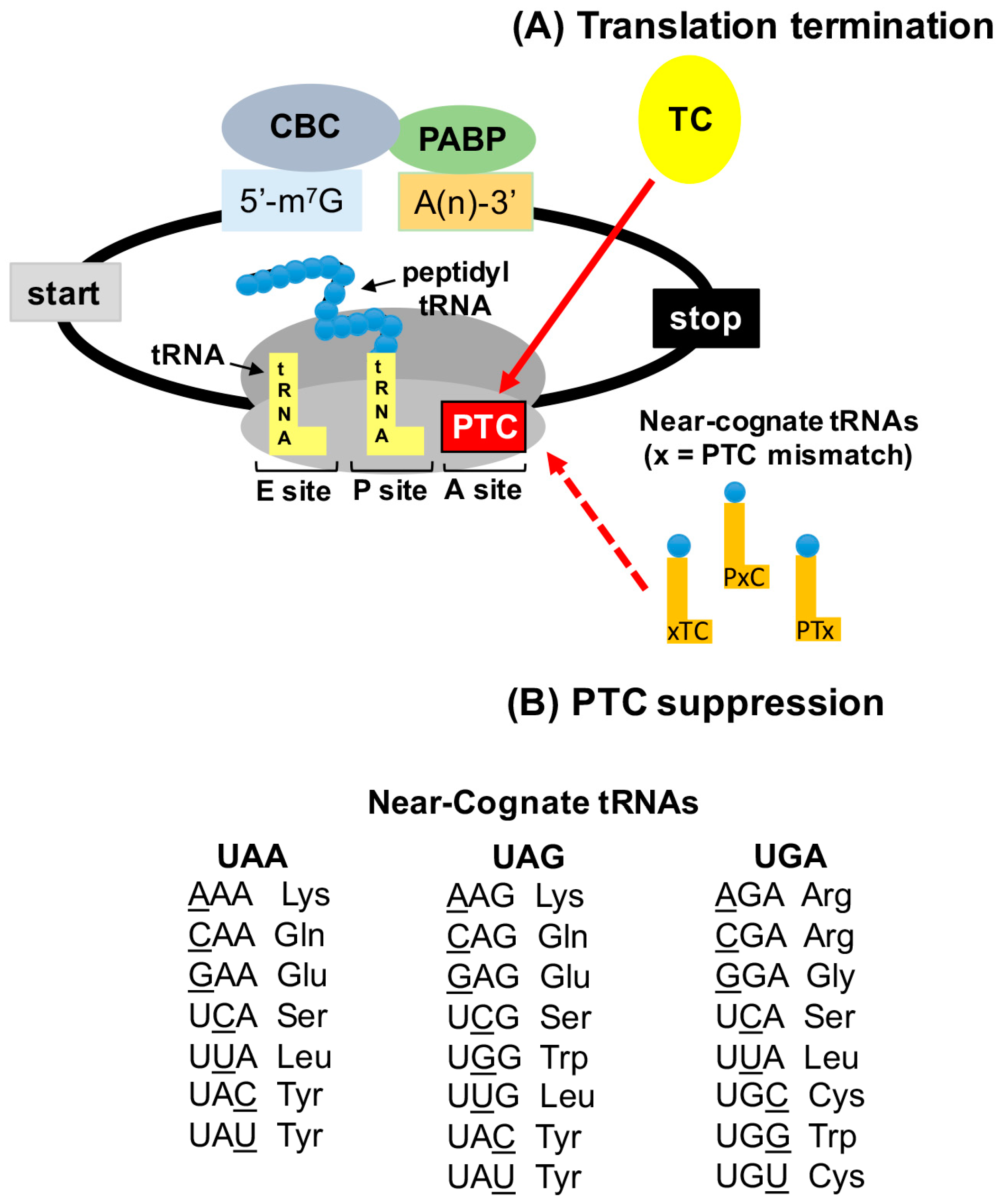
2.2. Termination Suppression
3. Nonsense Suppression as a Treatment for Lysosomal Storage Diseases
3.1. Aminoglycosides
3.2. PTC124
3.3. Other Readthrough Compounds
3.4. Inhibition of Nonsense-Mediated mRNA Decay (NMD)
4. Consideration of Personalized Medicine Approaches for LSDs
Acknowledgments
Conflicts of Interest
References
- Linde, L.; Kerem, B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008, 24, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Xue, X.; Gunn, G.; Bedwell, D.M. Therapeutics based on stop codon readthrough. Annu. Rev. Genom. Hum. Genet. 2014, 15, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Dougherty, J.P. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol. Ther. 2012, 136, 227–266. [Google Scholar] [CrossRef] [PubMed]
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Green, R. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb. Perspect. Biol. 2012, 4, a013706. [Google Scholar] [CrossRef] [PubMed]
- Zhouravleva, G.; Frolova, L.; Le Goff, X.; Le Guellec, R.; Inge-Vechtomov, S.; Kisselev, L.; Philippe, M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eER1 and eRF3. EMBO J. 1995, 14, 4065–4072. [Google Scholar] [PubMed]
- Song, H.; Mugnier, P.; Das, A.K.; Webb, H.M.; Evans, D.R.; Tuite, M.F.; Hemmings, B.A.; Barford, D. The crystal structure of human eukaryotic release factor eER1-- mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell 2000, 100, 311–321. [Google Scholar] [CrossRef]
- Taylor, D.; Unbehaun, A.; Li, W.; Das, S.; Lei, J.; Liao, H.Y.; Grassucci, R.A.; Pestova, T.V.; Frank, J. Cryo-EM structure of the mammalian eukaryotic release factor eER1-eER3-associated termination complex. Proc. Natl. Acad. Sci. USA 2012, 109, 18413–18418. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Saito, K.; Pisarev, A.V.; Wada, M.; Pisareva, V.P.; Pestova, T.V.; Gajda, M.; Round, A.; Kong, C.; Lim, M.; et al. Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev. 2009, 23, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Salhi, S.; Le Goff, C.; Viranaicken, W.; Diop, D.; Jean-Jean, O. Involvement of human release factors eER3a and eRF3b in translation termination and regulation of the termination complex formation. Mol. Cell. Biol. 2005, 25, 5801–5811. [Google Scholar] [CrossRef] [PubMed]
- Salas-Marco, J.; Bedwell, D.M. GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. Mol. Cell. Biol. 2004, 24, 7769–7778. [Google Scholar] [CrossRef] [PubMed]
- Frolova, L.; Le Goff, X.; Zhouravleva, G.; Davydova, E.; Philippe, M.; Kisselev, L. Eukaryotic polypeptide chain release factor eRF3 is an eRF1- and ribosome-dependent guanosine triphosphatase. RNA 1996, 2, 334–341. [Google Scholar] [PubMed]
- Velichutina, I.V.; Hong, J.Y.; Mesecar, A.D.; Chernoff, Y.O.; Liebman, S.W. Genetic interaction between yeast Saccharomyces cerevisiae release factors and the decoding region of 18S rRNA. J. Mol. Biol. 2001, 305, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Shao, S.; Murray, J.; Hegde, R.S.; Ramakrishnan, V. Structural basis for stop codon recognition in eukaryotes. Nature 2015, 524, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Gross, T.; Siepmann, A.; Sturm, D.; Windgassen, M.; Scarcelli, J.J.; Seedorf, M.; Cole, C.N.; Krebber, H. The dead-box RNA helicase DBP5 functions in translation termination. Science 2007, 315, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Roman, A.R.; Bolger, T.A.; Wente, S.R. Control of mRNA export and translation termination by inositol hexakisphosphate requires specific interaction with GLE1. J. Biol. Chem. 2010, 285, 16683–16692. [Google Scholar] [CrossRef] [PubMed]
- Pisarev, A.V.; Skabkin, M.A.; Pisareva, V.P.; Skabkina, O.V.; Rakotondrafara, A.M.; Hentze, M.W.; Hellen, C.U.; Pestova, T.V. The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol. Cell 2010, 37, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Preis, A.; Heuer, A.; Barrio-Garcia, C.; Hauser, A.; Eyler, D.E.; Berninghausen, O.; Green, R.; Becker, T.; Beckmann, R. Cryoelectron microscopic structures of eukaryotic translation termination complexes containing eRF1-eRF3 or eRF1-ABCE1. Cell Reports 2014, 8, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Khoshnevis, S.; Gross, T.; Rotte, C.; Baierlein, C.; Ficner, R.; Krebber, H. The iron-sulphur protein RNase l inhibitor functions in translation termination. EMBO Rep. 2010, 11, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, C.J.; Green, R. Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc. Natl. Acad. Sci. USA 2011, 108, E1392–E1398. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Salas-Marco, J.; Osherovich, L.Z.; Bedwell, D.M. Tpa1p is part of an mRNP complex that influences translation termination, mRNA deadenylation, and mRNA turnover in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006, 26, 5237–5248. [Google Scholar] [CrossRef] [PubMed]
- Henri, J.; Rispal, D.; Bayart, E.; van Tilbeurgh, H.; Seraphin, B.; Graille, M. Structural and functional insights into Saccharomyces cerevisiae Tpa1, a putative prolylhydroxylase influencing translation termination and transcription. J. Biol. Chem. 2010, 285, 30767–30778. [Google Scholar] [CrossRef] [PubMed]
- Singleton, R.S.; Liu-Yi, P.; Formenti, F.; Ge, W.; Sekirnik, R.; Fischer, R.; Adam, J.; Pollard, P.J.; Wolf, A.; Thalhammer, A.; et al. OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation. Proc. Natl. Acad. Sci. USA 2014, 111, 4031–4036. [Google Scholar] [CrossRef] [PubMed]
- Loenarz, C.; Sekirnik, R.; Thalhammer, A.; Ge, W.; Spivakovsky, E.; Mackeen, M.M.; McDonough, M.A.; Cockman, M.E.; Kessler, B.M.; Ratcliffe, P.J.; et al. Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc. Natl. Acad. Sci. USA 2014, 111, 4019–4024. [Google Scholar] [CrossRef] [PubMed]
