
A Prospective Treatment Option for Lysosomal Storage Diseases: CRISPR/Cas9 Gene Editing Technology for Mutation Correction in Induced Pluripotent Stem Cells

Abstract
:1. Lysosomal Storage Diseases
1.1. Current LSD Treatments
1.2. Drawbacks to Current LSD Treatments
2. iPSCs: Autologous Stem Cell Transplantation
3. CRISPR/Cas9 Gene Editing
3.1. Current Gene Editing Systems
3.2. CRISPR/Cas9: Molecular Design
4. Proof of Concept Studies: CRISPR/Cas9 for Correction of Genetic Disease
4.1. Compound Heterozygous Mutation Correction in β-Thalassemia
4.2. Mutation Correction in Other Inherited Monogenic Diseases
5. Clinical Potential, Prospective Applications, and Challenges Using CRISPR/Cas9
5.1. CRISPR/Cas9 in Clinical Trials
5.2. Off-Targeting Risks: CRISPR/Cas9 Mechanistic Hurdles
5.3. Suppression of NHEJ and Induction of HDR
5.4. CRISPR/Cas9 Application to LSDs
5.5. LSD-Specific Gene Editing Hurdles
6. Conclusions
Acknowledgment
Conflicts of Interest
References
- Harlan, F.K.; Lusk, J.S.; Mohr, B.M.; Guzikowski, P.; Batchelor, R.H.; Jiang, Y.; Naleway, J.J. Fluorgenic substrates for visualizing acidic organelle enzyme activities. PLoS ONE 2016, 11, e0156312. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Meikle, P.J.; Hopwood, J.J. Epidemiology of lysosomal storage diseases: An overview. In Fabry Disease: Perspective from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; Chapter 2. [Google Scholar]
- Dahl, S.; Mengel, E. Lysosomal storage diseases as differential diagnosis of hepatosplenomegaly. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Bedini, G.; Parati, E.A.; Bersano, A. Fabry Disease: Recognition; diagnosis; and treatment of neurological features. Curr. Treat. Options Neurol. 2016, 18, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Staretz-Chacham, O.; Lang, T.C.; LaMarca, M.E.; Krasnewich, D.; Sidransky, E. Lysosomal storage disorders in the newborn. Pediatrics 2009, 123, 1191–1207. [Google Scholar] [CrossRef] [PubMed]
- Mueller, P.; Jost, C.A.; Rohrbach, M.; Buechel, E.V.; Seifert, B.; Balmer, C.; Kretschmar, O.; Baumgartner, M.R.; Weber, R. Cardiac disease in children and young adults with various lysosomal storage diseases: Comparison of echocardiography and ECG changes among clinical groups. IJC Heart Vessels 2014, 2, 1–7. [Google Scholar] [CrossRef]
- Pastores, G.M. Musculoskeletal complications encountered in the lysosomal storage disorders. Best Pract. Res. Clin. Rheumatol. 2008, 22, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Goldman, S.M. Epidemiology of Parkinson’s disease. Neurol. Clin. 1996, 14, 317–335. [Google Scholar] [CrossRef]
- Foo, J.N.; Liany, H.; Bei, J.X.; Yu, X.Q.; Liu, J.; Au, W.L.; Prakash, K.M.; Tan, L.C.; Tan, E.K. Rare lysosomal enzyme gene smpd1 variant (p.R591c) associates with Parkinson’s disease. Neurobiol. Aging 2013, 34, 2890.e13–2890.e15. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Ozelius, L.J.; Bar-Shira, A.; Saunders-Pullman, R.; Mirelman, A.; Kornreich, R.; Gana-Weisz, M.; Raymond, D.; Rozenkrantz, L.; Deik, A.; et al. The p.L302p mutation in the lysosomal enzyme gene smpd1 is a risk factor for Parkinson disease. Neurology 2013, 80, 1606–1610. [Google Scholar] [CrossRef] [PubMed]