- Amrani, N.; Ganesan, R.; Kervestin, S.; Mangus, D.A.; Ghosh, S.; Jacobson, A. A faux 3’-UTR promotes aberrant termination and triggers nonsense-mediated mrna decay. Nature 2004, 432, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Amrani, N.; Ghosh, S.; Mangus, D.A.; Jacobson, A. Translation factors promote the formation of two states of the closed-loop mRNP. Nature 2008, 453, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Roque, S.; Cerciat, M.; Gaugue, I.; Mora, L.; Floch, A.G.; de Zamaroczy, M.; Heurgue-Hamard, V.; Kervestin, S. Interaction between the poly(A)-binding protein Pab1 and the eukaryotic release factor eRF3 regulates translation termination but not mRNA decay in Saccharomyces cerevisiae. RNA 2015, 21, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Bossi, L.; Roth, J.R. The influence of codon context on genetic code translation. Nature 1980, 286, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Martin, R. On the relationship between preferred termination codon contexts and nonsense suppression in human cells. Nucleic Acids Res. 1994, 22, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Mottagui-Tabar, S.; Bjornsson, A.; Isaksson, L.A. The second to last amino acid in the nascent peptide as a codon context determinant. EMBO J. 1994, 13, 249–257. [Google Scholar] [PubMed]
- Bonetti, B.; Fu, L.; Moon, J.; Bedwell, D.M. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J. Mol. Biol. 1995, 251, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Jones, M.K.; Hill, L.S.; Atkinson, J.; Martin, R. Context effects on misreading and suppression at UAG codons in human cells. Mol. Cell. Biol. 1995, 15, 6593–6600. [Google Scholar] [CrossRef] [PubMed]
- Mottagui-Tabar, S.; Tuite, M.F.; Isaksson, L.A. The influence of 5’ codon context on translation termination in Saccharomyces cerevisiae. Eur. J. Biochem. 1998, 257, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, M.Y.; Freistroffer, D.V.; Dincbas, V.; MacDougall, J.; Buckingham, R.H.; Ehrenberg, M. A direct estimation of the context effect on the efficiency of termination. J. Mol. Biol. 1998, 284, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Cassan, M.; Rousset, J.P. UAG readthrough in mammalian cells: Effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol. Biol. 2001, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Namy, O.; Hatin, I.; Rousset, J.P. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2001, 2, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Harrell, L.; Melcher, U.; Atkins, J.F. Predominance of six different hexanucleotide recoding signals 3’ of read-through stop codons. Nucleic Acids Res. 2002, 30, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Jones, M.K.; Watson, F.J.; Martin, R. The 3’ codon context effect on uag suppressor tRNA is different in Escherichia coli and human cells. J. Mol. Biol. 1993, 233, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tate, W.P.; Poole, E.S.; Horsfield, J.A.; Mannering, S.A.; Brown, C.M.; Moffat, J.G.; Dalphin, M.E.; McCaughan, K.K.; Major, L.L.; Wilson, D.N. Translational termination efficiency in both bacteria and mammals is regulated by the base following the stop codon. Biochem. Cell Biol. 1995, 73, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Frischmeyer, P.A.; van Hoof, A.; O’Donnell, K.; Guerrerio, A.L.; Parker, R.; Dietz, H.C. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 2002, 295, 2258–2261. [Google Scholar] [CrossRef] [PubMed]
- Arribere, J.A.; Cenik, E.S.; Jain, N.; Hess, G.T.; Lee, C.H.; Bassik, M.C.; Fire, A.Z. Translation readthrough mitigation. Nature 2016, 534, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Eswarappa, S.M.; Potdar, A.A.; Koch, W.J.; Fan, Y.; Vasu, K.; Lindner, D.; Willard, B.; Graham, L.M.; DiCorleto, P.E.; Fox, P.L. Programmed translational readthrough generates antiangiogenic VEGF-ax. Cell 2014, 157, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Chittum, H.S.; Lane, W.S.; Carlson, B.A.; Roller, P.P.; Lung, F.D.; Lee, B.J.; Hatfield, D.L. Rabbit beta-globin is extended beyond its UGA stop codon by multiple suppressions and translational reading gaps. Biochemistry (Mosc.) 1998, 37, 10866–10870. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Hayashi, A.; Campagnoni, C.W.; Kimura, A.; Inuzuka, T.; Baba, H. L-MPZ, a novel isoform of myelin p0, is produced by stop codon readthrough. J. Biol. Chem. 2012, 287, 17765–17776. [Google Scholar] [CrossRef] [PubMed]
- Schueren, F.; Lingner, T.; George, R.; Hofhuis, J.; Dickel, C.; Gartner, J.; Thoms, S. Peroxisomal lactate dehydrogenase is generated by translational readthrough in mammals. eLife 2014, 3, e03640. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.J.; Gandara, L.; De Lella Ezcurra, A.L.; Wappner, P. Hydroxylation and translational adaptation to stress: Some answers lie beyond the stop codon. Cell. Mol. Life Sci. 2016, 73, 1881–1893. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; McClendon, V.; Bonetti, B.; Bedwell, D.M. Premature translation termination mutations are efficiently suppressed in a highly conserved region of yeast Ste6p, a member of the ATP-binding cassette (ABC) transporter family. J. Biol. Chem. 1994, 269, 17802–17808. [Google Scholar] [PubMed]
- Blanchet, S.; Cornu, D.; Argentini, M.; Namy, O. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res. 2014, 42, 10061–10072. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Leszyk, J.D.; Mangus, D.A.; Jacobson, A. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc. Natl. Acad. Sci. USA 2015, 112, 3038–3043. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Frizzell, R.A.; Bedwell, D.M. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 1996, 2, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Nagel-Wolfrum, K.; Moller, F.; Penner, I.; Baasov, T.; Wolfrum, U. Targeting nonsense mutations in diseases with translational readthrough-inducing drugs (TRIDs). BioDrugs 2016, 30, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E. Pharmacologic therapy for stop mutations: How much CFTR activity is enough? Curr. Opin. Pulm. Med. 2004, 10, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Muses, S.; McClorey, G.; Wells, K.E.; Coursindel, T.; Terry, R.L.; Betts, C.; Hammond, S.; O’Donovan, L.; Hildyard, J.; et al. How much dystrophin is enough: The physiological consequences of different levels of dystrophin in the mdx mouse. Hum. Mol. Genet. 2015, 24, 4225–4237. [Google Scholar] [CrossRef] [PubMed]
- Oussoren, E.; Keulemans, J.; van Diggelen, O.P.; Oemardien, L.F.; Timmermans, R.G.; van der Ploeg, A.T.; Ruijter, G.J. Residual alpha-L-iduronidase activity in fibroblasts of mild to severe mucopolysaccharidosis type I patients. Mol. Genet. Metab. 2013, 109, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Brooks, D.A.; Hopwood, J.J.; Li, P.; Thompson, J.N.; Bedwell, D.M. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum. Mol. Genet. 2001, 10, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Sohar, I.; Gin, R.M.; Lobel, P. Aminoglycoside-mediated suppression of nonsense mutations in late infantile neuronal ceroid lipofuscinosis. Eur. J. Paediatr. Neurol. 2001, 5 (Suppl. SA), 57–62. [Google Scholar] [CrossRef] [PubMed]
- Helip-Wooley, A.; Park, M.A.; Lemons, R.M.; Thoene, J.G. Expression of CTNS alleles: Subcellular localization and aminoglycoside correction in vitro. Mol. Genet. Metab. 2002, 75, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Bedwell, D.M. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and TP53 cDNAs in a mammalian translation system. J. Mol. Med. 2002, 80, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.K.; Bawden, M.; Muller, V.J.; Sillence, D.; Hopwood, J.J.; Brooks, D.A. Alpha-L-iduronidase premature stop codons and potential read-through in mucopolysaccharidosis type I patients. J. Mol. Biol. 2004, 338, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, I.; Rebibo-Sabbah, A.; Cherniavsky, M.; Belakhov, V.; Hainrichson, M.; Chen, F.; Schacht, J.; Pilch, D.S.; Ben-Yosef, T.; Baasov, T. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J. Med. Chem. 2009, 52, 2836–2845. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, I.; Glikin, D.; Smolkin, B.; Hainrichson, M.; Belakhov, V.; Baasov, T. Repairing faulty genes by aminoglycosides: Development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg. Med. Chem. 2010, 18, 3735–3746. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Zhang, Z.; Mukherjee, A.B. Stop codon read-through with PTC124 induces palmitoyl-protein thioesterase-1 activity, reduces thioester load and suppresses apoptosis in cultured cells from incl patients. Mol. Genet. Metab. 2011, 104, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, J.; Atia-Glikin, D.; Belakhov, V.; Baasov, T. Repairing faulty genes by aminoglycosides: Identification of new pharmacophore with enhanced suppression of disease-causing nonsense mutations. Med. Chem. Commun. 2011, 2, 165–171. [Google Scholar] [CrossRef]
- Kandasamy, J.; Atia-Glikin, D.; Shulman, E.; Shapira, K.; Shavit, M.; Belakhov, V.; Baasov, T. Increased selectivity toward cytoplasmic versus mitochondrial ribosome confers improved efficiency of synthetic aminoglycosides in fixing damaged genes: A strategy for treatment of genetic diseases caused by nonsense mutations. J. Med. Chem. 2012, 55, 10630–10643. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Belakhov, V.; Kandasamy, J.; Baasov, T.; Li, S.C.; Li, Y.T.; Bedwell, D.M.; Keeling, K.M. The designer aminoglycoside NB84 significantly reduces glycosaminoglycan accumulation associated with MPS I-H in the Idua-W392X mouse. Mol. Genet. Metab. 2012, 105, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Bartolomeo, R.; Polishchuk, E.V.; Volpi, N.; Polishchuk, R.S.; Auricchio, A. Pharmacological read-through of nonsense ARSB mutations as a potential therapeutic approach for mucopolysaccharidosis VI. J. Inherit. Metab. Dis. 2013, 36, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Wang, D.; Dai, Y.; Murugesan, S.; Chenna, B.; Clark, J.; Belakhov, V.; Kandasamy, J.; Velu, S.E.; Baasov, T.; et al. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS ONE 2013, 8, e60478. [Google Scholar] [CrossRef] [PubMed]
- Gunn, G.; Dai, Y.; Du, M.; Belakhov, V.; Kandasamy, J.; Schoeb, T.R.; Baasov, T.; Bedwell, D.M.; Keeling, K.M. Long-term nonsense suppression therapy moderates MPS I-H disease progression. Mol. Genet. Metab. 2013, 111, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Thada, V.; Miller, J.N.; Kovacs, A.D.; Pearce, D.A. Tissue-specific variation in nonsense mutant transcript level and drug-induced read-through efficiency in the Cln1 mouse model of INCL. J. Cell. Mol. Med. 2016, 20, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Grau, M.; Garrido, E.; Cozar, M.; Rodriguez-Sureda, V.; Dominguez, C.; Arenas, C.; Gatti, R.A.; Cormand, B.; Grinberg, D.; Vilageliu, L. Evaluation of aminoglycoside and non-aminoglycoside compounds for stop-codon readthrough therapy in four lysosomal storage diseases. PLoS ONE 2015, 10, e0135873. [Google Scholar] [CrossRef] [PubMed]