- Kolodny, E.H.; Ullman, M.D.; Mankin, H.J.; Raghavan, S.S.; Topol, J.; Sulivan, J.L. Phenotypic manifestations of Gaucher disease: Clinical features in 48 biochemically verified type I patients and comment on type II patients. Prog. Clin. Biol. Res. 1982, 95, 33–65. [Google Scholar] [PubMed]
- Simonaro, C.M.; Desnick, R.J.; McGovern, M.M.; Wasserstein, M.P.; Schuchman, E.H. The demographics and distribution of type B Niemann-Pick disease: Novel mutations lead to new genotype/phenotype correlations. Am. J. Hum. Genet. 2002, 71, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Landels, E.C.; Ellis, I.H.; Fensom, A.H.; Green, P.M.; Bobrow, M. Frequency of the Tay-Sachs disease splice and insertion mutations in the UK Ashkenazi Jewish population. J. Med. Genet. 1991, 28, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, L.; Dong, J.; Desnick, R.J.; Kornreich, R. Carrier screening for mucolipidosis type IV in the American Ashkenazi Jewish population. Am. J. Hum. Genet. 2002, 70, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Van Gelder, C.M.; Vollebregt, A.A.M.; Plug, I.; van der Ploeg, A.T.; Reuser, A.J.J. Treatment options for lysosomal storage disorders: Developing insights. Expert Opin. Pharmacother. 2012, 13, 2281–2299. [Google Scholar] [CrossRef] [PubMed]
- Narita, A.; Shirai, K.; Itamura, S.; Matsuda, A.; Ishihara, A.; Matsushita, K.; Fukuda, C.; Kubota, N.; Takayama, R.; Shigematsu, H.; et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study. Ann. Clin. Transl. Neurol. 2016, 3, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C.; Vecchio, D.; Prady, H.; Abel, L.; Wraith, J.E. Miglustat for treatment of Niemann-Pick C disease: A randomized controlled study. Lancet Neurol. 2007, 6, 765–772. [Google Scholar] [CrossRef]
- Machaczka, M.; Hast, R.; Dahlman, I.; Lerner, R.; Klimkowska, M.; Engvall, M.; Hagglund, H. Substrate reduction therapy with miglustat for type 1 Gaucher disease: A retrospective analysis from a single institution. Upsala J. Med. Sci. 2012, 117, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Jeyakumar, M. Substrate reduction therapy. Acta Paediatr. 2008, 97, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Hawkins-Salsbury, J.A.; Reddy, A.S.; Sands, M.S. Combination therapies for lysosomal storage disease: Is the whole greater than the sum of its parts? Hum. Mol. Genet. 2011, 20, R54–R60. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Sphingolipid lysosomal storage disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Sands, M.S.; Davidson, B.L. Gene therapy for lysosomal storage diseases. Mol. Ther. 2006, 13, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Ashton, L.J.; Brooks, D.A.; McCourt, P.A.; Muller, V.J.; Clements, P.R.; Hopwood, J.J. Immunoquantification and enzyme kinetics of alpha-l-iduronidase in cultured fibroblasts from normal control and mucopolysaccharidosis type I patients. Am. J. Hum. Genet. 1992, 50, 787–794. [Google Scholar] [PubMed]