- Sabbavarapu, N.M.; Shavit, M.; Degani, Y.; Smolkin, B.; Belakhov, V.; Baasov, T. Design of novel aminoglycoside derivatives with enhanced suppression of diseases-causing nonsense mutations. ACS Med. Chem. Lett. 2016, 7, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Fourmy, D.; Recht, M.I.; Blanchard, S.C.; Puglisi, J.D. Structure of the a site of escherichia coli 16S ribosomal rRNA complexed with an aminoglycoside antibiotic. Science 1996, 274, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Recht, M.I.; Fourmy, D.; Blanchard, S.C.; Dahlquist, K.D.; Puglisi, J.D. RNA sequence determinants for aminoglycoside binding to an A-site rRNA model oligonucleotide. J. Mol. Biol. 1996, 262, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.R.; Puglisi, J.D. Structure of a eukaryotic decoding region A-site RNA. J. Mol. Biol. 2001, 306, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Bedwell, D.M. Recoding therapies for genetic diseases. In Recoding: Expansion of decoding rules enriches gene expression; Atkins, J.F., Gesteland, R.F., Eds.; Springer Publishing: New York, NY, USA, 2010; pp. 123–146. [Google Scholar]
- Recht, M.I.; Douthwaite, S.; Puglisi, J.D. Basis for prokaryotic specificity of action of aminoglycoside antibiotics. EMBO J. 1999, 18, 3133–3138. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.R.; Puglisi, J.D. Structural origins of aminoglycoside specificity for prokaryotic ribosomes. J. Mol. Biol. 2001, 306, 1037–1058. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.; Wilhelm, J.M. Mistranslation in a eucaryotic organism. Cell 1978, 13, 329–334. [Google Scholar] [CrossRef]
- Wilhelm, J.M.; Jessop, J.J.; Pettitt, S.E. Aminoglycoside antibiotics and eukaryotic protein synthesis: Stimulation of errors in the translation of natural messengers in extracts of cultured human cells. Biochemistry (Mosc.) 1978, 17, 1149–1153. [Google Scholar] [CrossRef]
- Wilhelm, J.M.; Pettitt, S.E.; Jessop, J.J. Aminoglycoside antibiotics and eukaryotic protein synthesis: Structure--function relationships in the stimulation of misreading with a wheat embryo system. Biochemistry (Mosc.) 1978, 17, 1143–1149. [Google Scholar] [CrossRef]
- Wilschanski, M.; Yahav, Y.; Yaacov, Y.; Blau, H.; Bentur, L.; Rivlin, J.; Aviram, M.; Bdolah-Abram, T.; Bebok, Z.; Shushi, L.; et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N. Engl. J. Med. 2003, 349, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Bebok, Z.; Ruiz, F.; King, C.; Jones, J.; Walker, L.; Greer, H.; Hong, J.; Wing, L.; Macaluso, M.; et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2001, 163, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.T.; Shirts, B.H.; Petros, L.M.; Flanigan, K.M.; Gesteland, R.F.; Atkins, J.F. Sequence specificity of aminoglycoside-induced stop codon readthrough: Potential implications for treatment of Duchenne muscular dystrophy. Ann. Neurol. 2000, 48, 164–169. [Google Scholar] [CrossRef]
- Wagner, K.R.; Hamed, S.; Hadley, D.W.; Gropman, A.L.; Burstein, A.H.; Escolar, D.M.; Hoffman, E.P.; Fischbeck, K.H. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann. Neurol. 2001, 49, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Politano, L.; Nigro, G.; Nigro, V.; Piluso, G.; Papparella, S.; Paciello, O.; Comi, L.I. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003, 22, 15–21. [Google Scholar] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Lewis, S.; Shilling, C.J.; Kota, J.; Serrano-Munuera, C.; et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Baradaran-Heravi, A.; Balgi, A.D.; Zimmerman, C.; Choi, K.; Shidmoossavee, F.S.; Tan, J.S.; Bergeaud, C.; Krause, A.; Flibotte, S.; Shimizu, Y.; et al. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016, 44, 6583–6598. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.H.; Hottendorf, G.H. Poly-L-aspartic acid protects cultured human proximal tubule cells against aminoglycoside-induced electrophysiological alterations. Toxicol. Lett. 1997, 90, 217–221. [Google Scholar] [CrossRef]
- Beauchamp, D.; Laurent, G.; Maldague, P.; Abid, S.; Kishore, B.K.; Tulkens, P.M. Protection against gentamicin-induced early renal alterations (phospholipidosis and increased DNA synthesis) by coadministration of poly-l-aspartic acid. J. Pharmacol. Exp. Ther. 1990, 255, 858–866. [Google Scholar] [PubMed]
- Du, M.; Keeling, K.M.; Fan, L.; Liu, X.; Bedwell, D.M. Poly-L-aspartic acid enhances and prolongs gentamicin-mediated suppression of the Cftr-G542X mutation in a cystic fibrosis mouse model. J. Biol. Chem. 2009, 284, 6885–6892. [Google Scholar] [CrossRef] [PubMed]
- Hutchin, T.; Cortopassi, G. Proposed molecular and cellular mechanism for aminoglycoside ototoxicity. Antimicrob. Agents Chemother. 1994, 38, 2517–2520. [Google Scholar] [CrossRef] [PubMed]
- Dehne, N.; Rauen, U.; de Groot, H.; Lautermann, J. Involvement of the mitochondrial permeability transition in gentamicin ototoxicity. Hear. Res. 2002, 169, 47–55. [Google Scholar] [CrossRef]
- Guthrie, O.W. Aminoglycoside induced ototoxicity. Toxicology 2008, 249, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Warchol, M.E. Cellular mechanisms of aminoglycoside ototoxicity. Curr. Opin. Otolaryngol. Head Neck Surg. 2010, 18, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Huth, M.E.; Ricci, A.J.; Cheng, A.G. Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. Int. J. Otolaryngol. 2011, 2011, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mingeot-Leclercq, M.P.; Tulkens, P.M. Aminoglycosides: Nephrotoxicity. Antimicrob. Agents Chemother. 1999, 43, 1003–1012. [Google Scholar] [PubMed]
- Lopez-Novoa, J.M.; Quiros, Y.; Vicente, L.; Morales, A.I.; Lopez-Hernandez, F.J. New insights into the mechanism of aminoglycoside nephrotoxicity: An integrative point of view. Kidney Int. 2011, 79, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Pagkalis, S.; Mantadakis, E.; Mavros, M.N.; Ammari, C.; Falagas, M.E. Pharmacological considerations for the proper clinical use of aminoglycosides. Drugs 2011, 71, 2277–2294. [Google Scholar] [CrossRef] [PubMed]
- Hobbie, S.N.; Akshay, S.; Kalapala, S.K.; Bruell, C.M.; Shcherbakov, D.; Bottger, E.C. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 20888–20893. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Guan, M.X. Interaction of aminoglycosides with human mitochondrial 12S rRNA carrying the deafness-associated mutation. Antimicrob. Agents Chemother. 2009, 53, 4612–4618. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; Carlier, M.B.; Rollman, B.; Van Hoof, F.; Tulkens, P. Mechanism of aminoglycoside-induced lysosomal phospholipidosis: in vitro and in vivo studies with gentamicin and amikacin. Biochem. Pharmacol. 1982, 31, 3861–3870. [Google Scholar] [CrossRef]
- Carlier, M.B.; Laurent, G.; Claes, P.J.; Vanderhaeghe, H.J.; Tulkens, P.M. Inhibition of lysosomal phospholipases by aminoglycoside antibiotics: in vitro comparative studies. Antimicrob. Agents Chemother. 1983, 23, 440–449. [Google Scholar] [CrossRef] [PubMed]
- De Broe, M.E.; Paulus, G.J.; Verpooten, G.A.; Roels, F.; Buyssens, N.; Wedeen, R.; van Hoof, F.; Tulkens, P.M. Early effects of gentamicin, tobramycin, and amikacin on the human kidney. Kidney Int. 1984, 25, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Mingeot-Leclercq, M.P.; Piret, J.; Tulkens, P.M.; Brasseur, R. Effect of acidic phospholipids on the activity of lysosomal phospholipases and on their inhibition induced by aminoglycoside antibiotics--ii. Conformational analysis. Biochem. Pharmacol. 1990, 40, 499–506. [Google Scholar] [CrossRef]
- Mingeot-Leclercq, M.P.; Piret, J.; Brasseur, R.; Tulkens, P.M. Effect of acidic phospholipids on the activity of lysosomal phospholipases and on their inhibition by aminoglycoside antibiotics--i. Biochemical analysis. Biochem. Pharmacol. 1990, 40, 489–497. [Google Scholar] [CrossRef]
- Nudelman, I.; Rebibo-Sabbah, A.; Shallom-Shezifi, D.; Hainrichson, M.; Stahl, I.; Ben-Yosef, T.; Baasov, T. Redesign of aminoglycosides for treatment of human genetic diseases caused by premature stop mutations. Bioorg. Med. Chem. Lett. 2006, 16, 6310–6315. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Hainrichson, M.; Nudelman, I.; Shallom-Shezifi, D.; Barbieri, C.M.; Pilch, D.S.; Westhof, E.; Baasov, T. Differential selectivity of natural and synthetic aminoglycosides towards the eukaryotic and prokaryotic decoding a sites. Chembiochem 2007, 8, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Hainrichson, M.; Nudelman, I.; Baasov, T. Designer aminoglycosides: The race to develop improved antibiotics and compounds for the treatment of human genetic diseases. Org. Biomol. Chem. 2008, 6, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Sloane, P.; Tang, L.P.; Backer, K.; Mazur, M.; Buckley-Lanier, J.; Nudelman, I.; Belakhov, V.; Bebok, Z.; Schwiebert, E.; et al. Suppression of CFTR premature termination codons and rescue of CFTR protein and function by the synthetic aminoglycoside NB54. J. Mol. Med. (Berl.) 2011, 89, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Mutyam, V.; Tang, L.; Biswas, S.; Du, M.; Jackson, L.A.; Dai, Y.; Belakhov, V.; Shalev, M.; Chen, F.; et al. Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. Am. J. Respir. Cell Mol. Biol. 2014, 50, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Rebibo-Sabbah, A.; Nudelman, I.; Ahmed, Z.M.; Baasov, T.; Ben-Yosef, T. In vitro and ex vivo suppression by aminoglycosides of PCDH15 nonsense mutations underlying type 1 Usher syndrome. Hum. Genet. 2007, 122, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Rebibo-Sabbah, A.; Overlack, N.; Nudelman, I.; Belakhov, V.; Baasov, T.; Ben-Yosef, T.; Wolfrum, U.; Nagel-Wolfrum, K. Beneficial read-through of a USH1C nonsense mutation by designed aminoglycoside NB30 in the retina. Invest. Ophthalmol. Vis. Sci. 2010, 51, 6671–6680. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Overlack, N.; Moller, F.; Belakhov, V.; van Wyk, M.; Baasov, T.; Wolfrum, U.; Nagel-Wolfrum, K. A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med. 2012, 4, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Belakhov, V.; Werner, H.; Wegener, E.; Gartner, J.; Nudelman, I.; Baasov, T.; Huppke, P. Readthrough of nonsense mutations in Rett syndrome: Evaluation of novel aminoglycosides and generation of a new mouse model. J. Mol. Med. 2010, 89, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Vecsler, M.; Ben Zeev, B.; Nudelman, I.; Anikster, Y.; Simon, A.J.; Amariglio, N.; Rechavi, G.; Baasov, T.; Gak, E. Ex vivo treatment with a novel synthetic aminoglycoside NB54 in primary fibroblasts from Rett syndrome patients suppresses MECP2 nonsense mutations. PLoS ONE 2011, 6, e20733. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shukla, C.; Liu, X.; Schoeb, T.R.; Clarke, L.A.; Bedwell, D.M.; Keeling, K.M. Characterization of an MPS I-H knock-in mouse that carries a nonsense mutation analogous to the human IDUA-W402X mutation. Mol. Genet. Metab. 2010, 99, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Whitley, C.B.; Belani, K.G.; Chang, P.N.; Summers, C.G.; Blazar, B.R.; Tsai, M.Y.; Latchaw, R.E.; Ramsay, N.K.; Kersey, J.H. Long-term outcome of Hurler syndrome following bone marrow transplantation. Am. J. Med. Genet. 1993, 46, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E. The first 5 years of clinical experience with laronidase enzyme replacement therapy for mucopolysaccharidosis I. Expert Opin. Pharmacother. 2005, 6, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Laraway, S.; Breen, C.; Mercer, J.; Jones, S.; Wraith, J.E. Does early use of enzyme replacement therapy alter the natural history of mucopolysaccharidosis I? Experience in three siblings. Mol. Genet. Metab. 2013, 109, 315–316. [Google Scholar] [CrossRef] [PubMed]