- Krause, D.S.; Theise, N.D.; Collector, M.I.; Henegariu, O.; Hwang, S.; Gardner, R.; Neutzel, S.; Sharkis, S.J. Multi-organ; multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 2001, 105, 369–377. [Google Scholar] [CrossRef]
- Priller, J.; Flugel, A.; Wehner, T.; Boentert, M.; Haas, C.A.; Prinz, M.; Fernandez-Klett, F.; Prass, K.; Bechmann, I.; de Boer, B.A.; et al. Targeting gene-modified hematopoietic cells to the central nervous system: Use of green fluorescent protein uncovers microglial engraftment. Nat. Med. 2011, 7, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, T.G.; da Silveira Matte, U.; Giugliani, R.; Baldo, G. Genome editing: Potential treatment for lysosomal storage disease. Curr. Stem Cell Rep. 2015, 1, 9–15. [Google Scholar] [CrossRef]
- Lund, T.C.; Cathey, S.S.; Miller, W.P.; Eapen, M.; Andreansky, M.; Dvorak, C.C.; Davis, J.H.; Dalal, J.D.; Devine, S.M.; Eames, G.M.; et al. Outcomes after hematopoietic stem cell transplantation for children with I-Cell disease. Biol. Blood Marrow Transplant. 2014, 20, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T. Emerging therapies and therapeutic concepts for lysosomal storage diseases. Expert Opin. Orphan Drugs 2013, 1, 385–404. [Google Scholar] [CrossRef]
- Rastall, D.P.; Amalfitano, A. Recent advances in gene therapy for lysosomal storage disorders. Appl. Clin. Genet. 2015, 8, 157–169. [Google Scholar] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of scid-x1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Smid, B.E.; Ferraz, M.J.; Verhoek, M.; Mirzanian, M.; Wisse, P.; Overkleeft, H.S.; Hollak, C.E.; Aerts, J.M. Biochemical response to substrate reduction therapy versus enzyme replacement therapy in Gaucher disease type 1 patients. Orphanet J. Rare Dis. 2016, 11, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.W.; Furbish, F.S.; Murray, G.J.; Garfield, M.; Brady, R.O. Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc. Natl. Acad. Sci. USA 1990, 87, 1913–1916. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: A report from the Gaucher Registry. Am. J. Med. 2002, 118, 112–119. [Google Scholar] [CrossRef]
- Ibrahim, J.; Underhill, L.H.; Taylor, J.S.; Angell, J.; Peterschmitt, J. Clinical response to eliglustat in treatment-naïve patients with Gaucher disease type 1: Post-hoc comparison to imiglucerase-treated patients enrolled in the International Collaborative Gaucher Group Gaucher Registry. Mol. Genet. Metab. Rep. 2016, 8, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Altarescu, G.; Elstein, D. Pilot study using ambroxol as a pharamacological chaperone in type 1 Gaucher disease. Blood Cells Mol. Dis. 2013, 50, 134–137. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, J.; Valstar, M.J.; Narajczyk, M.; Kulik, W.; Ljlst, L.; Wagemans, T.; van der Wal, W.M.; Wijburg, F.A. Genistein in Sanfilippo disease: A randomized controlled crossover trial. Ann. Neurol. 