- Dickson, P.I.; Hanson, S.; McEntee, M.F.; Vite, C.H.; Vogler, C.A.; Mlikotic, A.; Chen, A.H.; Ponder, K.P.; Haskins, M.E.; Tippin, B.L.; et al. Early versus late treatment of spinal cord compression with long-term intrathecal enzyme replacement therapy in canine mucopolysaccharidosis type I. Mol. Genet. Metab. 2010, 101, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; El Hachem, C.; Harchaoui, S.; Ribault, V.; Amiour, M.; Guillot, M.; Maire, I.; Froissart, R.; Guffon-Fouilhoux, N. [Hurler syndrome. Early diagnosis and successful enzyme replacement therapy: A new therapeutic approach. Case report]. Arch. Pediatr. 2008, 15, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hirawat, S.; Northcutt, V.J.; Welch, E.M.; Elfring, G.L.; Hwang, S.; Almstead, N.G.; Ju, W.; Miller, L.L. Phase 1 safety and PK study of PTC124 for nonsense-mutation suppression therapy of cystic fibrosis. Pediatr. Pulmonol. 2004, 38, 248. [Google Scholar]
- Hirawat, S.; Welch, E.M.; Elfring, G.L.; Northcutt, V.J.; Paushkin, S.; Hwang, S.; Leonard, E.M.; Almstead, N.G.; Ju, W.; Peltz, S.W.; et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J. Clin. Pharmacol. 2007, 47, 430–444. [Google Scholar] [CrossRef] [PubMed]
- Peltz, S.W.; Morsy, M.; Welch, E.M.; Jacobson, A. Ataluren as an agent for therapeutic nonsense suppression. Annu. Rev. Med. 2013, 64, 407–425. [Google Scholar] [CrossRef] [PubMed]
- Bedwell, D.M.; Wang, D.; Welch, E.; Keeling, K.M. The nonsense suppression drug PTC124 restored alpha-L-iduronidase activity and reduces glycosaminoglycan accumulation is MPS-IH mice carrying the Idua-W402X mutation. Mol. Genet. Metab. 2015, 114, S20. [Google Scholar] [CrossRef]
- Du, M.; Liu, X.; Welch, E.M.; Hirawat, S.; Peltz, S.W.; Bedwell, D.M. PTC124 is an orally bioavailable compound that promotes suppression of the human Cftr-G542X nonsense allele in a CF mouse model. Proc. Natl. Acad. Sci. USA 2008, 105, 2064–2069. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bonnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation duchenne muscular dystrophy. PLoS ONE 2013, 8, e81302. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.S.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Florence, J.; Eagle, M.; Gappmaier, E.; Glanzman, A.M.; Spiegel, R.; Barth, J.; Elfring, G.; et al. The 6-minute walk test and other clinical endpoints in Duchenne muscular dystrophy: Reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve 2013, 48, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Damoiseaux, R.; Nahas, S.; Gao, K.; Hu, H.; Pollard, J.M.; Goldstine, J.; Jung, M.E.; Henning, S.M.; Bertoni, C.; et al. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J. Exp. Med. 2009, 206, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Kayali, R.; Ku, J.M.; Khitrov, G.; Jung, M.E.; Prikhodko, O.; Bertoni, C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum. Mol. Genet. 2012, 21, 4007–4020. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Ku, J.M.; Du, L.; Hu, H.; Gatti, R.A. Synthesis and evaluation of compounds that induce readthrough of premature termination codons. Bioorg. Med. Chem. Lett. 2011, 21, 5842–5848. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, A.; Lahav, L.; Rosin-Arbesfeld, R. Restoration of APC gene function in colorectal cancer cells by aminoglycoside- and macrolide-induced read-through of premature termination codons. Gut 2010, 59, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Caspi, M.; Firsow, A.; Rajkumar, R.; Skalka, N.; Moshkovitz, I.; Munitz, A.; Pasmanik-Chor, M.; Greif, H.; Megido, D.; Kariv, R.; et al. A flow cytometry-based reporter assay identifies macrolide antibiotics as nonsense mutation read-through agents. J. Mol. Med. (Berl.) 2015, 94, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, M.; Nakayama, Y.; Hara, T.; Shiozuka, M.; Takeda, S.; Kaga, K.; Kondo, S.; Morita, S.; Kitamura, T.; Matsuda, R. Negamycin can restore dystrophin in mdx skeletal muscle. Acta Myol. 2001, 20, 154–158. [Google Scholar]
- Arakawa, M.; Shiozuka, M.; Nakayama, Y.; Hara, T.; Hamada, M.; Kondo, S.; Ikeda, D.; Takahashi, Y.; Sawa, R.; Nonomura, Y.; et al. Negamycin restores dystrophin expression in skeletal and cardiac muscles of mdx mice. J. Biochem. (Tokyo) 2003, 134, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Allamand, V.; Bidou, L.; Arakawa, M.; Floquet, C.; Shiozuka, M.; Paturneau-Jouas, M.; Gartioux, C.; Butler-Browne, G.S.; Mouly, V.; Rousset, J.P.; et al. Drug-induced readthrough of premature stop codons leads to the stabilization of laminin alpha2 chain mRNA in CMD myotubes. J. Gene Med. 2008, 10, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Floquet, C.; Rousset, J.P.; Bidou, L. Readthrough of premature termination codons in the adenomatous polyposis coli gene restores its biological activity in human cancer cells. PLoS ONE 2011, 6, e24125. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Deprez, B.; Lejeune, F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Pibiri, I.; Lentini, L.; Melfi, R.; Gallucci, G.; Pace, A.; Spinello, A.; Barone, G.; Di Leonardo, A. Enhancement of premature stop codon readthrough in the CFTR gene by ataluren (PTC124) derivatives. Eur. J. Med. Chem. 2015, 101, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Moosajee, M.; Tracey-White, D.; Smart, M.; Weetall, M.; Torriano, S.; Kalatzis, V.; da Cruz, L.; Coffey, P.; Webster, A.R.; Welch, E. Functional rescue of REP1 following treatment with PTC124 and novel derivative PTC-414 in human choroideremia fibroblasts and the nonsense-mediated zebrafish model. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gregory-Evans, C.Y. Nonsense suppression therapies in ocular genetic diseases. Cell. Mol. Life Sci. 2015, 72, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Fatscher, T.; Boehm, V.; Gehring, N.H. Mechanism, factors, and physiological role of nonsense-mediated mRNA decay. Cell. Mol. Life Sci. 2015, 72, 4523–4544. [Google Scholar] [CrossRef] [PubMed]
- Khajavi, M.; Inoue, K.; Lupski, J.R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 2006, 14, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Bhuvanagiri, M.; Schlitter, A.M.; Hentze, M.W.; Kulozik, A.E. Nmd: RNA biology meets human genetic medicine. Biochem. J. 2010, 430, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Moriniere, M.; Delhommeau, F.; Calender, A.; Ribeiro, L.; Delaunay, J.; Baklouti, F. Nonsense-mediated mRNA decay (NMD) blockage promotes nonsense mRNA stabilization in protein 4.1r deficient cells carrying the 4.1r coimbra variant of hereditary elliptocytosis. Blood Cells. Mol. Dis. 2010, 45, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Yamashita, A.; Kashima, I.; Higuchi, I.; Osame, M.; Ohno, S. Specific inhibition of nonsense-mediated mRNA decay components, SMG-1 or UPF1, rescues the phenotype of Ullrich disease fibroblasts. Mol. Ther. 2006, 14, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Yamashita, A.; Higuchi, I.; Ohnishi, T.; Shiraishi, T.; Osame, M.; Ohno, S. Inhibition of nonsense-mediated mRNA decay rescues the phenotype in Ullrich’s disease. Ann. Neurol. 2004, 55, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Linde, L.; Boelz, S.; Nissim-Rafinia, M.; Oren, Y.S.; Wilschanski, M.; Yaacov, Y.; Virgilis, D.; Neu-Yilik, G.; Kulozik, A.E.; Kerem, E.; et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J. Clin. Invest. 2007, 117, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, F. Triple effect of nonsense-mediated mRNA decay inhibition as a therapeutic approach for cancer. Single Cell Biol. 2016, 5, 136. [Google Scholar] [CrossRef]
- Mendell, J.T.; Sharifi, N.A.; Meyers, J.L.; Martinez-Murillo, F.; Dietz, H.C. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 2004, 36, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Maquat, L.E. Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: To die or not to die, that is the question. Curr. Opin. Genet. Dev. 2011, 21, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Isken, O.; Maquat, L.E. The multiple lives of NMD factors: Balancing roles in gene and genome regulation. Nat. Rev. Genet. 2008, 9, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, P.S.; Lucy Raymond, F.; Nguyen, L.S.; Rodriguez, J.; Hackett, A.; Vandeleur, L.; Smith, R.; Shoubridge, C.; Edkins, S.; Stevens, C.; et al. Mutations in UPF3b, a member of the nonsense-mediated mrRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet. 2007, 39, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.S.; Kim, H.G.; Rosenfeld, J.A.; Shen, Y.; Gusella, J.F.; Lacassie, Y.; Layman, L.C.; Shaffer, L.G.; Gecz, J. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum. Mol. Genet. 2013, 22, 1816–1825. [Google Scholar] [CrossRef] [PubMed]
- Viegas, M.H.; Gehring, N.H.; Breit, S.; Hentze, M.W.; Kulozik, A.E. The abundance of RNPS1, a protein component of the exon junction complex, can determine the variability in efficiency of the nonsense mediated decay pathway. Nucleic Acids Res. 2007, 35, 4542–4551. [Google Scholar] [CrossRef] [PubMed]
- Seoighe, C.; Gehring, C. Heritability in the efficiency of nonsense-mediated mRNA decay in humans. PLoS ONE 2010, 5, e11657. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.S.; Wilkinson, M.F.; Gecz, J. Nonsense-mediated mRNA decay: Inter-individual variability and human disease. Neurosci. Biobehav. Rev. 2014, 46, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B.A. Enzymes approved for human therapy: Indications, mechanisms and adverse effects. BioDrugs 2015, 29, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2010, 363, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; de Boeck, K.; Flume, P.A.; et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del CFTR. N. Engl. J. Med. 2015, 373, 1783–1784. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less is more: Substrate reduction therapy for lysosomal storage disorders. Int. J. Mol. Sci. 2016, 17, 1065. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological chaperone therapy: Preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Year | LSD | Gene | Model | Mutation | Drug | Major Findings | Reference |
|---|---|---|---|---|---|---|---|
| 2001 | MPS I-H | IDUA | -Cell-free translation system -Cultured patient fibroblasts | -Q70X (UAG) -W402X (UAG) | -Gentamicin | -Q70X more susceptible to RT than W402X in cell free system -3% of normal enzyme activity restored in fibroblasts -GAG storage normalized -Lysosomal proliferation normalized | Keeling et al. [57] |
| 2001 | Infantile neuronal ceroid lipofuscinosis | TPP1 or CLN2 | -Cultured patient fibroblasts | -Q66X (UAG) -R127X (UGA) -R208X (UGA) | -Gentamicin | -7% of normal enzyme activity restored for R127X allele -0.5% restored for R208X allele -None restored at Q66X allele | Sleat et al. [58] |
| 2002 | Cystinosis | CTNS | -Cultured patient fibroblasts -Reporter in HEK293 cells | W138X (UGA) | -Gentamicin | -Cysteine levels significantly reduced compared to controls -Detection of reporter protein in the presence of drug | Helip-Wooley et al. [59] |
| 2002 | MPS I-H | IDUA | -Cell-free translation system | W402X (UAG) | -Amikacin -Gentamicin -Tobramycin | -Tobramycin least effective -W402X context more resistant to RT than other UAG contexts | Keeling et al. [60] |
| 2004 | MPS I-H | IDUA | -Cultured patient fibroblasts -cDNAs in CHO-K1 cells | -Q70X (UAG) -W180X (UGA) -Y343X (UAG) -Q400X (UAG) -W402X (UAG) -R628X (UGA) | -Gentamicin | -Significant increases in enzyme activity observed in all treated fibroblasts except Y343X -All CHO-K1 cell lines responded with an increase in enzyme activity (UGA>UAG>UAA) | Hein et al. [61] |
| 2009 | MPS I-H | IDUA | -Cell-free translation system | -Q70X (UAG) -W402X (UGA) | -Gentamicin -NB30 -NB54 -Paramomycin | -NB30 & NB54 more effective at suppressing PTCs than paramomycin & gentamicin -New drugs shown to be less toxic than traditional aminoglycosides | Nudelman et al. [62] |