2012, 71, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Banikazemi, M.; Gordon, R.E.; Goldman, M.; Phelps, R.; Kim, L.; Glass, A.; Winston, J.; Dikman, S.; Fallon, J.T. A phase ½ clinical trial of enzyme replacement in Fabry disease: Pharmacokinetic; substrate clearance; and safety studies. Am. J. Hum. Genet. 2012, 68, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Charrow, J.; Desnick, R.J.; Guffon, N.; Kempf, J.; Lachmann, R.H.; Lemay, R.; Linthorst, G.E.; Packman, S.; Scott, C.R. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J. Med. Genet. 2015, 52, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Guce, A.I.; Clark, N.E.; Salgado, E.N.; Ivanen, D.R.; Kulminskaya, A.A.; Brumer, H., III; Garman, S.C. Catalytic mechanism of human alpha-galactosidase. J. Biol. Chem. 2009, 285, 3625–3632. [Google Scholar] [CrossRef] [PubMed]
- Dersh, D.; Iwamoto, Y.; Argon, Y. Tay Sachs disease mutations in HEXA target the alpha chain of hexosaminidase A to ER-associated degradation. Mol. Biol. Cell 2016, 27, 3813–3827. [Google Scholar] [CrossRef] [PubMed]
- Hartung, J.; Potschick, K.; Kunz, J.; Kalache, K.; Braulke, T. P44.13: Prenatal diagnosis of mucolipidosis II using genetic testing. Ultrasound Obsterics Gynecol. 2007, 30, 621. [Google Scholar] [CrossRef]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2001, 290, 2298–2301. [Google Scholar] [CrossRef] [PubMed]
- Zech, M.; Nubling, G.; Castrop, F.; Jochim, A.; Schulte, E.C.; Mollenhauer, B.; Lichtner, P.; Peters, A.; Gieger, C.; Marquardt, T.; et al. Niemann-Pick C disease gene mutations and age-related neurodegenerative disorders. PLoS ONE 2013, 8, e82879. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Rojas, V.M.; Martins, A.M.; Valadares, E.R.; Clarke, J.T.R.; Goes, J.E.C.; Kakkis, E.D.; Worden, M.A.; Sidman, M.; Cox, G.F. A dose-optimization trial of laronidase (Aldurazyme) patients with mucopolysaccharidosis I. Mol. Genet. Metab. 2009, 96, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.B.; Cho, S.Y.; Park, S.W.; Kim, S.J.; Ko, A.R.; Kwon, E.K.; Han, S.J.; Jin, D.K. Phase I/II clinical trial of enzyme replacement therapy with idursulfase beta in patients with mucopolysaccharidosis II (Hunter Syndrome). Orphanet J. Rare Dis. 2013, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Braunlin, E.; Rosenfeld, H.; Kampmann, C.; Johnson, J.; Beck, M.; Giugliani, R.; Guffon, N.; Scarpa, M.; Schwartz, I.V.; Leao Teles, E.; et al. Enzyme replacement therapy for mucopolysaccharidosis VI: Long-term effects of galsulfase (Naglazyme) therapy. J. Inherit. Metab. Dis. 2013, 36, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Lu, I.L.; Yin, C.Y.; Lin, S.B.; Chen, S.T.; Yeh, L.Y.; Yang, F.Y.; Au, L.C. Correction/mutation of acid alpha-d-glucosidase gene by modified single-stranded oligonucleotides: In vitro and in vivo studies. Gene Ther. 2003, 10, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Merk, T.; Wibmer, T.; Schumann, C.; Kruger, S. Enzyme replacement therapy in Pompe’s disease. Med. Klin. (Munich) 2007, 102, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H.; Desnick, R.J. Types a and b niemann-pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H.; Levran, O.; Pereira, L.V.; Desnick, R.J. Structural organization and complete nucleotide sequence of the gene encoding human acid sphingomyelinase (smpd1). Genomics 1992, 12, 197–205. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Ohm, T.G.; Treiber-Held, S.; Distl, R.; Glockner, F.; Schonheit, B.; Tamanai, M.; Meske, V. Cholesterol and tau protein—Findings in Alzheimer’s and Niemann-Pick C’s disease. Pharmacopsychiatry 2003, 36, S120–S126. [Google Scholar] [PubMed]
- Yang, J.; Li, S.; He, X.B.; Cheng, C.; Le, W. Induced pluripotent stem cells in Alzheimer’s disease: Applications for disease modeling and cell-replacement therapy. Mol. Neurodegener. 2016, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Byers, D.; Lee, H.L.; Reijo, P.R. Modeling Parkinson’s disease using induced pluripotent stem cells. Curr. Neurol. Neurosci. Rep. 2012, 12, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Wernig, M.; Markoulaki, S.; Sun, C.W.; Meissner, A.; Cassady, J.P.; Beard, C.; Brambrink, T.; Wu, L.C.; Townes, T.M.; et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 2007, 318, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-derived oligodendrocyte progenitors can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Shihabuddin, L.S.; Numan, S.; Huff, M.R.; Dodge, J.C.; Clarke, J.; Macauley, S.L.; Yang, W.; Taksir, T.V.; Parsons, G.; Passini, M.A.; et al. Intracerebral transplantation of adult mouse neural progenitor cells into the niemann-pick-a mouse leads to a marked decrease in lysosomal storage pathology. J. Neurosci. 2004, 24, 10642–10651. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Ye, L.; Chang, J.C.; Beyer, A.I.; Wang, J.; Muench, M.O.; Kan, Y.W. Seamless gene correction of beta-thalassemia mutations in patient specific iPSCs using CRISPR/Cas9 and piggybac. Genome Res. 2014, 24, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.R.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liang, D.; Wang, Y.; Bai, M.; Tang, W.; Bao, S.; Yan, Z.; Li, D.; Li, J. Correction of a genetic disease in mouse via use of CRISPR/Cas9. Cell Stem Cell 2013, 13, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; Fisher, J.; O’Rourke, K.P.; Muley, A.; Kastenhuber, E.R.; Livshits, G.; Tschaharganeh, D.F.; Socci, N.D.; Lowe, S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Ma, Y.; Li, Q.; Sun, Z.; Ma, L.; Wu, L.; Wang, L.; Zeng, L.; Shao, Y.; Chen, Y.; et al. CRISPR/Cas9—Mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol. Med. 2016, 8, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Hoban, M.; Lumaquin, D.; Kuo, C.Y.; Romero, Z.; Long, J.; Ho, M.; Young, C.S.; Mojadidi, M.; Fitz-Gibbon, S.; Cooper, A.R.; et al. CRISPR/Cas9-mediated correction of the sickle mutation in human CD34+ cells. Mol. Ther. 2016, 24, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Belfort, M.; Bonocora, R.P. Homing endonucleases: From genetic anomalies to programmable genomic clippers. Methods Mol. Biol. 2015, 1123, 1–26. [Google Scholar]