| 2010 | MPS I-H | IDUA | -Cell-free translation system | Q70X (UAG) | -Gentamicin -NB30 -NB54 -NB74 -NB84 | -Novel aminoglycoside derivative NB84 is most effective at suppressing PTC -NB84 also less toxic than traditional aminoglycosides | Nudelman et al. [63] |
| 2011 | Infantile neuronal ceroid lipofuscinosis | PPT1 or CLN1 | -Cultured patient fibroblasts -Cultured patient lymphoblasts | -L10X (UAG) -R151X (UGA) -R164X (UGA) -Q291X (UAG) | -Gentamicin -PTC124 | -Both drugs restore enzyme activity in fibroblasts (~1%) and lymphoblasts (~0.3%) -Cell toxicity observed with gentamicin, but not PTC124 -PTC124 treatment restored full-length PPT1 protein, decreased ceroid levels & granular deposits, suppressed apoptosis | Sarkar et al. [64] |
| 2011 | MPS I-H | IDUA | -Cell-free translation system -Reporter in HEK293 cells | Q70X (UAG) | -Gentamicin -NB30 -NB54 -Other NB derivatives | -New aminoglycoside derivatives more effective than gentamicin at suppressing Q70X -Derivatives show less cell toxicity than gentamicin | Kandasamy et al. [65] |
| 2012 | MPS I-H | IDUA | -Cell-free translation system | Q70X (UAG) | -G418 -Gentamicin -NB124 -Other NB derivatives | -New synthetic aminoglycosides suppressed the Q70X mutation much more effectively than gentamicin or G418 -Derivatives showed less cell toxicity than traditional aminoglycosides | Kandasamy et al. [66] |
| 2012 | MPS I-H | IDUA | -Idua-W402X mouse | W402X (UAG) mouse locus | -Amikacin -G418 -Gentamicin -* NB54 -* NB84 -Paramomycin * 30 mg/kg administered SQ once for 2 weeks | -MEF studies showed that NB84 and NB54, are more effective at suppressing W402X than traditional aminoglycosides tested (more enzyme; lower GAGs; improved lysosomal morphology) -2-week in vivo treatment with NB54 and NB84 showed while both drugs led to a significant GAG decrease in multiple tissues (spleen, heart, brain) -NB84 was more efficient. -While brain GAGs were reduced, brain GM2 and GM3 gangliosides were not. | Wang et al. [67] |
| 2012 | MPS VI | ARSB | -cultured patient fibroblasts | -R315X (UGA) -R327X (UGA) -Q456X (UAA) -Q503X (UAG) | -Gentamicin -PTC124 | -No increase in enzyme activity observed with gentamicin treatment -Significant increase in enzyme activity observed with PTC124 treatment in all cells with the exception of the Q503X cell line -Lysosome size reduced with PTC124 treatment -ARSB protein undetectable by western blotting | Bartolomeo et al. [68] |
| 2013 | MPS I-H | IDUA | Idua-W402X mouse | W402X (UAG) mouse locus | -Gentamicin -NB84 NMD inhibitors: -Caffeine -NMDI-1 (30 mg/mL gentamicin administered SQ once daily for 14 days +/− 5 mg/mL NMDI-1 administered SQ once daily on days 12–14 | -Combining NMD inhibitors NMD-1 or caffeine with either gentamicin or NB84 enhanced enzyme activity and GAG reduction in MEFs -NMDI-1 also enhanced the ability of gentamicin to restore enzyme activity, normalize lysosome enzyme proliferation, and reduce GAG accumulation in the brain and spleen -No ill effects were observed with NMDI-1 administration | Keeling et al. [69] |
| 2014 | MPS I-H | IDUA | -Idua-W402X mouse | W402X (UAG) mouse locus | -NB84 (30 mg/kg administered SQ twice weekly for 28 weeks) | -Significant increase in enzyme activity in multiple tissues (more activity obtained when treatment was initiated early) -Tissue GAG accumulation reduced -neuroinflammation reduced -Improved heart morphology & function -Improved bone morphology -Improved activity levels | Gunn et al. [70] |
| 2015 | Infantile neuronal ceroid lipofuscinosis | PPT1 or CLN1 | -Cln1-R151X mouse | R151X (UGA) mouse locus | PTC124 (10 mg/kg administered 4 times daily for 2 days) | Significant increase in PPT1 activity in liver and muscle, but not in brain, heart, lung, or kidney | Thada et al. [71] |
| 2015 | a MPS VI b MPS IIIB c MPS IIIC d Niemann-Pick A/B | a ARSB b NAGLU c HGSNAT d SMPD1 | -Cultured patient fibroblasts -Cell-free translation system -cDNAs in COS7 cells | -a W146X (UGA) -a W322X (UGA) -a Q503X(UAG) -b W168X (UAG) -b Q566X (UAG) -c R203X (UGA) -c R384X (UGA) -c W403X (UGA) -d W168X (UAG) -d Y313X (UAA) -d R441X (UGA) | -BZ6 -BZ16 -G418 -Gentamicin -PTC124 -RTC13 -RTC14 | -1%-4% of WT enzyme levels measured in W322X fibroblasts treated with gentamicin & ARSB protein detected by immunofluorescence -Enzyme restoration was not observed in MPS IIIB and IIIC fibroblasts with any of the drugs, but an increase in mRNA abundance was detected -Cell free systems showed RT stimulation of the three SMDP1 mutations with G418 and gentamicin, but not the other drugs -RT was also observed with the HGSNAT mutations -No RT was observed with the ARSB mutations -In transfected COS7 cells G418, gentamicin, and PTC124 induced RT of the W146X, W168X, and Y313X mutations. | Gomez-Grau et al. [72] |
| 2016 | MPS I-H | IDUA | -Cell-free translation system | Q70X (UAG) | -Gentamicin -NB74 -NB124 -NB156 -NB157 | -New synthetic compounds NB156 and NB157 were compared to their parent compounds and gentamicin. -Both new compounds showed an improved ability to suppress the Q70X mutation compared to the parent compounds and gentamicin. | Sabbavarapu et al. [73] |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keeling, K.M. Nonsense Suppression as an Approach to Treat Lysosomal Storage Diseases. Diseases 2016, 4, 32. https://doi.org/10.3390/diseases4040032
Keeling KM. Nonsense Suppression as an Approach to Treat Lysosomal Storage Diseases. Diseases. 2016; 4(4):32. https://doi.org/10.3390/diseases4040032
Chicago/Turabian StyleKeeling, Kim M. 2016. "Nonsense Suppression as an Approach to Treat Lysosomal Storage Diseases" Diseases 4, no. 4: 32. https://doi.org/10.3390/diseases4040032
APA StyleKeeling, K. M. (2016). Nonsense Suppression as an Approach to Treat Lysosomal Storage Diseases. Diseases, 4(4), 32. https://doi.org/10.3390/diseases4040032