- Isalan, M. Zinc-finger nucleases: How to play two good hands. Nat. Methods 2012, 9, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C.; Rosenberg, J.M.; Rich, A. Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl. Acad. Sci. USA 1976, 73, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 2579–2586. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.K.; Haber, J.E. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair f double strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Szostak, J.W.; Orr-Weaver, T.L.; Rothstein, R.J. The double-strand-break repair model for recombination. Cell 1983, 33, 25–35. [Google Scholar] [CrossRef]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR/Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Drousiotou, A.; Ionanou, P.; Georgiou, T.; Mavrikiou, E.; Christopoulos, G.; Kyriakides, T.; Voyasianos, M.; Argyriou, A.; Middleton, L. Neonatal screening for Duchenne muscular dystrophy: A novel semiquantitative application of the bioluminescence test for creatine kinase in a pilot national program in Cyprus. Genet. Test. 1998, 2, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Batsevich, V.V.; Lee, Y.L.; et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. NEJM 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S. First CRISPR clinical trial gets green light from US panel. Nat. News 2016. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. NCT02863913; NCT02867345; NCT02867332; NCT02793856. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02793856?term=crispr&rank=4 (accessed on 27 November 2016).
- Kim, E.; Kim, S.; Kim, D.H.; Choi, B.S.; Choi, I.Y.; Kim, J.S. Precision genome engineering with programmable DNA-nicking enzymes. Genome Res. 2012, 22, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinsten, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Wi, X.; Wang, J.; Wang, Y.; Qiu, Z.; Chang, T.; Huang, H.; Lin, R.J.; Yee, J.K. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 2015, 33, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 54, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.; Hungerford, D. A minute chromosome in human chronic granulocytic leukemia. Science 1960, 132, 1497. [Google Scholar]
- Bishop, M.J. Molecular themes in oncogenesis. Cell 1991, 64, 235–248. [Google Scholar] [CrossRef]
- Tomlinson, I.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Rahman, S.; Roylance, R.R.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids; skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.; Wu, X.; Jiang, W.; Marrffini, L.A.; Zhang, F. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Li, A.Y.J.; Boo, L.M.; Wang, S.Y.; Lin, H.H.; Wang, C.C.C.; Yen, Y.; Chen, B.P.C.; Chen, D.J.; Ann, D.K. Suppression of non-homologous end joining repair by overexpressing of HMGA2. Cancer Res. 2009, 69, 5699–5706. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRIPSR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.B.; Boix, C.; Charpentier, M.; de Cian, A.; Cochennec, J.; Duvernois-Berthet, E.; Perrouault, L.; Tesson, L.; Edouard, J.; Thinard, R.; et al. Improved genome editing efficiency and flexibility using modified oligonucleotides with talen and crispr-cas9 nucleases. Cell. Rep. 2016, 14, 2263–2272. [Google Scholar] [CrossRef] [PubMed]
- Mianne, J.; Chessum, L.; Kumar, S.; Aguilar, C.; Codner, G.; Hutchison, M.; Parker, A.; Mallon, A.M.; Wells, S.; Simon, M.M.; et al. Correction of the auditory phenotype in C57BL/6N mice via CRISPR/Cas9-mediated homology directed repair. Genome Med. 2016, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fedele, A.O. Sanfilippo syndrome: Causes, consequences, and treatments. Appl. Clin. Genet. 2015, 8, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Van der Ploeg, A.T.; Reuser, A.J. Pompe’s disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Van Til, N.P.; Stok, M.; Aerts Kaya, F.S.; de Waard, M.C.; Farahbakhshian, E.; Visser, T.P.; Kroos, M.A.; Jacobs, E.H.; Willart, M.A.; van der Wegen, P.; et al. Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the pompe disease phenotype. Blood 2010, 115, 5329–5337. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Arias, R.; Calafell, F.; Mateu, E.; Comas, D.; Andres, A.; Bertranpetit, J. Sequence variability of a human pseudogene. Genome Res. 2001, 11, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Long, G.L.; Winfield, S.; Adolph, K.W.; Ginns, E.I.; Bornstein, P. Structure and organization of the human metaxin gene (MTX) and pseudogene. Genomics 1996, 33, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.C.; Komiya, T.; Bergman, B.E.; Mihara, K.; Bornstein, P. Metaxin is a component of a preprotein import complex in the outer membrane of the mammalian mitochondrion. J. Biol. Chem. 1997, 272, 6510–6518. [Google Scholar] [CrossRef] [PubMed]
- Lacorazza, H.D.; Flax, J.D.; Snyder, E.Y.; Jendoubi, M. Expression of human beta-hexosaminidase alpha-subunit gene (the gene defect of Tay-Sachs disease) in mouse brains upon engraftment of transduced progenitor cells. Nat. Med. 1996, 2, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; Emiliani, C.; Tancini, B.; Severini, G.M.; Chigorno, V.; Bordignon, C.; Sonnino, S.; Orlacchio, A. Absence of metabolic cross-correction in Tay-Sachs cells: Implications for gene therapy. J. Biol. Chem. 2002, 277, 20177–20184. [Google Scholar] [CrossRef] [PubMed]
- Sinici, I.; Yonekawa, S.; Tkachyova, I.; Gray, S.J.; Samulski, R.J.; Wakarchuk, W.; Mark, B.L.; Mahuran, D.J. In cellulo examination of a beta-alpha hybrid construct of beta-hexosaminidase a subunits, reported to interact with the GM2activator protein and hydrolyze GM2ganglioside. PLoS ONE 2013, 8, e57908. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Tamura, T.; Tsuji, D.; Dohzono, Y.; Kitakaze, K.; Ohno, K.; Saito, S.; Sakuraba, H.; Itoh, K. Therapeutic potential of intracerebroventricular replacement of modified human β-hexosaminidase b for GM2gangliosidosis. Mol. Ther. 2011, 19, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Cachón-González, M.B.; Wang, S.Z.; Ziegler, R.; Cheng, S.H.; Cox, T.M. Reversibility of neuropathology in Tay-Sachs-related diseases. Hum. Mol. Genet. 2014, 23, 730–748. [Google Scholar] [CrossRef] [PubMed]
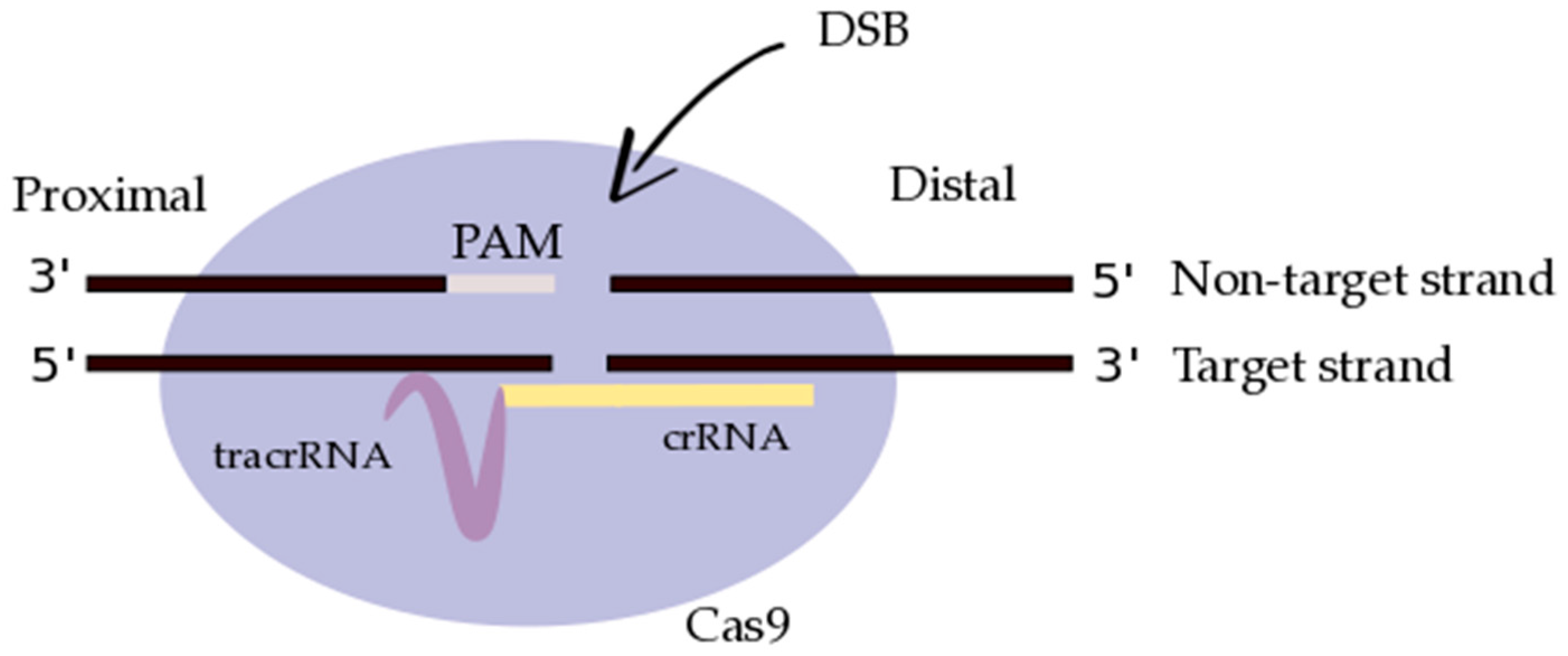

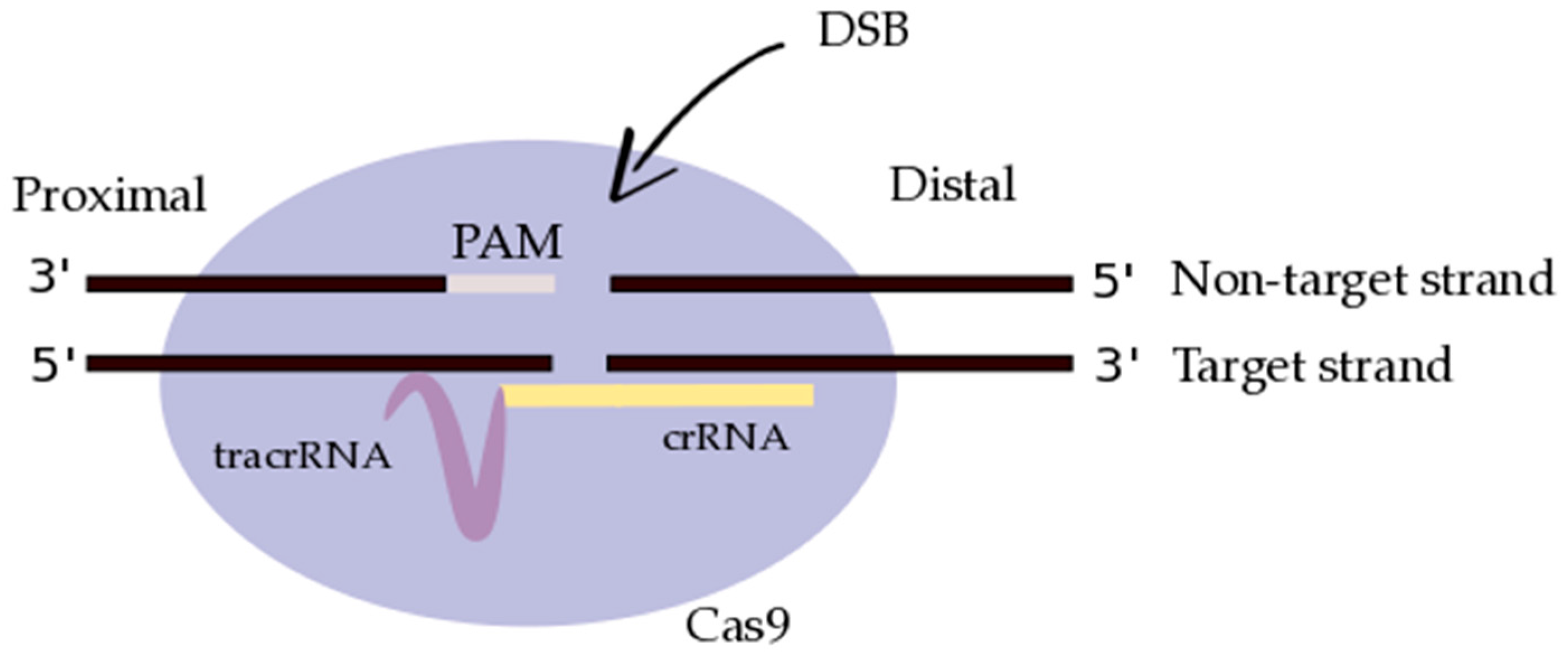{kind=link}
| LSD | Gene Affected | Current Treatment Options * | Example Drugs Available (Drug, Company) | Overall CRISPR/Cas9 Suitability (+/−) | References | |
|---|---|---|---|---|---|---|
| Gaucher Disease (GD) | GBA1 | ERT; SRT; PCT | Ceredase®, Genzyme (Cambridge, MA, USA); Cerdelga®, Genzyme (Cambridge, MA, USA); Mucoslovan®; Boehringer Ingelheim (Biberach, Germany) | + † | [23,33,34,35,36,37] | |
| Sanfilippo Syndrome (MPS III) | A | SGSH | SRT | Genistein ‡ | + | |
| B | Naglu | [38] | ||||
| C | HGSNAT | |||||
| D | GNS | |||||
| Fabry | alpha-Gal A | ERT | Fabrazyme®, Genezyme (Cambridge, MA, USA) | + | [39,40,41] | |
| Tay Sachs | HexA | - | - | − | [42] | |
| I-cell disease | GNPTAB | - | - | − § | [43] | |
| Niemann-Pick C Disease (NPC) | NPC1 or NPC2 | SRT | Zavesca®, Actelion (Allschwil, Switzerland) | + | [19,44,45,46] | |
| MPS I | IDUA | ERT | Aldurazyme®, Genzyme (Cambridge, MA, USA) | + | [47] | |
| MPS II | IDS | ERT | Hunterase®, CytoBioteck (Bogota, Colombia) | + | [48] | |
| MPS VI | ARSB | ERT | Naglazyme®, Biomarin (San Rafael, CA, USA) | + | [49] | |
| Pompe disease | GAA | ERT | Myozyme® Genzyme (Cambridge, MA, USA) | + | [50,51] | |
| Niemann-Pick A disease | SMPD1 | - | - | + | [52,53] | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christensen, C.L.; Choy, F.Y.M. A Prospective Treatment Option for Lysosomal Storage Diseases: CRISPR/Cas9 Gene Editing Technology for Mutation Correction in Induced Pluripotent Stem Cells. Diseases 2017, 5, 6. https://doi.org/10.3390/diseases5010006
Christensen CL, Choy FYM. A Prospective Treatment Option for Lysosomal Storage Diseases: CRISPR/Cas9 Gene Editing Technology for Mutation Correction in Induced Pluripotent Stem Cells. Diseases. 2017; 5(1):6. https://doi.org/10.3390/diseases5010006
Chicago/Turabian StyleChristensen, Chloe L., and Francis Y. M. Choy. 2017. "A Prospective Treatment Option for Lysosomal Storage Diseases: CRISPR/Cas9 Gene Editing Technology for Mutation Correction in Induced Pluripotent Stem Cells" Diseases 5, no. 1: 6. https://doi.org/10.3390/diseases5010006
APA StyleChristensen, C. L., & Choy, F. Y. M. (2017). A Prospective Treatment Option for Lysosomal Storage Diseases: CRISPR/Cas9 Gene Editing Technology for Mutation Correction in Induced Pluripotent Stem Cells. Diseases, 5(1), 6. https://doi.org/10.3390/diseases5010006